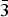
I. INTRODUCTION
Paar et al. (Reference Paar, Roberts, Criddle and Topa1998) reported a discovery of a Pd2HgSe3 phase in the form of a small inclusion of approximately 20 µm in size from carbonate veins in Hope's Nose, Torquay, Devon, England. This phase was first synthetized by Drábek et al. (Reference Drábek, Vymazalová and Laufek2014) in his systematic investigation of phase relations in the Hg–Pd–Se system. Drábek et al. (Reference Drábek, Vymazalová and Laufek2014) described the trigonal symmetry of the Pd2HgSe3 compound and indicated its unit-cell parameters. However, its crystal structure has been hitherto unknown. Besides the phase Pd2HgSe3, two other ternary phases were reported in the Hg–Pd–Se system: mineral tischendorfite Pd8Hg3Se9 (Stanley et al., Reference Stanley, Criddle, Förster and Roberts2002) and a synthetic phase Pd5HgSe (Laufek et al., Reference Laufek, Vymazalová, Drábek, Navrátil, Plecháček and Drahokoupil2012). A Pt-bearing analogue, a phase Pt2HgSe3, was described as a mineral jacutingaite by Vymazalová et al. (Reference Vymazalová, Laufek, Drábek, Cabral, Haloda, Sidorinová, Lehmnn, Galbiatti and Drahokoupil2012).
In this contribution, we present a Rietveld crystal structure analysis of the phase Pd2HgSe3. Powder diffraction data up to 130° 2θ (CoKα) are also reported. Part of this work was presented in a conference abstract (Laufek et al., Reference Laufek, Vymazalová, Drábek and Drahokoupil2011).
II. EXPERIMENTAL
The ternary Pd2HgSe3 phase was prepared using silica glass tube technique. Stoichiometric amounts of palladium (Aldrich 99.99%), mercury (Lachema, 99.999%), and selenium (Aldrich, 99.999%) were used as starting materials for synthesis. The following amounts of reagents were used for synthesis: 0.0658 g of palladium (0.6180 mmol), 0.0620 g of mercury (0.3090 mmol), and 0.0732 g of selenium (0.9270 mmol). The evacuated tube with the carefully weighted charge was at first heated at 800 °C for 24 h. After cooling in a cold-water bath, the charge was removed from the tube and gently ground to powder in acetone using a pestle and an agate mortar. Afterwards, the pulverized charge was again sealed in an evacuated silica glass tube and heated at 400 °C for 41 days. After heating, the sample was quenched in a cold-water bath.
Chemical analyses (EMPA) of Pd2HgSe3 phase were performed with a Cameca SX-100 electron microprobe using the wavelength-dispersion mode. Accelerating voltage was set to 15 kV, and the beam current was 10 nA. X-ray lines and standards were as follows: SeLα (pure Se), PdLα (pure Pd), and HgMα (HgS). Data were acquired (11 analyses) from different spots on several grains and then averaged. The chemical data yielded an overall composition Pd 32.53 wt.%, Hg 30.19 wt.%, Se 36.52 wt.%, and total 99.24 wt.% with empirical formula Pd2.00Hg0.98Se3.02 calculated on six atoms per formula unit.
Ideally, the experimental product should consist only of title phase Pd2HgSe3. The subsequent phase and Rietveld analysis of diffraction data revealed small admixture of Pd8Hg3Se9 (~12 wt.%) and HgSe (~2 wt.%) in the experimental product. These admixtures have very likely occurred because of possible presence of small amounts of selenium and mercury in a vapour phase during the initial stages of solid-state reactions.
III. STRUCTURE REFINEMENT
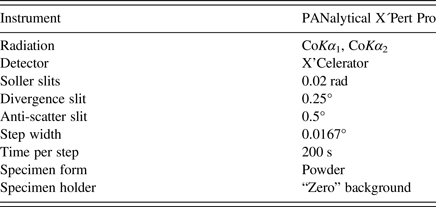
The powder X-ray diffraction pattern of the sample was collected in the Bragg–Brentano geometry on an X'Pert Pro PANalytical diffractometer, equipped with an X'Celerator detector using CoKα radiation. An iron filter, placed in a diffracted beam, was used to produce pseudo-monochromatic radiation. To minimize background, a flat low-background silicon wafer was used as the specimen holder. The data were collected in the range from 6 to 130° 2θ. A full-width at half maximum of 0.106° 2θ was obtained at 19.497° 2θ indicating well-crystalized sample of Pd2HgSe3. The experimental details are summarized in Table I. With the exception of a few peaks attributable to Pd8Hg3Se9 (~12 wt.%) and HgSe (~2 wt.%), the diffraction pattern of Pd2HgSe3 was indexed using the DICVOL06 programme (Louër and Boultif, Reference Louër and Boultif2007) on the basis of a trigonal cell listed in Table II. The unit cell suggested by Drábek et al. (Reference Drábek, Vymazalová and Laufek2014) was confirmed. The crystal structure of Pd2HgSe3 was subsequently refined by the Rietveld method for powder X-ray diffraction data using the FullProf programme (Rodriguez-Carvajal, Reference Rodriguez-Carvajal1990). The starting structural model for Pd2HgSe3 was derived from the structural data of Pt2HgSe3 phase, which is known as a mineral jacutingaite (Vymazalová et al., Reference Vymazalová, Laufek, Drábek, Cabral, Haloda, Sidorinová, Lehmnn, Galbiatti and Drahokoupil2012). This mineral phase has very similar unit-cell parameters [a = 7.3477(2), c = 5.2955(1) Å] and analogous stoichiometry. Only the Pt atoms were replaced by Pd atoms and the structure was subsequently refined. The structure data for Pd8Hg3Se9 and HgSe were taken from the papers of Laufek et al. (Reference Laufek, Vymazalová, Drábek, Navrátil and Drahokoupil2014) and Earley (Reference Earley1950), respectively. During the Rietveld refinement, the scale factors and unit-cell parameters of these two phases were refined, while their fractional coordinates and displacement parameters were fixed.
