Introduction
The majority of Metagonimus Katsurada, 1912 species (eight of nine known species), including causative agents for human metagonimiasis, occur in East Asia (Chai et al., Reference Chai, Murrell and Lymbery2005). Most of them have been reported in Japan: M. yokogawai Katsuradai, 1912, M. takahashii Suzuki, 1930, M. miyatai Saito, Chai, Kim, Lee & Rim, 1997, M. hakubaensis Shimazu, 1999, M. katsuradai Izumi, 1935, M. otsurui Shimazu & Urabe, 2002. Metagonimus minutus Katzuta, 1932 has been reported in Taiwan, and M. takahashii and M. miyatai have been found in Korea (Morozov, Reference Morozov and Skrjabin1952; Ito, Reference Ito1964b; Komiya, Reference Komiya1965; Saito et al., Reference Saito, Chai, Kim, Lee and Rim1997; Shimazu, Reference Shimazu1999, Reference Shimazu2003; Shimazu & Urabe, Reference Shimazu and Urabe2002; Kino et al., Reference Kino, Suzuki, Oishi, Suzuki, Yamagiwa and Ishiguro2006; Yu & Chai, Reference Yu, Chai and Liu2012; Pornruseetairatn et al., Reference Pornruseetairatn, Kino, Shimazu, Nawa, Scholz, Ruangsittichai, Saralamba and Thaenkham2016). For most of these species, the genetic data based on three DNA markers (the 28S rRNA gene, the rDNA internal transcribed spacer 2 (ITS2) region and the cytochrome c oxidase I (cox1) gene of mitochondrial DNA (mtDNA)) were provided (Pornruseetairatn et al., Reference Pornruseetairatn, Kino, Shimazu, Nawa, Scholz, Ruangsittichai, Saralamba and Thaenkham2016). The most widespread species is M. yokogawai, which, in addition to Japan, has been found in Taiwan, Korea and China. Previously, it was believed that M. yokogawai was also common in the southern part of the Russian Far East (Skrjabin et al., Reference Skrjabin, Podjapolskaya and Schultz1930; Оshmarin, Reference Оshmarin1963; Shatrov, Reference Shatrov1974; Besprozvannykh et al., Reference Besprozvannykh, Ermolenko and Dvorjadkin1987; Besprozvannykh, Reference Besprozvannykh2000). However, based on studies of the life cycle, morphology of the developmental stages and genetic data (two nuclear markers), it was found that the flukes known in Russia as M. yokogawai are a new species of the genus, M. suifunensis Shumenko, Tatonova & Besprozvannykh, 2017 (Shumenko et al., Reference Shumenko, Tatonova and Besprozvannykh2017). In addition to this species, it was previously established that another Metagonimus species exists in the Russian southern Far East, M. katsuradai (Besprozvannykh et al., Reference Besprozvannykh, Ermolenko and Dvorjadkin1987). We discovered that the adult worms were morphologically identical to flukes from Anas platyrhynchos dom. duckling fed with Rhodeus sericeus sericeus (Pallas) fish from the Bolshaya Ussurka River of the Russian southern Far East. The genetic study showed that flukes obtained in the Russian southern Far East and previously referred to as M. katsuradai (Besprozvannykh et al., Reference Besprozvannykh, Ermolenko and Dvorjadkin1987) represent a new species. The results of this investigation are presented in this study. We also confirmed the validity of a previously described species, M. suifunensis (Shumenko et al., Reference Shumenko, Tatonova and Besprozvannykh2017), using the cox1 gene of mDNA, which was previously applied for the Japanese representatives of the Metagonimus genus (Pornruseetairatn et al., Reference Pornruseetairatn, Kino, Shimazu, Nawa, Scholz, Ruangsittichai, Saralamba and Thaenkham2016).
Materials and methods
Sample collection and morphology
The material used for this study included R. sericeus sericeus fish which were taken from the Bolshaya Ussurka River in the Russian southern Far East. Five fish were fed to ducklings (A. platyrhynchos dom.) that had been hatched from eggs in the laboratory. Within 10 days after infection, eight adult Metagonimus worms were isolated from the small intestines of the ducklings. These worms, identified by microscopic examination, were rinsed in saline, killed in hot distilled water and preserved in 70% ethanol. After fixation, they were placed in 96% ethanol. Whole mounts were prepared for examination by staining the specimens with alum carmine, dehydrating the specimens in a series of graded ethanol concentrations and then clearing in xylene, followed by mounting on a slide using Canada balsam under a coverslip. All measurements are given in millimetres (mm).
