I. INTRODUCTION
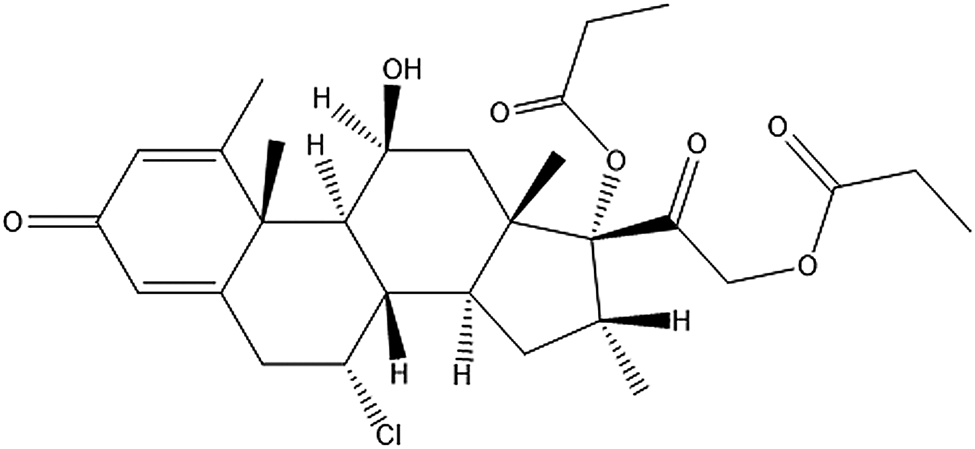
Alclometasone dipropionate (original brand name: Aclovate; generic names: Aclosone, Almeta, Delonal, Legederm, Modrasone, Perderm, etc.) is a synthetic steroid used for topical dermatological applications. It is a prodrug, meaning after being administered, it metabolizes into a pharmacologically active drug. Like other topical corticosteroids, alclometasone dipropionate has anti-inflammatory, anti-itching, and vasoconstrictive properties to treat skin dermatoses such as dermatitis (allergic, contact, actinic, etc.), eczema, and psoriasis. Alclometasone dipropionate is insoluble in water and is applied to the skin as a cream (propylene glycol-based solvent) or an ointment (hexylene glycol-based solvent). The IUPAC name (CAS Registry number 66734-13-2) is [2-[(7R,8S,9S,10R,11S,13S,14S,16R,17R)-7-chloro-11-hydroxy-10,13,16-trimethyl-3-oxo-17-propanoyloxy-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthren-17-yl]-2-oxoethyl] propanoate. A two-dimensional molecular diagram is shown in Figure 1.
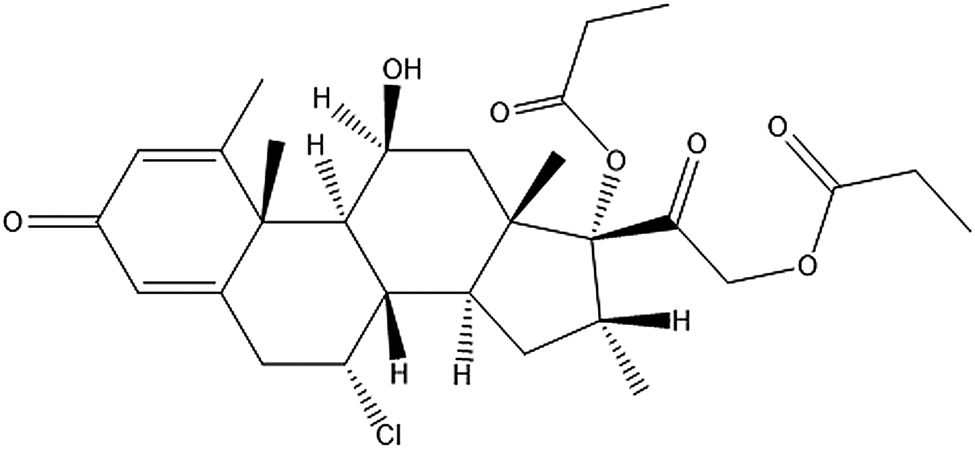
Figure 1. Molecular structure of alclometasone dipropionate.
This work was carried out as a part of a project (Kaduk et al., Reference Kaduk, Crowder, Zhong, Fawcett and Suchomel2014) to determine the crystal structures of large-volume commercial pharmaceuticals and include high-quality powder diffraction data for these pharmaceuticals in the Powder Diffraction File (Fawcett et al., Reference Fawcett, Kabekkodu, Blanton and Blanton2017).