Table I. Experimental conditions.
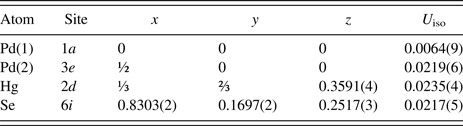
Table II. Refined parameters for Pd2HgSe3 [room temperature, space group P
 $\bar 3$
m1. a = 7.3096(2) Å, c = 5.2829(1) Å, V = 244.45(1) Å3, D
c = 8.84 g/cm3, Z = 2, R
p = 1.53%, R
wp = 2.21%, and R
B = 4.37%, F(000) = 502, μ = 206.3 mm−1].
$\bar 3$
m1. a = 7.3096(2) Å, c = 5.2829(1) Å, V = 244.45(1) Å3, D
c = 8.84 g/cm3, Z = 2, R
p = 1.53%, R
wp = 2.21%, and R
B = 4.37%, F(000) = 502, μ = 206.3 mm−1].
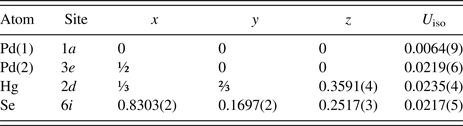
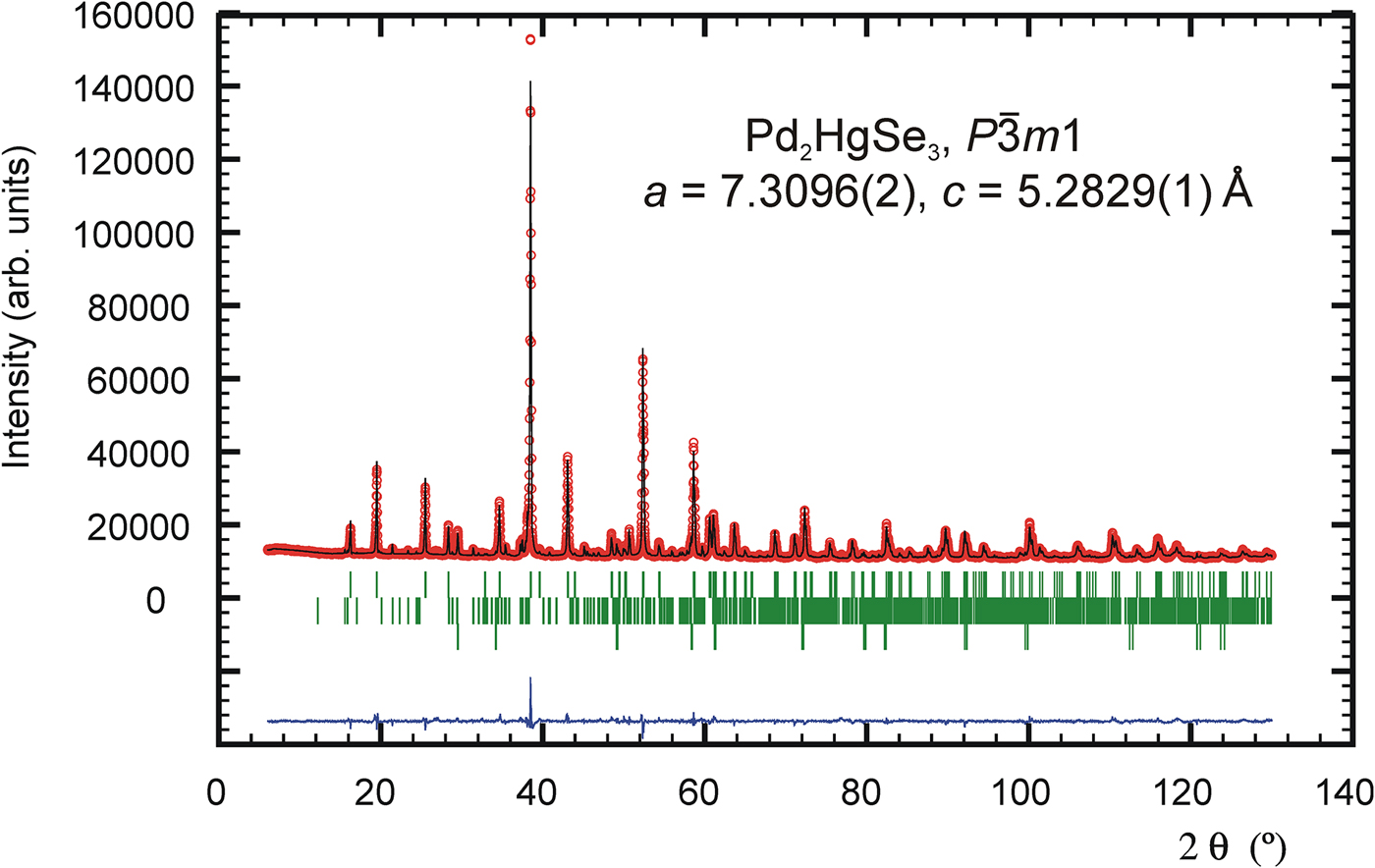
The pseudo-Voigt function was used to generate the shape of the diffraction peaks in the subsequent Rietveld refinement. The background was modelled by the linear interpolation between consecutive breakpoints in the pattern. The refined parameters of Pd2HgSe3 include those describing the peak shape and width, peak asymmetry, unit-cell parameters, fractional coordinates, March–Dollase model of preferred orientation along [001] and isotropic displacement parameters for all atoms. Peak asymmetry was modelled using the correction of Bérar and Baldinozzi (Reference Bérar and Baldinozzi1993); two parameters were refined. In total, 24 parameters were refined; 138 reflections were observed. The refinement of preferred orientation correction significantly improved the Rietveld fit; the R wp factor decreased from 4.78 to 2.21%. The G 1 parameter of the March–Dollase correction model was refined to 0.761(1). The final cycles of refinement converged to the residual factors: R B = 4.37%, R wp = 2.21%, and R p = 1.53%. The refined atomic coordinates are given in Table II. Figure 1 shows the final Rietveld plot.