DNA extraction, amplification and sequencing
Genomic DNA of M. pusillus sp. nov. was isolated using the HotSHOT method (Truett et al., Reference Truett, Heeger, Mynatt, Truett, Walker and Warman2000). The complete nucleotide sequences of the ITS1–5.8S–ITS2 rDNA region and the partial sequences of the 28S rRNA gene for M. pusillus sp. nov. were amplified using the following primers: BD1 (5′-GTC GTA ACA AGG TTT CCG TA-3′) (Morgan & Blair, Reference Morgan and Blair1995), 28S1R (5′-AAG TAT TTA GCC TTG GAT GGA GTT T-3′) (Shumenko et al., Reference Shumenko, Tatonova and Besprozvannykh2017) for the ITS region; and DIGl2 (5′-AAG CAT ATC ACT AAG CGG-3′), 1500R (5′-GCT ATC CTG AGG GAA ACT TCG-3′) (Tkach et al., Reference Tkach, Littlewood, Olson, Kinsella and Swiderski2003) for the 28S rRNA gene. The composition of the polymerase chain reaction (PCR) mixture, as well as amplification and sequencing conditions, were as described previously (Shumenko et al., Reference Shumenko, Tatonova and Besprozvannykh2017). In addition, for M. pusillus sp. nov. and for M. suifunensis specimens from the Russian southern Far East (described previously by Shumenko et al., Reference Shumenko, Tatonova and Besprozvannykh2017), the partial sequences of the сох1 gene were amplified and sequenced using JB3 (5′-TTT TTT GGG CAT CCT GAG GTT TAT-3′) and JB4.5 (5′-TAA AGA AAG AAC ATA ATG AAA ATG-3′) primers (Bowles & McManus, Reference Bowles and McManus1994) and conditions that were identical to those used for ribosomal markers, with the exception of a lower annealing temperature (52°C).
Alignments and the phylogenetic analysis
Sequences were visualized in Finch version 1.4.0 (www.geospiza.com) and then assembled and aligned manually using the ClustalW option in MEGA version 5.03 (Tamura et al., Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011). For the analysis of interspecific phylogenetic relationships, we used the sequences of the superfamily Opisthorchioidea from GenBank. Nucleotide sequences used in this study are listed in table 1. Genetic p-distances, nucleotide composition and types of nucleotide substitutions were estimated using MEGA version 5.03. The GTR + G, TVM + I + G and GTR + I + G models were determined as optimal for tree-building based on the ITS2 region, the 28S rRNA gene and cox1 gene, respectively, according to Akaike information criterion in jModelTest version 2.1.5 (Darriba et al., Reference Darriba, Taboada, Doallo and Posada2012). Phylogenetic analysis was conducted using the Maximum Likelihood (ML) method in PhyML version 3.1 (Guindon & Gascuel, Reference Guindon and Gascuel2003) with 100 bootstrap replications. The Bayesian inference (BI) method was performed using MrBayes version 3.1.2 (Ronquist & Huelsenbeck, Reference Ronquist and Huelsenbeck2003). For the 28S rRNA gene, ITS2 region and cox1 gene, BI analysis was performed using 700,000, 2,200,000 and 400,000 generations of MCMC chains, respectively. This number of generations was sufficient, because the SD value fell below 0.01. Of the samples, 25% were excluded to construct the consensus trees. The chain was sampled every 100th generation.
Table 1. Sequences analysed in this study.
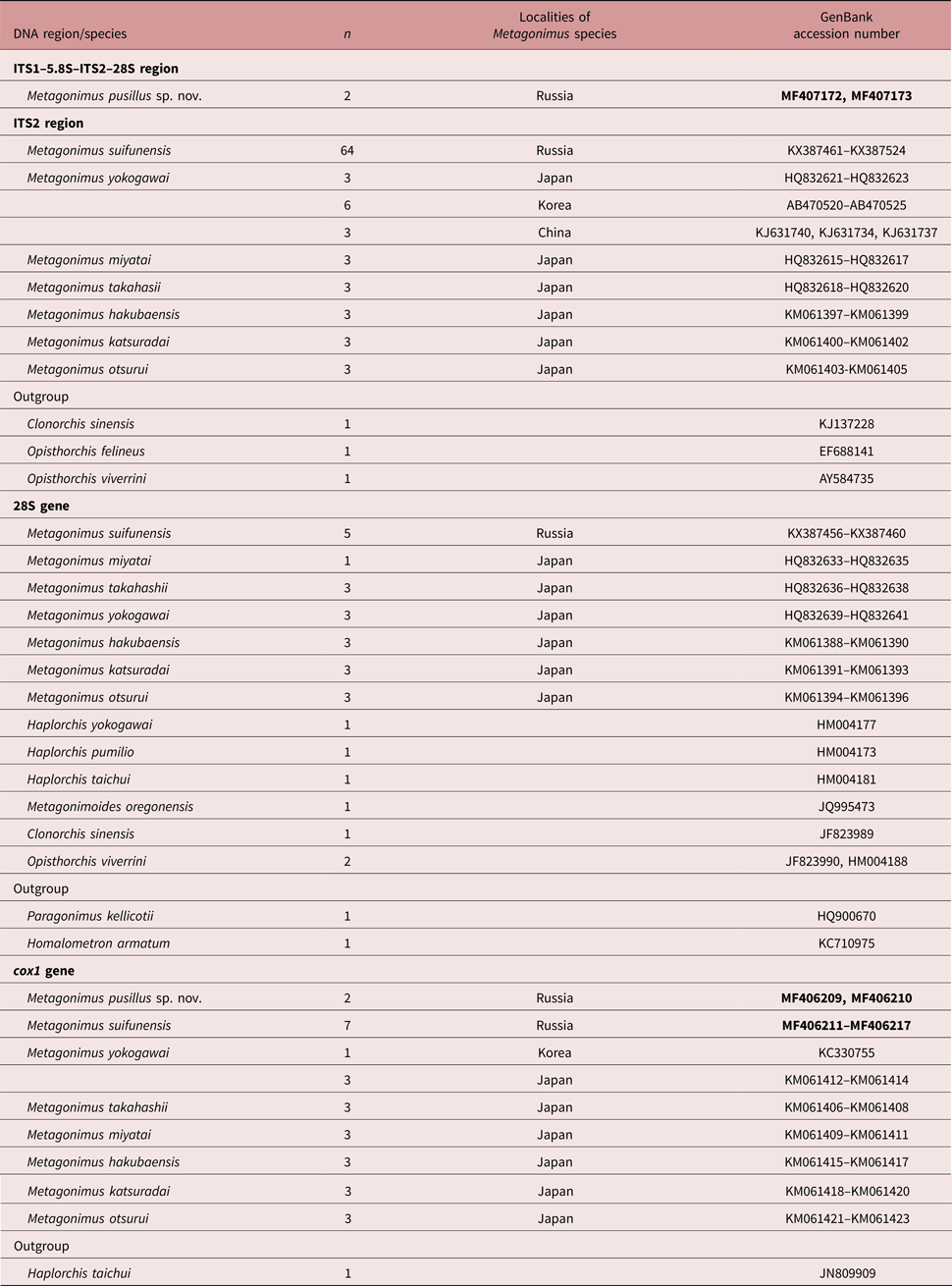
Accession numbers in bold are newly determined sequences.