II. EXPERIMENTAL
Alclometasone dipropionate was a commercial reagent, purchased from USP (Lot #R044G0), and was used as-received. The white powder was packed into a 1.5 mm diameter Kapton capillary and rotated during the measurement at ~50 Hz. The powder pattern was measured at 295 K at beamline 11-BM (Lee et al., Reference Lee, Shu, Ramanathan, Preissner, Wang, Beno, Von Dreele, Ribaud, Kurtz, Antao, Jiao and Toby2008; Wang et al., Reference Wang, Toby, Lee, Ribaud, Antao, Kurtz, Ramanathan, Von Dreele and Beno2008) of the Advanced Photon Source at Argonne National Laboratory using a wavelength of 0.412826 Å from 0.5 to 50° 2θ with a step size of 0.001° and a counting time of 0.1 s step−1.
The pattern was difficult to index. Difficulty in indexing a high-resolution pattern from 11-BM is a sign that the sample may be a mixture. The strategy that was finally successful was to use DICVOL14 (Louër and Boultif, Reference Louër and Boultif2014), allowing up to three unindexed lines and a tolerance of ±0.03°. This yielded a primitive orthorhombic unit cell with a = 10.5881, b = 14.6695, c = 17.2491 Å, V = 2679.07 Å3, and Z = 4. Analysis of the systematic absences using EXPO2014 (Altomare et al., Reference Altomare, Cuocci, Giacovazzo, Moliterni, Rizzi, Corriero and Falcicchio2013) suggested the space group P212121. A reduced cell search in the Cambridge Structural Database (Groom et al., Reference Groom, Bruno, Lightfoot and Ward2016) combined with the chemistry C, H, Cl, and O only yielded no hits. An alclometasone dipropionate molecule was built using Spartan ‘18 (Wavefunction, 2018) and converted into .mol2 and .mop files using OpenBabel (O'Boyle et al., Reference O'Boyle, Banck, James, Morley, Vandermeersch and Hutchison2011). The same structural model was obtained by Monte Carlo simulated annealing techniques using FOX (Favre-Nicolin and Černý, Reference Favre-Nicolin and Černý2002) and EXPO2014.
The initial refinement using GSAS-II (Toby and Von Dreele, Reference Toby and Von Dreele2013) revealed the presence of >13 unindexed peaks. These were indexed on a high-quality (M/F = 47.6/415.9) primitive orthorhombic unit cell having a = 10.6933, b = 14.6540, c = 17.1724 Å, and V = 2690.90 Å3 using DICVOL14 (Louër and Boultif, Reference Louër and Boultif2014). The similarity of this cell to that of the initial unit cell (defined as Form 1 alclometasone dipropionate) led us to modify that cell and carry out a molecular mechanics geometry optimization using the Forcite module of Materials Studio (Dassault, 2018) to obtain an initial structural model for what was defined as Form 2 alclometasone dipropionate. The final refinement was begun using the results from the density functional theory (DFT) calculations.
The Rietveld refinement was carried out using GSAS-II (Toby and Von Dreele, Reference Toby and Von Dreele2013). Only the 2.0–20.0° portion of the pattern was included in the refinement (d min = 1.188 Å). All non-H bond distances and angles were subjected to restraints, based on a Mercury/Mogul Geometry Check (Bruno et al., Reference Bruno, Cole, Kessler, Luo, Motherwell, Purkis, Smith, Taylor, Cooper, Harris and Orpen2004; Sykes et al., Reference Sykes, McCabe, Allen, Battle, Bruno and Wood2011) of the molecule. The results were exported to a csv file. The Mogul average and standard deviation for each quantity were used as the restraint parameters and were incorporated using the new feature Restraints/Edit Restraints/Add MOGUL Restraints, which reads the bond distance and angle restraints from the csv file. The restraints contributed 4.2% to the final χ 2. The hydrogen atoms were included in calculated positions, which were recalculated during the refinement using Materials Studio (Dassault, 2018). A common U iso was refined for the non-H atoms of the ring system of Form 1 and another for the non-H atoms of the side chains. The U iso of these atoms in Form 2 were fixed to the values of Form 1. The U iso for each hydrogen atom was constrained to be 1.3× that of the heavy atom to which it is attached. The background was modeled using a 3-term shifted Chebyshev polynomial, and a peak at 5.26° to model the scattering from the Kapton capillary and any amorphous component.
The final refinement of 245 variables using 18 029 observations and 202 restraints yielded the residuals Rwp = 0.0852 and GOF = 1.33. The largest peak (1.64 Å from C67) and hole (1.60 Å from O66) in the difference Fourier map for Form 1 were 0.36 and −0.38(9) eÅ−3, and the largest peak (0.10 Å from C17) and hole (1.75 Å from O51) in the difference Fourier map for Form 2 were 0.38 and −0.45(11) eÅ−3, respectively. The Rietveld plot is included in Figure 2. The largest errors in the fit are in the shapes of some of the strong low-angle peaks and in the description of the amorphous background.