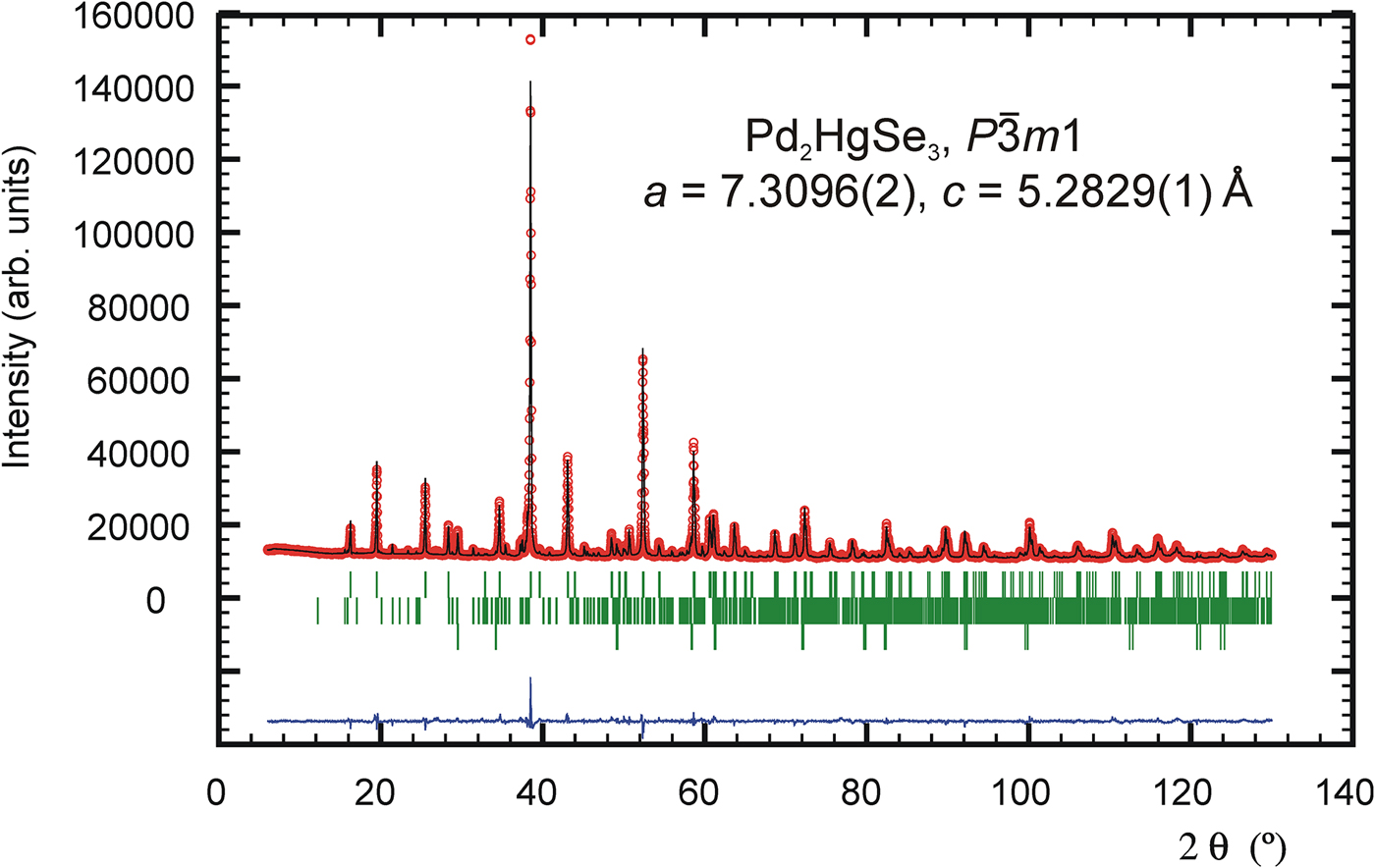
Figure 1. (Color online) The observed (circles), calculated (solid line), and difference Rietveld profiles for Pd2HgSe3. The upper reflections bars correspond to Pd2HgSe3; the middle and lower bars to 12 and 2 wt.% of Pd8Hg3Se9 and HgSe impurities, respectively.