Results
Metagonimus pusillus sp. nov.
Taxonomic summary
Synonym. Metagonimus katsuradai Katsuradai, 1912 according to Besprozvannykh et al. (Reference Besprozvannykh, Ermolenko and Dvorjadkin1987), Ermolenko & Besprozvannykh (Reference Ermolenko and Besprozvannykh1987), Besprozvannykh (Reference Besprozvannykh2000), Besprozvannykh & Ermolenko (Reference Besprozvannykh and Ermolenko2005).
Definitive host. Anas platyrhynchos dom. (experimentally, in present study), Rattus norvegicus (Besprozvannykh et al., Reference Besprozvannykh, Ermolenko and Dvorjadkin1987).
Site. Small intestine.
Intensity of infection. Eight specimens.
First intermediate host. Parajuga subtegulata Prozorova et Starobogatov, Parajuga subextensa Prozorova et Starobogatov (Besprozvannykh et al., Reference Besprozvannykh, Ermolenko and Dvorjadkin1987; Besprozvannykh, Reference Besprozvannykh2000).
Second intermediate host. Rhodeus sericeus sericeus (present study), predominately Cyprinidae fish (Ermolenko & Besprozvannykh, Reference Ermolenko and Besprozvannykh1987; Besprozvannykh & Ermolenko, Reference Besprozvannykh and Ermolenko2005).
Type locality. The Bolshaya Ussurka River, Primorsky Region, the Russian southern Far East; 45°57′N, 133°53′E.
Type deposition. Holotype No. 114-Tr, paratypes No. 115 − 118-Tr. This material is held in the parasitological collection of the Zoological Museum (Federal Scientific Center of the East Asia Terrestrial Biodiversity, Far East Branch of the Russian Academy of Sciences, Vladivostok, Russia); E-mail: petrova@ibss.dvo.ru. Deposited 20 November 2016.
Etymology. The species’ name reflects its small size.
Description
Adult worm, five specimens examined (fig. 1, table 2). Body oval–elongated with tapered anterior end. Maximum body width at level of testis. Surface covered by fine spines. Anterior end of body with numerous subdermal glands. Ducts of these glands open on ventral part of body. Oral sucker subterminal, transversely oval. Prepharynx short or, in some cases, pharynx adjacent to oral sucker. Pharynx spherical or transversely oval, oesophagus long. Caecum bifurcation on border between anterior and middle thirds of body. Caeca reach level of posterior margin of right testis. Ventral sucker elliptical, in middle third of body, at right of median line of body. Anterior end of ventral sucker protruded into thin-walled ventrogenital sac. Genital pore opens ventrally, at level of ventrogenital sac. Testes globular or oval, in posterior third of body, parallel or diagonal. Testes usually close to each other, but sometimes located a short distance from each other. Right testis some distance from posterior end of body. Seminal vesicle bipartite, in middle third of body, before ovary. Anterior part of seminal vesicle 2–3 times smaller than posterior one. Pars prostatica small, with prostatic cells. Ejaculatory duct short. Ovary globular, on border of middle and posterior thirds of body, on median line between left testis and seminal receptacle. Uterus dorsal to testes and ovary, from level of middle part of ventral sucker to level of posterior margin of right testis. One of the uterus loops at level of border of testes’ contact. Seminal receptacle at right of ovary. Eggs operculated, light yellow, with developed miracidia, in anterior loops of uterus. Vitelline follicles laterally, from level of middle part of ovary to posterior end of body, almost contiguous in post-testicular region. Vitelline reservoir at space between ovary, seminal receptacle and left testis. Excretory vesicle Y-shaped, bifurcation on anterior margin of left testis.