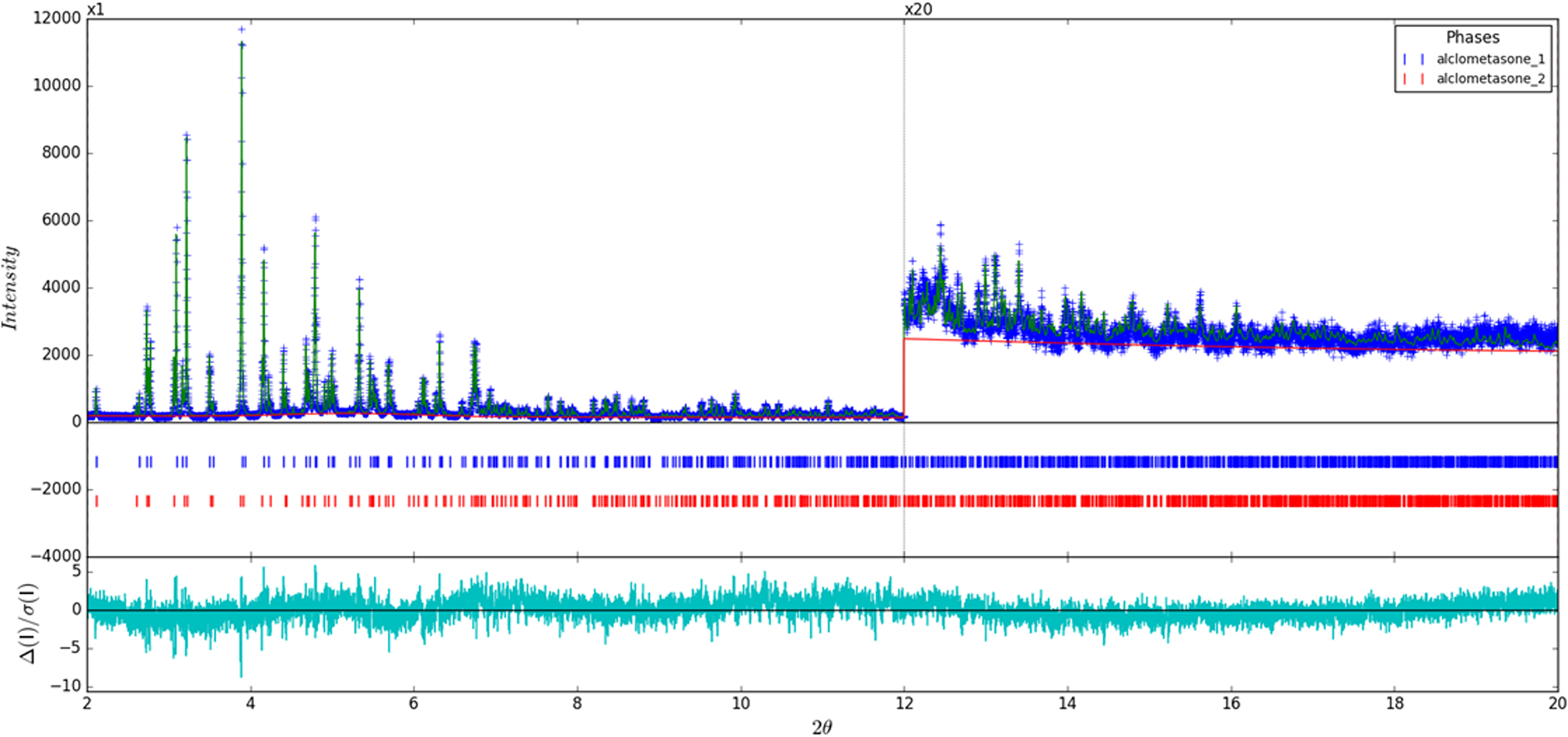
Figure 2. (Color online) Rietveld plot for the refinement of alclometasone dipropionate present in both Form 1 and Form 2. The blue crosses represent the observed data points, and the green line is the calculated pattern. The cyan curve is the normalized error plot. The vertical scale has been multiplied by a factor of 20× for 2θ> 12°.
Density functional geometry optimizations were carried out for both forms using CRYSTAL14 (Dovesi et al., Reference Dovesi, Orlando, Erba, Zicovich-Wilson, Civalleri, Casassa, Maschio, Ferrabone, De La Pierre, D-Arco, Noël, Causà and Kirtman2014). The basis sets for the H, C, N, and O atoms were those of Gatti et al. (Reference Gatti, Saunders and Roetti1994), and the basis set for Cl was that of Peintinger et al. (Reference Peintinger, Vilela Oliveira and Bredow2013). The calculations were run on eight 2.1 GHz Xeon cores (each with 6 GB RAM) of a 304-core Dell Linux cluster at IIT, using 8 k-points and the B3LYP functional, and took ~175 h (Form 1) and ~123 h (Form 2).