IV. RESULTS AND DISCUSSION
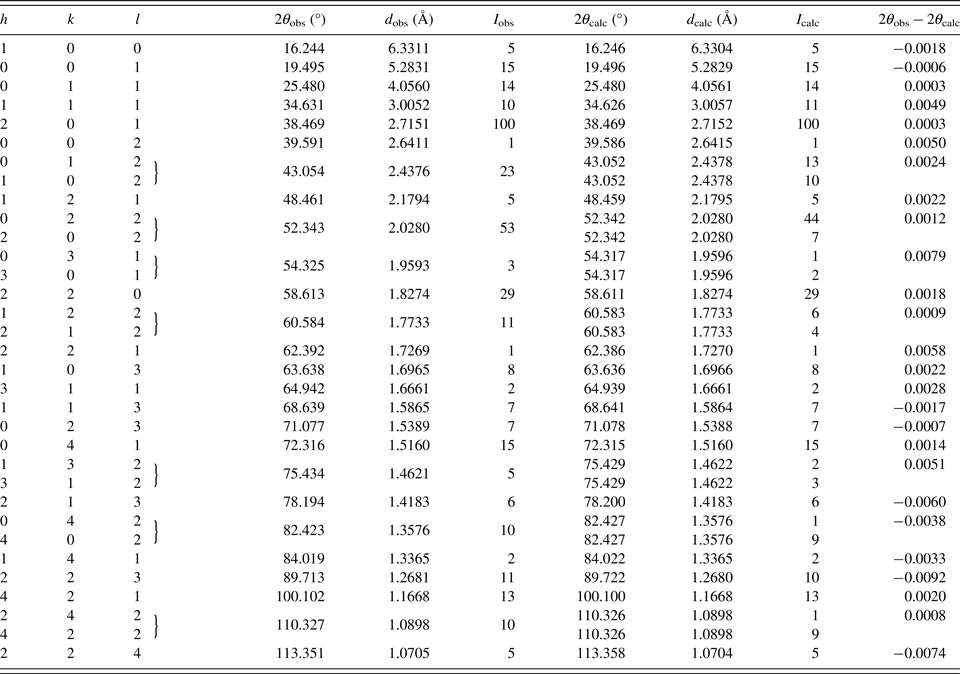
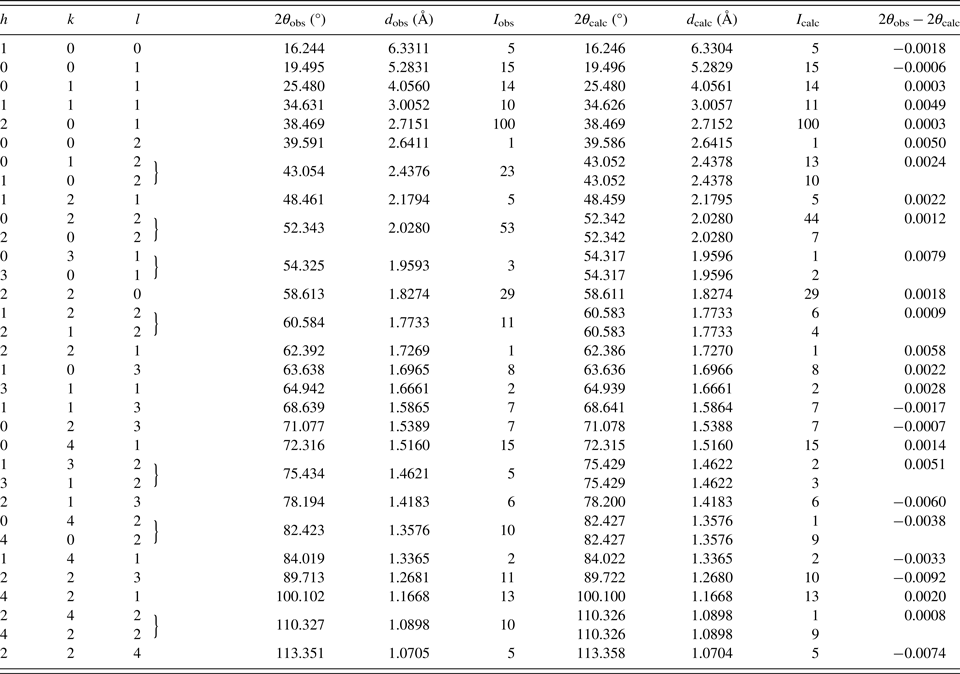
The powder diffraction data are listed in Table III. The observed values of diffraction positions, d-spacing, and intensities were extracted by the Topas programme (Bruker AXS, 2014) using the split Pearson VII profile function. The 2θ obs and d obs were corrected for the refined sample displacement correction. No significant diffraction overlap between diffraction patterns of Pd2HgSe3 and HgSe occurs. The phase Pd8Hg3Se9 (Pmmn, V = 782.5 Å) shows relatively complex diffraction pattern. Consequently, roughly above 60° of 2θ, there is a noticeable diffraction overlap between patterns of Pd2HgSe3 and Pd8Hg3Se9. However, considering the low content of Pd8Hg3Se9 in a sample and its complex diffraction pattern, its intensity contribution to the diffraction pattern of Pd2HgSe3 above 60° of 2θ is negligible.
Table III. Powder diffraction data for Pd2HgSe3 (CoKα radiation) (reflections with I obs and I calc < 1 are not shown in the table).
The crystal structure of Pd2HgSe3 is isostructural with mineral jacutingaite, Pt2HgSe3 (Vymazalová et al., Reference Vymazalová, Laufek, Drábek, Cabral, Haloda, Sidorinová, Lehmnn, Galbiatti and Drahokoupil2012), and synthetic phases Pt4Tl2S6, Pt4Tl2Se6, Pt4Tl2Te6, and Pd4Tl2Se6 (Bronger and Bonsmann, Reference Bronger and Bonsmann1995). The structure contains two independent Pd positions, one Hg and one Se position. The Pd(1) atom is surrounded by six Se atoms at 2.526(2) Å forming a slightly distorted octahedron with intra-octahedral Se–Pd(1)–Se angles 85.14(5)° and 94.85(5)° instead of 90° being for a regular octahedron. Unlike the Pd(1) atom, the Pd(2) atom forms a square planar configuration with four Se atoms at a distance of 2.478(2) Å. The coordination of the Pd(2) atom is further completed by two Hg atoms at 2.837(1) Å. The vector of Pd–Hg bonds is almost perpendicular to the [PdSe4] squares. This Pd–Hg distance is comparable to 2.82 Å found in PdHg (Terada and Cagle, Reference Terada and Cagle1960) indicating significant Pd–Hg bonding interactions.