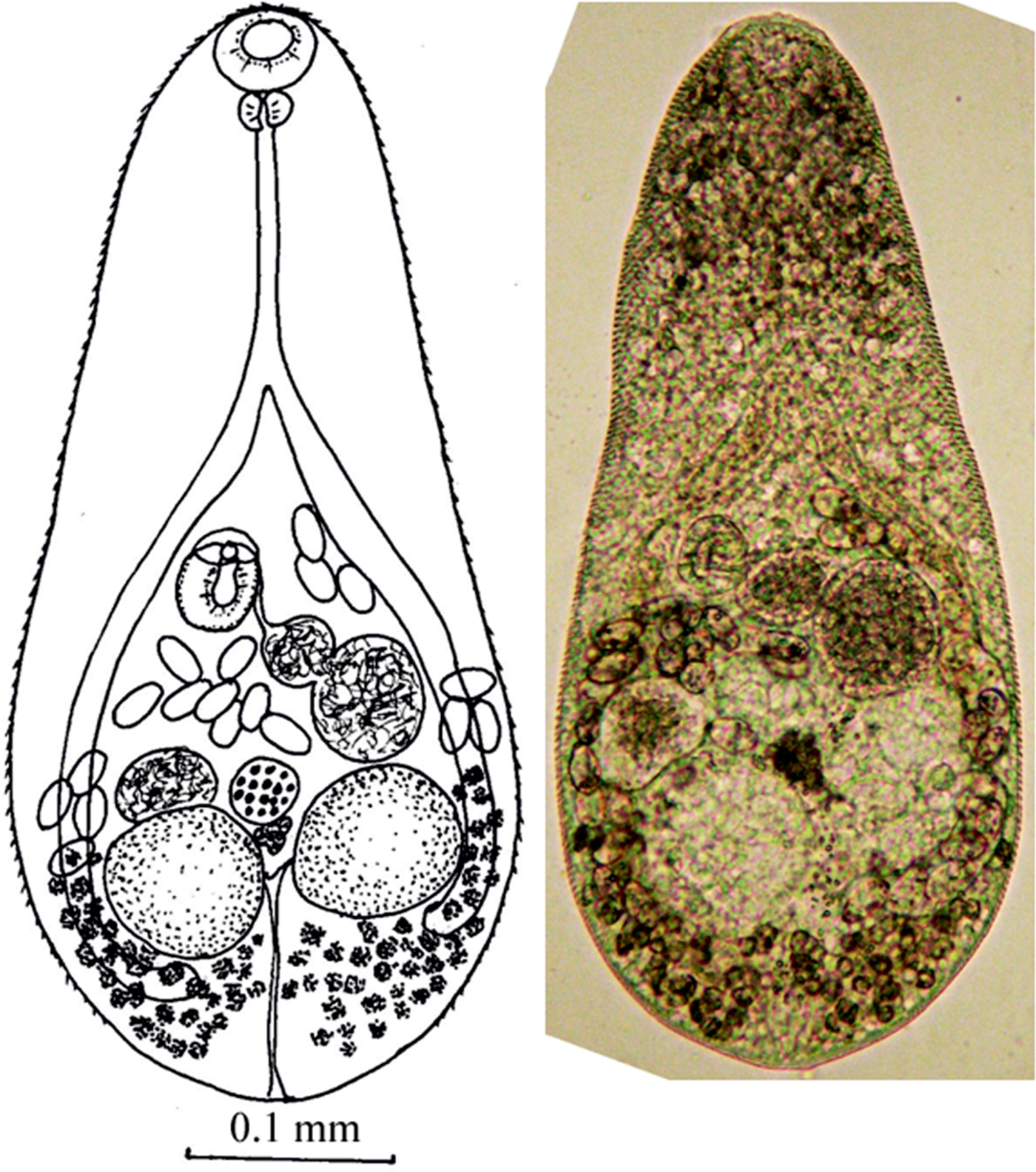
Fig. 1. Adult worm of M. pusillus sp. nov.
Genetic analysis
In total, the complete ITS1–5.8S–ITS2 (1144 bp), partial 28S rRNA gene (1403 bp) and partial с ox1 gene (339 bp) sequences were obtained for two specimens of M. pusillus sp. nov. from Russia, and seven partial sequences of the cox1 gene (339 bp) were obtained for M. suifunensis. There were no substitutions between sequences of all markers for both of the M. pusillus sp. nov. specimens, while the cox1 sequences of M. suifunensis were represented by two haplotypes, differing by T → C transition at the 73-bp position. For phylogenetic reconstructions and determination of p-distances between species, the aligned sequences of the ITS2 region, the 28S rRNA gene and the cox1 gene were used (297, 1151 and 339 bp, respectively).
The nucleotide composition of the ITS1–5.8S–ITS2 region for M. pusillus sp. nov. was: A, 19.1%; T, 30.5%; G, 26.7%; and C, 23.7%. The length of the ITS2 region was 284 bp. The genetic distances between M. pusillus sp. nov. from Russia and other representatives of Metagonimus varied from 0.7% (M. otsurui) to 5.5% (M. takahashii/yokogawai, Group 2) (see supplementary table S1). The nucleotide composition of the 28 rRNA gene was: A, 21.8%; T, 24.5%; G, 31.6%; and C, 22.1%. The differences between M. pusillus sp. nov. and other Metagonimus species were in the range 0.6% (M. katsuradai, M. otsurui) to 1.5% (M. takahashii) (see supplementary table S2).
According to distances based on сох 1 gene sequences, M. katsuradai and M. otsurui from Japan are the closest to M. pusillus sp. nov. (9.4 and 10.4%, respectively). The differences from the sequences of other Metagonimus representatives are slightly higher (13.4–16.7%). Metagonimus suifunensis differs from other species by 13.2–16.0% (see supplementary table S3). Nucleotide substitutions in the sequences are predominantly in the third codon position (88%). Amino acid sequences of the cox1 gene show no differences between M. pusillus sp. nov. and M. katsuradai from Japan, and only one non-synonymous substitution with M. otsurui (see supplementary fig. S1). Metagonimus yokogawai from Japan and M. suifunensis differ by two amino acid substitutions in the cox1 gene sequences.