III. RESULTS AND DISCUSSION
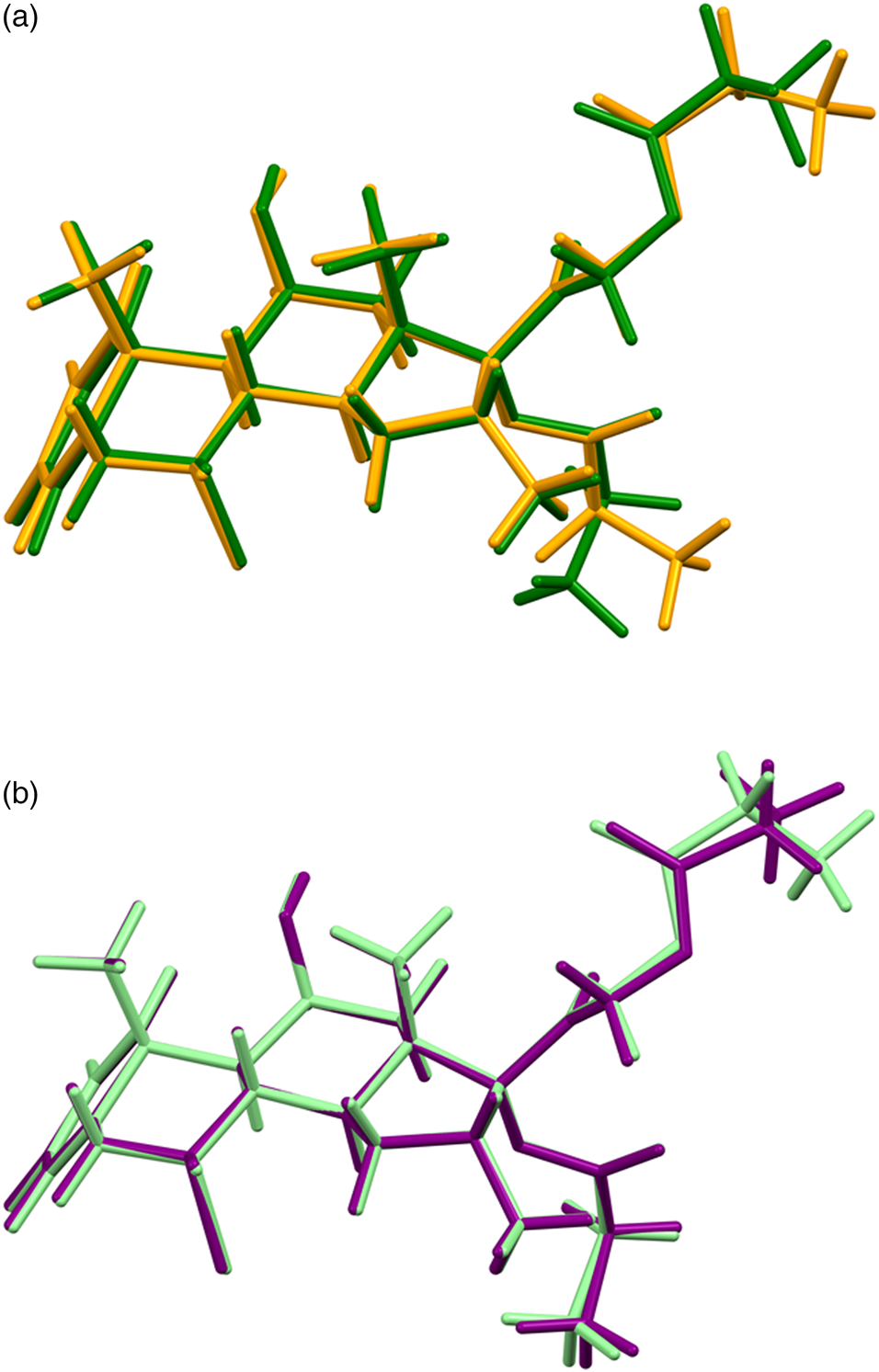
The refined atom coordinates of both forms of alclometasone dipropionate and the coordinates from the DFT optimizations are reported in the CIFs deposited with ICDD. The root-mean-square Cartesian displacements of the two forms are compared in Table I. The agreement between the refined and optimized structures of Form 1 is excellent [Figure 3(a)], confirming that the structure is correct (van de Streek and Neumann, Reference van de Streek and Neumann2014). The largest difference is at C61, the methyl group at the end of one propionate side chain. The agreement between the refined and optimized structures of Form 2 is not as good as observed for Form 1 [Figure 3(b)] and reflects differences in the conformations of both methyl groups C61 and C70 at the ends of the propionate side chains. The Form 2 polymorph is a minority phase (32.0(2) wt%) and is probably determined less accurately than Form 1. The largest differences between the Rietveld-refined structures of the two forms are at both methyl groups C61 and C70 [Figure 4(a)], while the DFT-optimized structures differ mainly at C61 [Figure 4(b)]. This discussion concentrates on the CRYSTAL-optimized structures. The asymmetric units (with the atom numbering) are illustrated in Figure 5(a), Form 1 and Figure 5(b), Form 2; and the crystal structures are presented in Figure 6(a), Form 1 and Figure 6(b), Form 2.