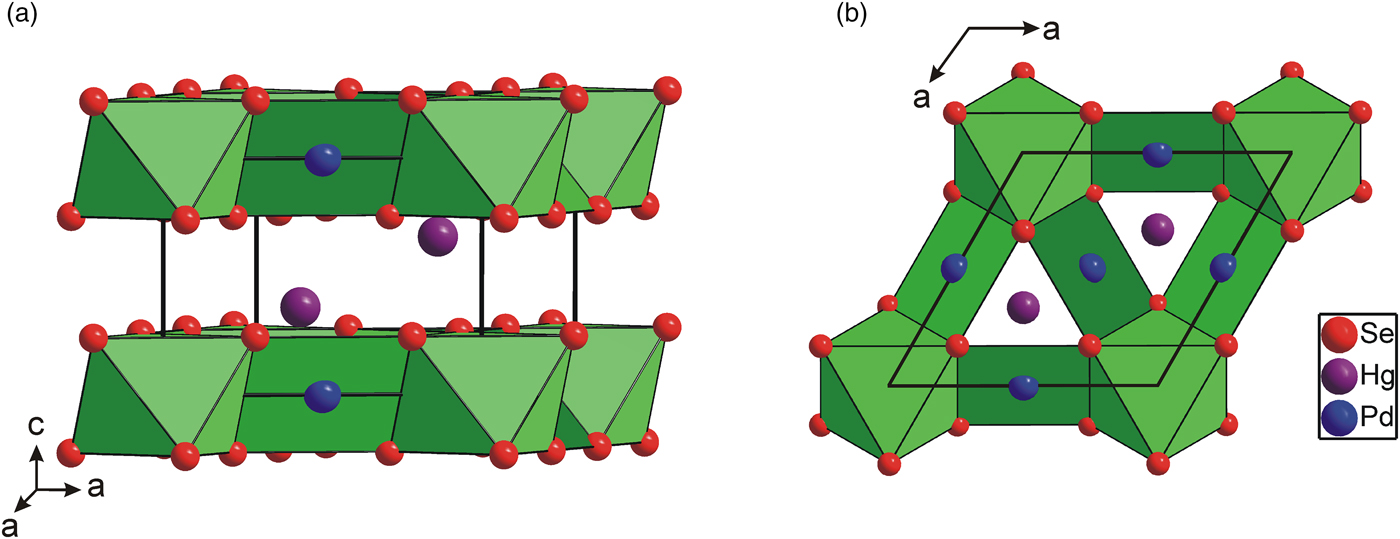
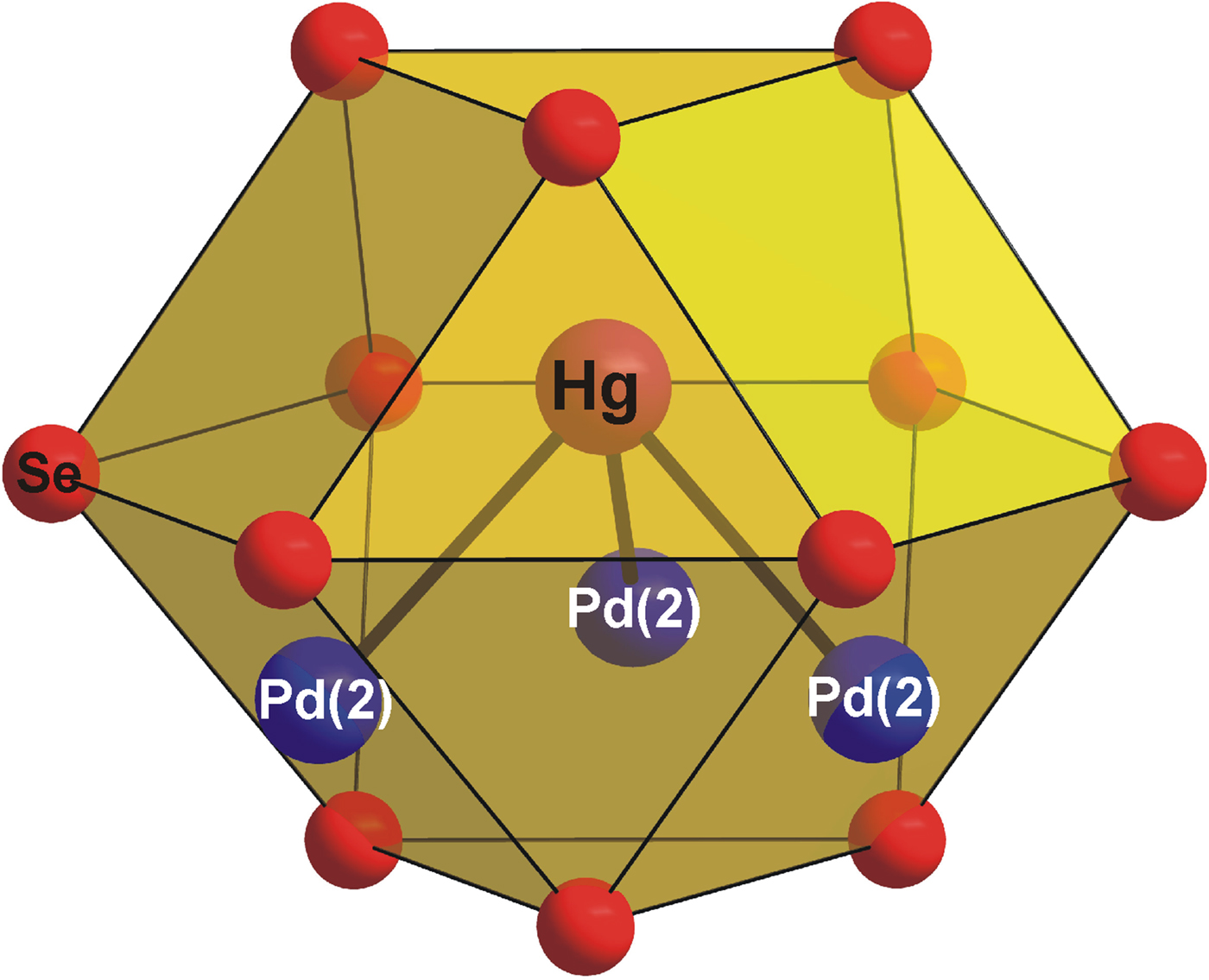
As is shown in Figure 2, the [PdSe6] octahedra shares six Se–Se edges with adjacent [PdSe4] squares forming layers parallel to the (001) plane. The layers are stacked along the c-axis and show the AA type of stacking. It is interesting to note that the [PdSe6] octahedra have a shared edge length [d Se–Se = 3.419(2) Å] considerably shorter than its unshared edge [d Se–Se = 3.721(2) Å], which is in agreement with the Pauling third rule (Pauling, Reference Pauling1929). The [PdSe4] squares also show two different Se–Se edge lengths: 3.419(2) and 3.588(2) Å for the shared and unshared edges, respectively. This brings the Pd–Pd separation to 3.655(1) Å and consequently reduces the Coulombic repulsion between cations. Hg atoms show three short Pd and Se contacts at 2.837(1) and 2.918(2) Å. For the sake of convenient structure description, Hg atoms can be considered to occupy the anti-cubooctahedral voids formed by Se atoms with the 3 + 6 + 3 bonding scheme. However, the next six and three Se atoms are at distances of 3.698(2) and 3.834(2) Å, respectively, and thus represent only weak Hg–Se bonds. In addition, Hg atom has three Pd contacts at 2.837(1) Å forming [HgPd3] trigonal pyramid (Figure 3). Selected interatomic distances compared with those found in isostructural Pt2HgSe3 are summarized in Table IV. The relatively high values of displacement parameters (U iso) of Pd(2), Hg, and Se atoms (see Table II) might be an indirect indication of possible structural disorder; however, no broad peaks or apparent peak asymmetry have been observed in powder diffraction pattern of Pd2HgSe3.