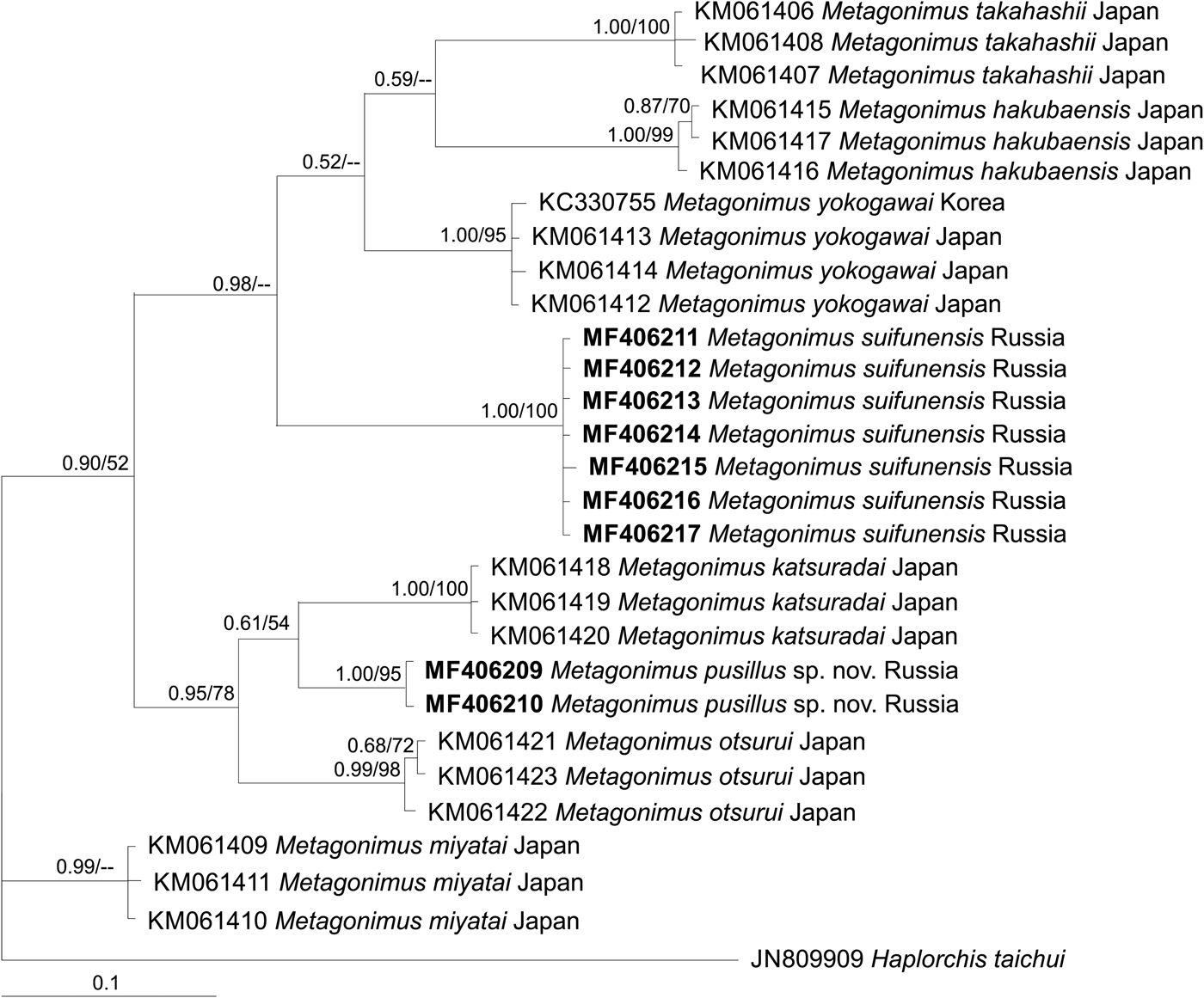
Phylogenetic trees based on all three markers show that M. pusillus sp. nov. specimens are included in a common cluster with M. otsurui and M. katsuradai from Japan, forming a sister group with high posterior probability/bootstrap support (figs 2, 3 and 4). Metagonimus suifunensis also formed a separate branch in the tree based on the cox1 gene.