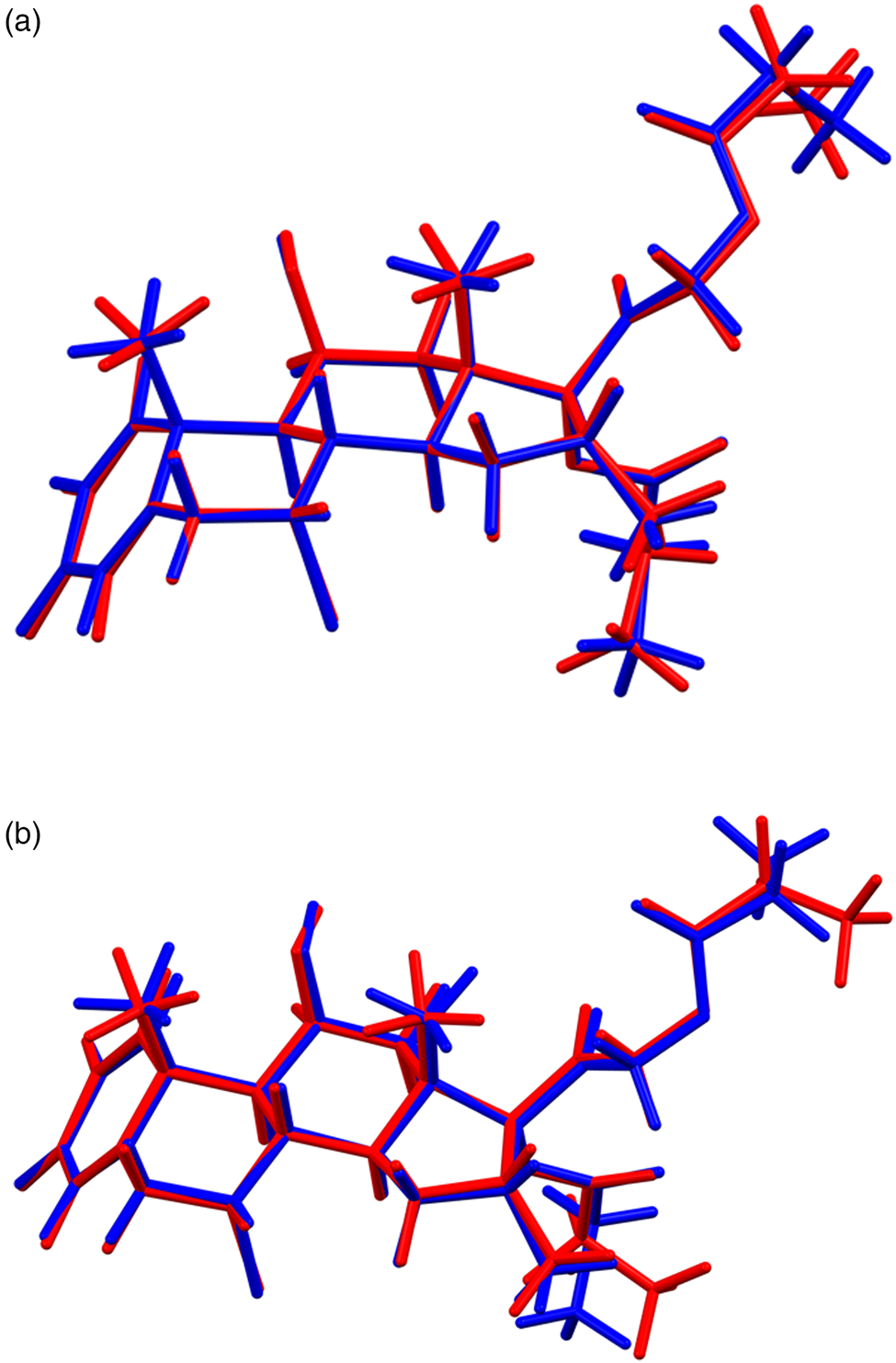
Figure 3. (Color online) (a) Comparison of the Rietveld-refined (red) and CRYSTAL14-optimized (blue) structures of alclometasone dipropionate, Form 1. (b) Comparison of the Rietveld-refined (red) and CRYSTAL14-optimized (blue) structures of alclometasone dipropionate, Form 2.

Figure 4. (Color online) (a) Comparison of the Rietveld-refined structures of alclometasone dipropionate Form 1 (green) and Form 2 (orange). (b) Comparison of the CRYSTAL14-optimized structures of alclometasone dipropionate Form 1 (light green) and Form 2 (purple).