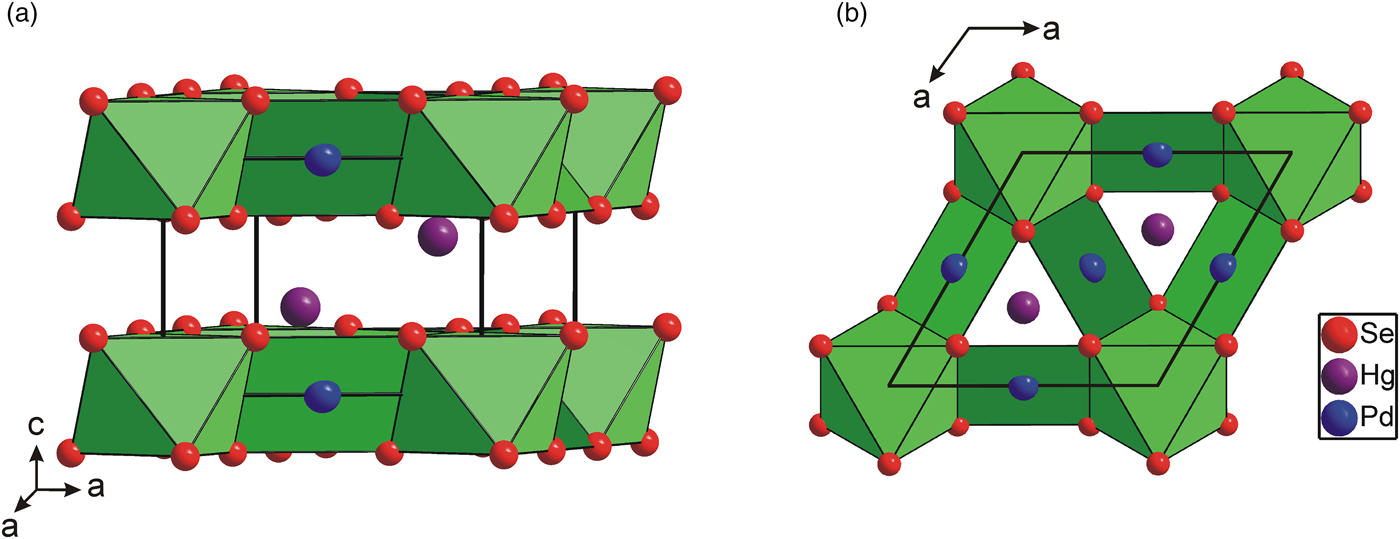
Figure 2. (Color online) Polyhedral representation of Pd2HgSe3 crystal structure showing the [Pd(1)Se6] octahedra and [Pd(2)Se4] squares. (a) Perspective view, (b) view along the c-axis.