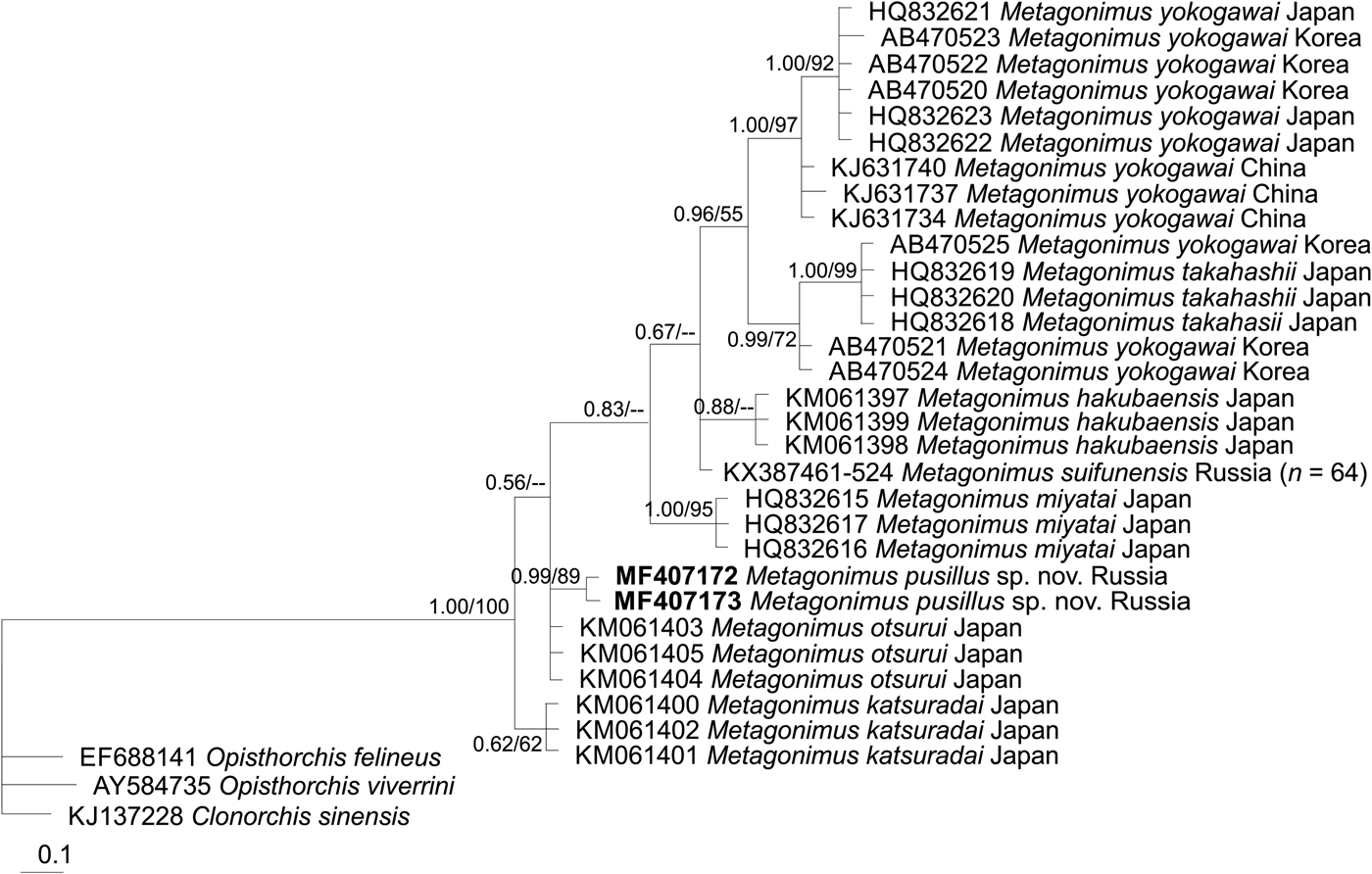
Fig. 2. The phylogeny based on the ITS2 rDNA sequences. Posterior probability ≥50 (nodal support of ≥0.50) is shown for BI/ML analyses, respectively. The outgroup species are listed in table 1.

Fig. 3. The phylogeny based on the 28S rRNA gene sequences. Posterior probability ≥50 (nodal support of ≥0.50) is shown for BI/ML analyses, respectively. The outgroup species are listed in table 1.
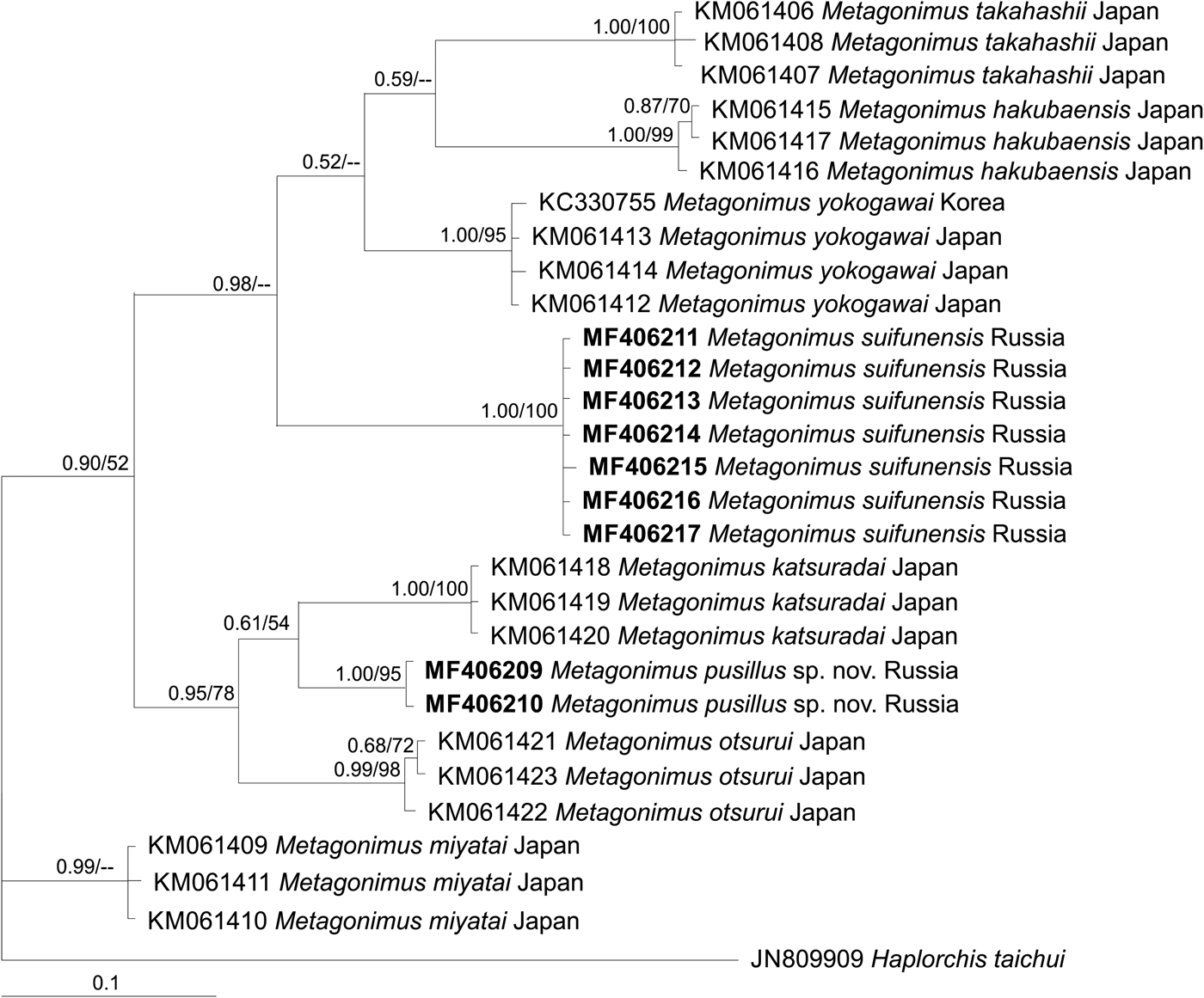
Fig. 4. The phylogeny based on cox1 mtDNA gene sequences. Posterior probability ≥50 (nodal support of ≥0.50) is shown for BI/ML analyses, respectively. The outgroup species are listed in table 1.
Discussion
Adult worms, obtained here as a result of an experiment, corresponded to the Metagonimus genus in all morphological parameters (the structure of the reproductive system and the topology of organs). They, like M. katsuradai, M. otsurui, M. hakubaensis and M. minutes (table 2), refer to Metagonimus worms in which the body size of mature individuals does not exceed 0.650 mm (Besprozvannykh et al., Reference Besprozvannykh, Ermolenko and Dvorjadkin1987; Saito et al., Reference Saito, Chai, Kim, Lee and Rim1997; Shimazu & Kino, Reference Shimazu and Kino2015; Shumenko et al., Reference Shumenko, Tatonova and Besprozvannykh2017). Therefore, we conducted a comparative morphometric analysis only for these species. Metagonimus pusillus sp. nov. differs from M. hakubaensis and M. minutes in the metric characters of adult worms (sizes of the body, suckers, pharynx, etc.) (table 2). According to most morphometric data, the samples of M. pusillus sp. nov. are most similar to M. katsuradai and M. otsurui. According to the figure presented in the paper of Shimazu & Urabe (Reference Shimazu and Urabe2002), M. pusillus sp. nov. differs from M. otsurui only in the ratio of the anterior and posterior parts of the seminal vesicle (the anterior part of the seminal vesicle in M. otsurui is larger than the posterior one, while in M. pusillus sp. nov. the anterior part of the seminal vesicle