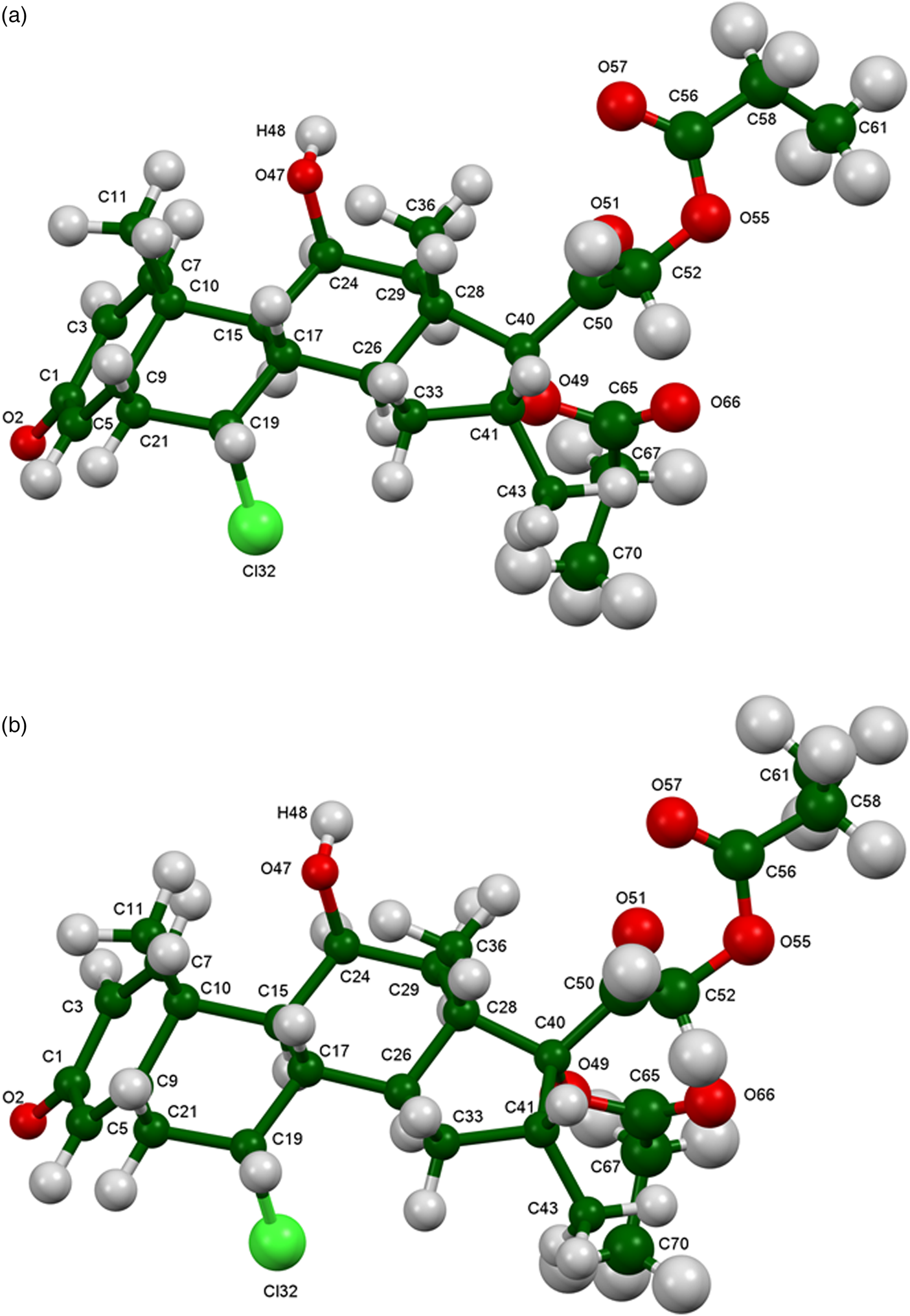
Figure 5. (Color online) (a) Asymmetric unit of alclometasone dipropionate, Form 1, with the atom numbering. The atoms are represented by 50% probability spheroids. (b) Asymmetric unit of alclometasone dipropionate, Form 2, with the atom numbering. The atoms are represented by 50% probability spheroids.

Figure 6. (Color online) (a) Crystal structure of alclometasone dipropionate, Form 1, viewed down the a-axis. (b) Crystal structure of alclometasone dipropionate, Form 2, viewed down the a-axis.
Table I. Root-mean-square Cartesian displacements (Å) between the two polymorphs of alclometasone dipropionate.

The two structures are nearly identical (Figure 7). The differences lie in the orientations of the methyl groups C61 at the ends of the propionate side chains and in subtle differences in the hydrogen bonds. The propionate side chains may be disordered. The general orientation of the molecules is in the ac-plane. The solid-state DFT calculations indicate that Form 2 is lower in energy by −0.04 kcal mol−1. The energy difference is well within the expected uncertainty of such calculations, so the two forms must be considered equivalent in energy. Compared to those of Form 1, the lattice parameters of Form 2 differ by +2.32%, −0.18%, and −0.82%, respectively; the cell volume of Form 2 is 1.30% larger than that of Form 1.