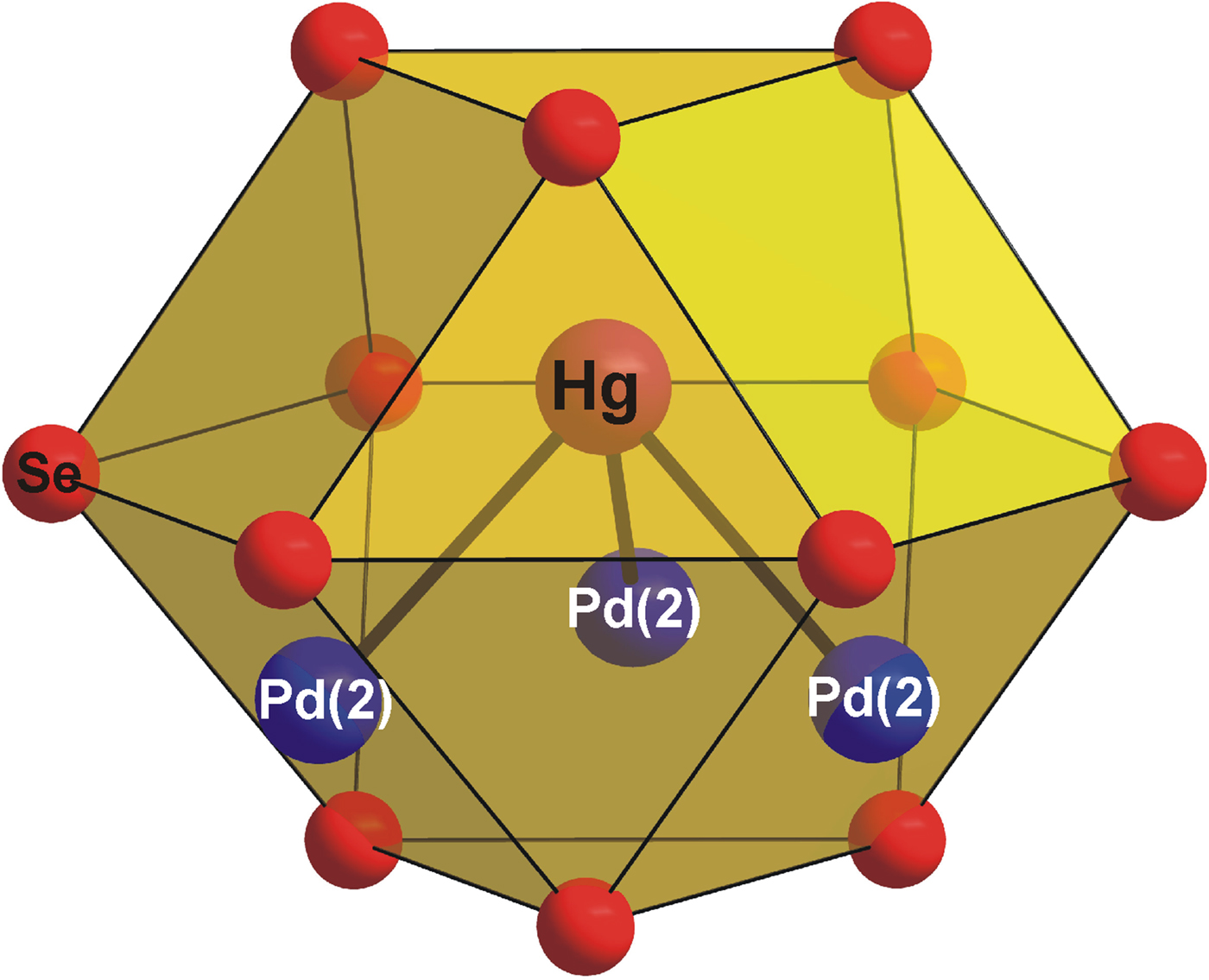
Figure 3. (Color online) The coordination polyhedra of Hg in the Pd2HgSe3 crystal structure.
Table IV. Selected interatomic distances (Å) in isostructural compounds Pd2HgSe3 and Pt2HgSe3.

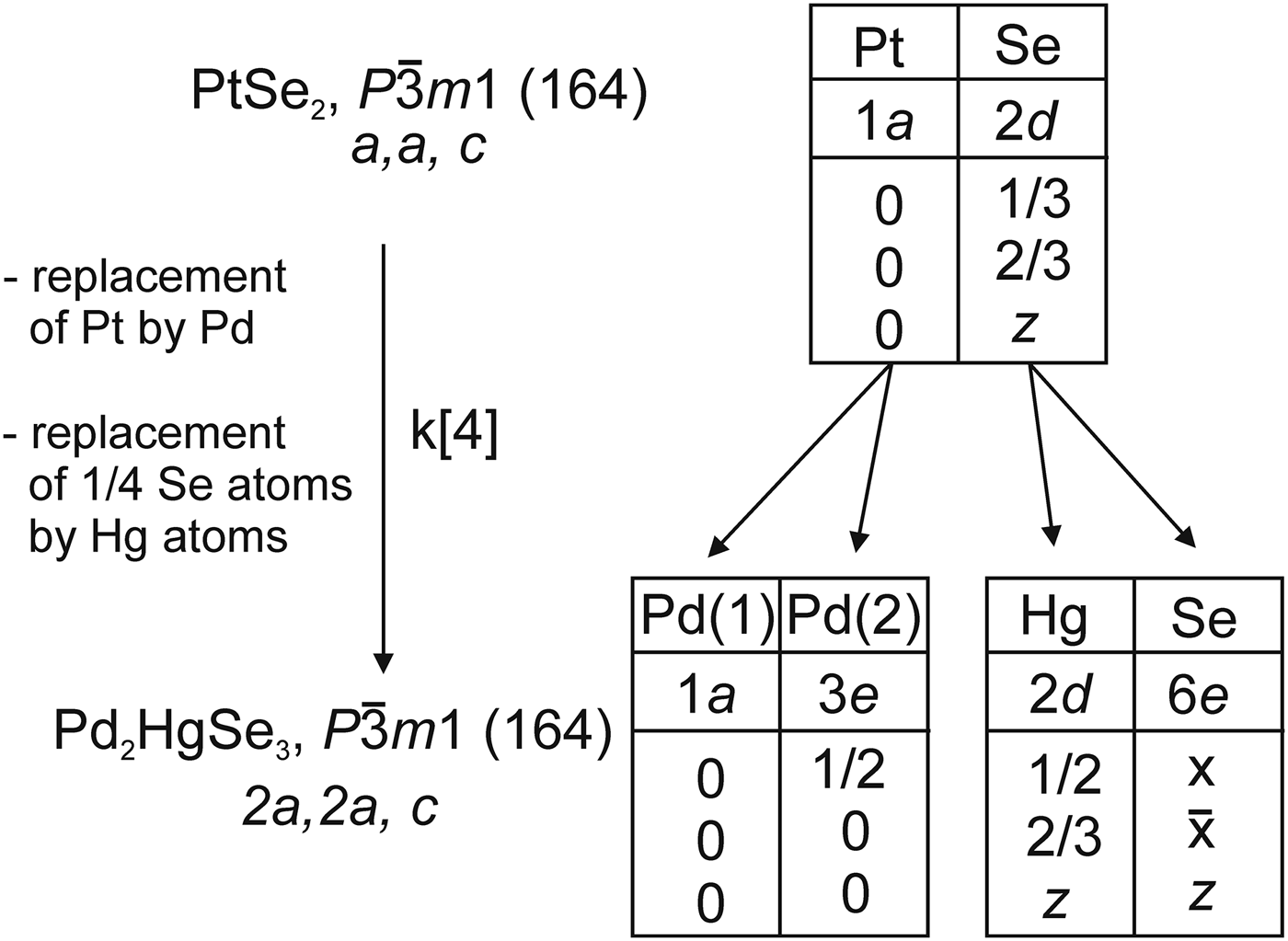
The crystal structure of Pd2HgSe3 can also be described as a 2a.2a.c superstructure of PtSe2 structure, where Pt atoms and one-quarter of Se atoms have been replaced by Pd and Hg atoms, respectively. PtSe2, which is also known as a mineral sudovikovite, adopts a layered structure of the CdI2-structure type (Furuseth et al., Reference Furuseth, Selte and Kjekshus1965). The corresponding group–subgroup relation is
 $P\bar 3m1(k4/2a.2a.c)P\bar 3m1$
and is shown in Figure 4. The splitting of Wyckoff position in the space group
$P\bar 3m1(k4/2a.2a.c)P\bar 3m1$
and is shown in Figure 4. The splitting of Wyckoff position in the space group
 $P\bar 3m1$
was constructed using the programme Wycksplit (Kroumova et al., Reference Kroumova, Perez-Mato and Aroyo1998).