is smaller). According to other criteria, such as the ratio of suckers, the position of the right testis, uterus and vitellaria, which are usually used to differentiate species of this genus (Pornruseetairatn et al., Reference Pornruseetairatn, Kino, Shimazu, Nawa, Scholz, Ruangsittichai, Saralamba and Thaenkham2016), M. pusillus sp. nov., M. katsuradai and M. otsurui have no differences. However, M. pusillus sp. nov. differs from M. otsurui by the maximum size of most organs; it differs from M. katsuradai by the width of the body, the length of the pharynx, and the maximum dimensions of the testes and the ovary (according to the description of Shimazu, Reference Shimazu2003), and the maximum body width according to Izumi (1935, cited in Ito, Reference Ito1964b). Adult M. pusillus sp. nov. worms are identical (except for small differences in sizes of testis and ovary) to the fluke previously identified in the same territory of the Russian southern Far East and referred to as M. katsuradai (Besprozvannykh et al., Reference Besprozvannykh, Ermolenko and Dvorjadkin1987). This evidence indicates that these worms belong to the same species. Moreover, proceeding from their morphometric differences with other Metagonimus representatives, we consider these worms to be a new species, which we designated as M. pusillus sp. nov. The validity of the species status is also confirmed by the morphometric differences of M. pusillus sp. nov. (= M. katsuradai (Besprozvannykh et al., Reference Besprozvannykh, Ermolenko and Dvorjadkin1987)), M. katsuradai and M. otsurui at the cercarial stage. Cercariae of M. pusillus sp. nov. differ from those of M. katsuradai by having smaller sizes of body, oral sucker and tail, as well as in the number of rows of hooks and the number of hooks in each row (table 3). Fewer differences were found between cercariae of M. pusillus sp. nov. and M. otsurui (only the pharynx length) (table 3). Unfortunately, for M. otsurui there are no data on the number of rows of hooks, and only the number of hooks in the first row is known.