Figure 7. (Color online) Comparison of Form 1 (green) and Form 2 (orange) alclometasone dipropionate structures.
All of the bond distances and angles in both forms fall within the normal ranges indicated by a Mercury–Mogul Geometry Check (Macrae et al., Reference Macrae, Bruno, Chisholm, Edington, McCabe, Pidcock, Rodriguez-Monge, Taylor, van de Streek and Wood2008). In both forms, the torsion angles involving rotation about the C40–C50 bond lie on the tails of the expected distributions or in minority populations. In Form 2, the O49–C65–C67–C70 torsion angle is flagged as unusual. These torsion angles reflect the orientations of the propionate side chains with respect to the steroid core. Presumably, molecular crowding results in unusual conformations. The O55–C56–C58–C61 torsion angle in Form 2 is also flagged as unusual. The two propionate chains in both forms have different conformations (Table II). Even though the O55–C56–C58–C61 and O49–C65–C67–C70 torsion angles in Form 2 are flagged as unusual and the comparable torsions in Form 1 are not; in both forms, these torsion angles lie in the extended tail of a distribution around the normal value of ~180° [Figures 8(a) and 8(b)]. This observation points out the importance of actually looking at the distributions of torsion angles.
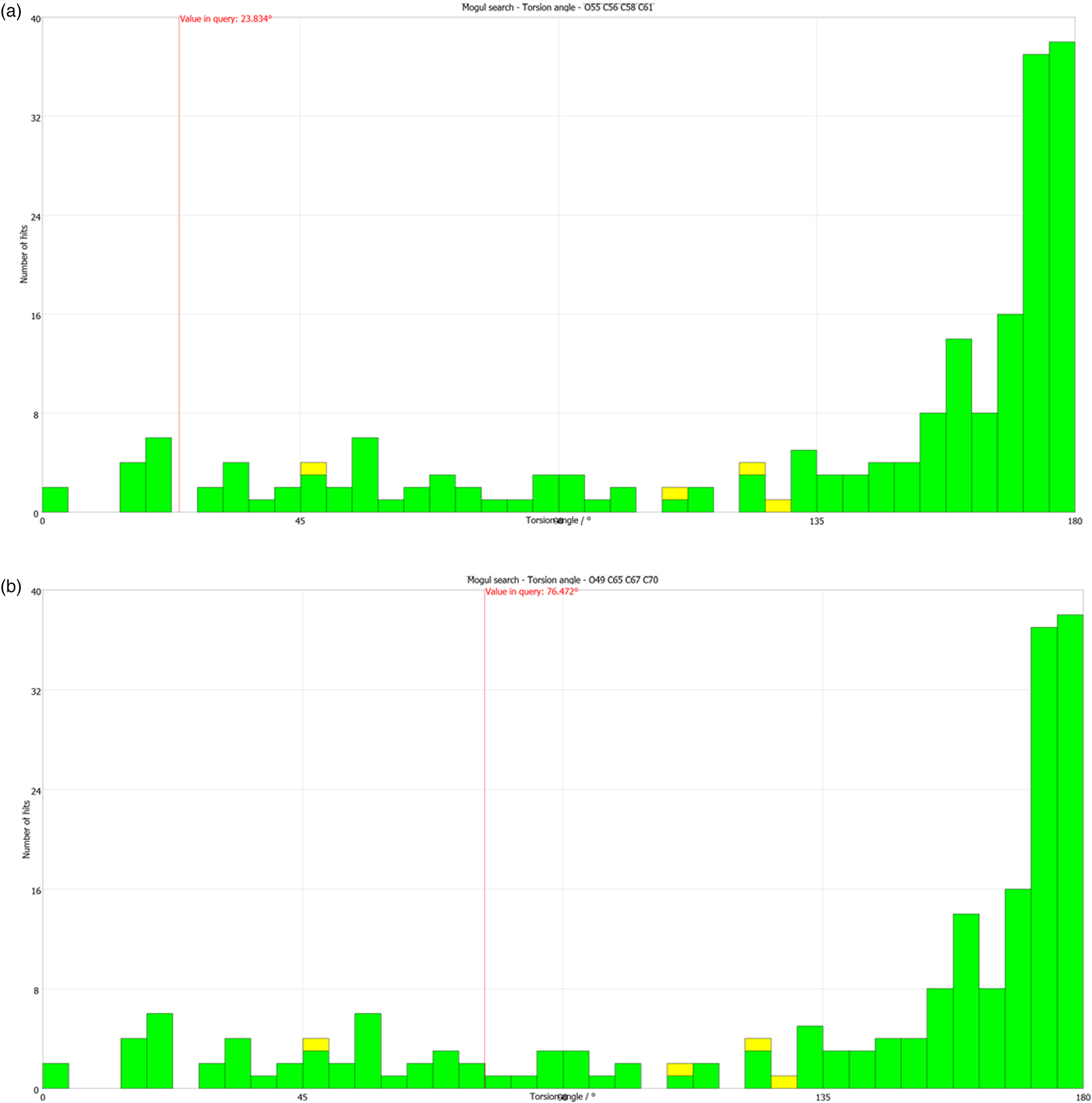
Figure 8. (Color online) (a) The “not unusual” O55–C56–C58–C61 torsion angle in alclometasone dipropionate Form 1 compared to the Mogul distribution of similar torsion angles. (b) The “unusual” O49–C65–C67–C70 torsion angle in alclometasone dipropionate Form 1 compared to the Mogul distribution of similar torsion angles.
Table II. Propionate torsion angles (°) in the DFT-optimized structures of the two polymorphs of alclometasone dipropionate.

Quantum chemical geometry optimizations (DFT/B3LYP/6-31G*/water) using Spartan ‘18 (Wavefunction, 2018) indicated that the conformation of molecule 2 is 0.1 kcal mol−1 lower in energy than that of molecule 1, and thus that the energies are indistinguishable. The minimum energy conformation has a third arrangement of the methyl groups at the ends of the propionate chains, which suggests that these chains may be flexible and/or disordered.
The analysis of the contributions to the total crystal energy in both forms using the Forcite module of Materials Studio (Dassault, 2018) indicates that Form 2 is also slightly lower in energy, and suggests that bond and angle distortion terms are significant in the intramolecular deformation energy, as might be expected for a fused-ring system. The intermolecular energy is dominated by electrostatic attractions, which in this force-field-based analysis include hydrogen bonds. The hydrogen bonds are better analyzed using the results of the DFT calculations.
The H-bond landscape is surprisingly rich (Tables III and IV). In both forms, the only traditional hydrogen bond is between the hydroxyl group O47 and the ketone O2. These discrete hydrogen bonds link pairs of molecules along the c-axis. The energies of these hydrogen bonds were calculated using the correlation of Rammohan and Kaduk (Reference Rammohan and Kaduk2018). In both forms, Cl32 acts as an acceptor in two intramolecular C–H⋯Cl hydrogen bonds involving the ring hydrogens H16 and H27, as well as in an intermolecular hydrogen bond involving the methyl group C36–H38. There are several C–H⋯O hydrogen bonds, mainly to ketone oxygens, but also to the hydroxyl group O47 and the ether oxygen O49. There is also an intramolecular C–H⋯C hydrogen bond from the methyl group C36 to the ketone carbon C50. The topologies of the hydrogen bonds in the two forms are the same, but the strengths differ subtly.
Table III. Hydrogen bonds (CRYSTAL14) in alclometasone dipropionate Form 1.

a Intramolecular.
Table IV. Hydrogen bonds (CRYSTAL14) in alclometasone dipropionate Form 2.

a Intramolecular.
The volumes enclosed by the Hirshfeld surfaces (Figure 9; Hirshfeld, Reference Hirshfeld1977; Turner, et al., Reference Turner, McKinnon, Wolff, Grimwood, Spackman, Jayatilaka and Spackman2017) of Form 1 and Form 2 are 655.63 and 664.06 Å3, 98.68% and 98.67% of 1/4 the unit cell volumes, respectively. The molecules, thus, exhibit typical packing density. All of the significant close contacts (red in Figure 9) involve the hydrogen bonds. The volume/non-H atom is 18.4 and 18.7 Å3.

Figure 9. (Color online) (a) Hirshfeld surface of alclometasone dipropionate, Form 1. Intermolecular contacts longer than the sums of the van der Waals radii are colored blue, and contacts shorter than the sums of the radii are colored red. Contacts equal to the sums of radii are white. (b) Hirshfeld surface of alclometasone dipropionate, Form 2. Intermolecular contacts longer than the sums of the van der Waals radii are colored blue, and contacts shorter than the sums of the radii are colored red. Contacts equal to the sums of radii are white.
The Bravais–Friedel–Donnay–Harker (Bravais, Reference Bravais1866; Friedel, Reference Friedel1907; Donnay and Harker, Reference Donnay and Harker1937) morphology suggests that we might expect blocky morphology for alclometasone dipropionate. The second-order spherical harmonic models were included in the refinement. The texture indices were 1.001 and 1.008, indicating that the preferred orientation was not significant in this rotated capillary specimen. The powder patterns of the two forms of alclometasone dipropionate from this synchrotron data set have been submitted to ICDD for inclusion in the Powder Diffraction File™.
DEPOSITED DATA
CIF and/or RAW data files were deposited with ICDD. You may request this data from ICDD at info@icdd.com.
ACKNOWLEDGEMENTS
We thank Lynn Ribaud and Saul Lapidus for their assistance in the data collection, and Andrey Rogachev for the use of computing resources at IIT.
FUNDING INFORMATION
The use of the Advanced Photon Source at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. This work was partially supported by the International Centre for Diffraction Data.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.