Table 2. Measurements (mm) for adult worms of Metagonimus species.

L, length; W, width.
Table 3. Measurements (mm) for Metagonimus cercariae.

The validity of M. pusillus sp. nov. is also based on genetic data. Using all three markers (ITS2 region, the 28S rRNA gene and the cox1 gene), the differences between M. pusillus sp. nov. and other representatives of the genus correspond to genetic distances within the Metagonimus genus (Pornruseetairatn et al., Reference Pornruseetairatn, Kino, Shimazu, Nawa, Scholz, Ruangsittichai, Saralamba and Thaenkham2016; Shumenko et al., Reference Shumenko, Tatonova and Besprozvannykh2017), which is also confirmed by analysis of phylogenetic reconstructions.
Closest to the M. pusillus sp. nov. species are M. otsurui and M. katsuradai from Japan. When using different markers, some differences in the p-distances were found between these species. Thus, using the nucleotide sequences of the 28S rRNA gene, M. pusillus sp. nov. differed from both Japanese species by 0.6%, whereas the difference between M. otsurui and M. katsuradai was threefold lower (0.2%). However, based on the ITS2 region, the genetic distance between M. pusillus sp. nov. and M. otsurui was not very large (0.7%) and equalled the distance between M. otsurui and M. katsuradai, while the level of difference between M. pusillus sp. nov. and M. katsuradai was twofold higher (1.5%). Other Metagonimus representatives had a much higher level of difference from M. pusillus sp. nov. for both nuclear markers. The highest value for the distance between these species was based on the sequences of the cox1 mtDNA gene (9.4–11.1%), compared to the values between other representatives of the genus Metagonimus (12.7–18.1%), including the previously described M. suifunensis, the validity of which was confirmed in this study using an additional genetic marker. Based on a previously published study (Pornruseetairatn et al., Reference Pornruseetairatn, Kino, Shimazu, Nawa, Scholz, Ruangsittichai, Saralamba and Thaenkham2016), it is possible to describe the representatives of M. pusillus sp. nov. as a separate species, since the genetic distances were equal to, or greater than, the distance between the two Japanese species, M. otsurui and M. katsuradai. Moreover, the interspecific phylogenetic reconstructions based on the three markers separated the M. pusillus sp. nov. samples in a cluster with high bootstrap support/posterior probability. Thus, the validity of the species is confirmed for all three markers.
It should be noted that there is a possibility that M. otsurui and M. katsuradai from Japan could be incorrectly identified as separate species. This situation could occur due to the identification of species at the intermediate parasite stage (metacercariae) (Pornruseetairatn et al., Reference Pornruseetairatn, Kino, Shimazu, Nawa, Scholz, Ruangsittichai, Saralamba and Thaenkham2016). This hypothesis is also confirmed by low values of genetic distances obtained by the latter authors using nuclear markers. If the ITS2 region exhibits a low level of divergence between closely related species of trematodes (Vilas et al., Reference Vilas, Criscione and Blouin2005), then the 28S rRNA gene has a higher degree of reliability (Thaenkham et al., Reference Thaenkham, Nawa, Blair and Pakdee2011; Shumenko et al., Reference Shumenko, Tatonova and Besprozvannykh2017), and this marker must separate M. otsurui and M. katsuradai species. However, the distances between M. otsurui and M. katsuradai obtained using this marker were 3–9 times lower than the values found for other Metagonimus representatives. For example, this value was three times lower than the distances between both these species and M. pusillus sp. nov. Distinguishing between M. otsurui and M. katsuradai from Japan is more significant using a mitochondrial marker, as noted previously by Pornruseetairatn et al. (Reference Pornruseetairatn, Kino, Shimazu, Nawa, Scholz, Ruangsittichai, Saralamba and Thaenkham2016). However, almost all substitutions in cox1 gene sequences are on the third position of the codon and are synonymous. Thus, this may be intra-species variability, because mitochondrial markers usually accumulate mutations more rapidly, and these synonymous mutations are not under the influence of selection. This hypothesis can only be supported by increasing the sample sizes from different geographical regions for each species of Metagonimus. However, M. pusillus sp. nov., M. otsurui and M. katsuradai may be cryptic species with recent ancestry, which are well distinguished only by the more sensitive mitochondrial marker.
Nevertheless, Pornruseetairatn et al. (Reference Pornruseetairatn, Kino, Shimazu, Nawa, Scholz, Ruangsittichai, Saralamba and Thaenkham2016) provided molecular data for two Metagonimus species from Japan, M. otsurui and M. katsuradai, and recognized them as valid species. Since the trematode described in this study has an equal number, or more, significant differences from these species in all three markers, we conclude that worms from Russia are a new species, M. pusillus sp. nov.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0022149X17001146
Financial support
This study was funded by the Russian Science Foundation, project RSF No. 17-65-00004.
Conflict of interest
None.
Ethical standards
Euthanasia of laboratory animals was carried out in accordance with the Committee on the Ethics of Animal Experiments of the Federal Scientific Center of the East Asia Terrestrial Biodiversity, Russia (Permit Number 3 of 2 June 2011).