$P\bar 3m1$
was constructed using the programme Wycksplit (Kroumova et al., Reference Kroumova, Perez-Mato and Aroyo1998).
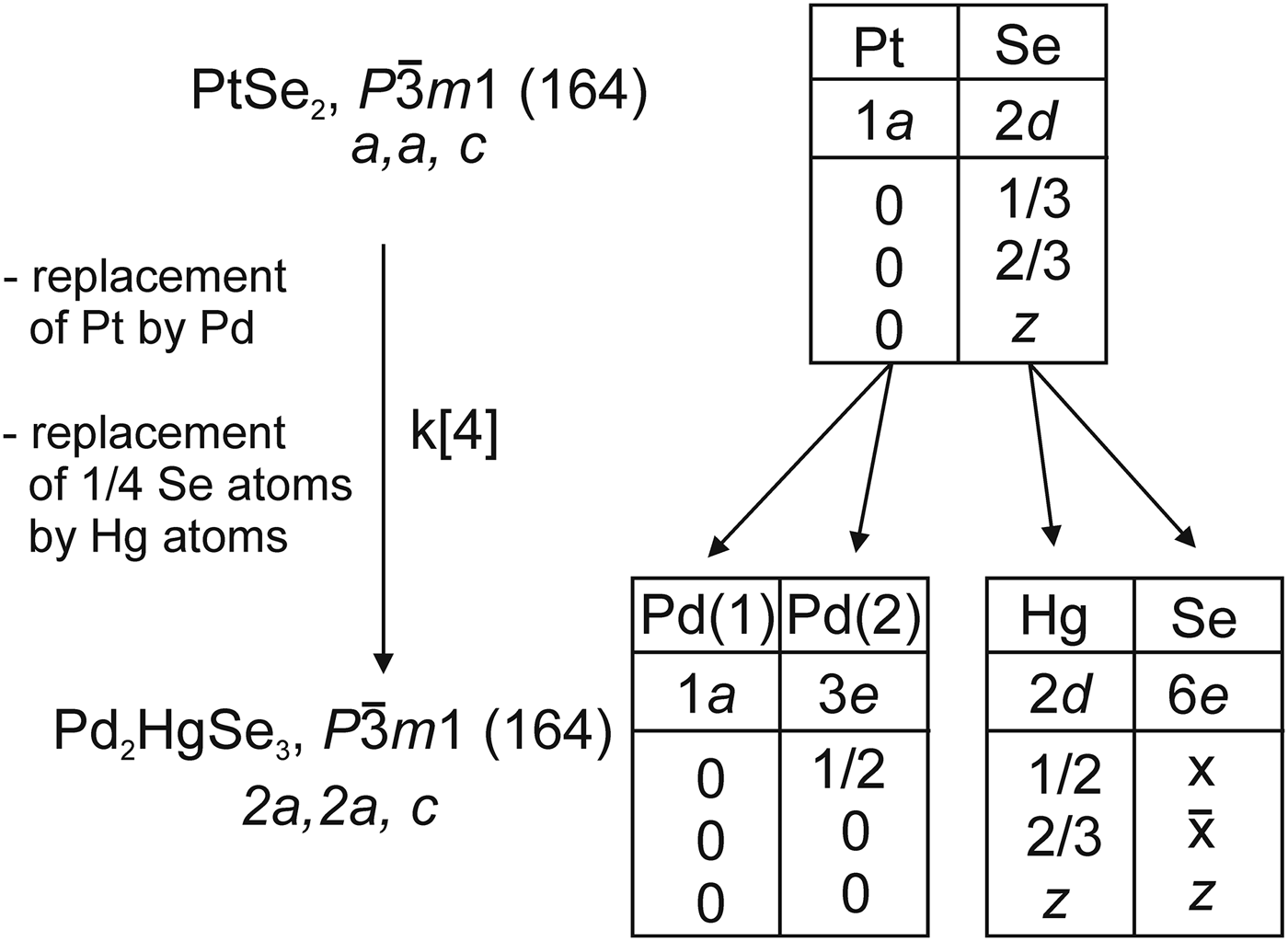
Figure 4. The group–subgroup relation between the PtSe2 and Pd2HgSe3 structures including splitting of the Wyckoff positions.
Apart from its isostructural compounds (see above), the most closely structural related phase to Pd2HgSe3 is the mineral temagamite, Pd3HgTe3 (Laufek et al., Reference Laufek, Vymazalová, Drábek, Dušek, Navrátil and Černošková2016). Both crystal structures show trigonal symmetry and Pd3HgTe3 shows approximately similar unit-cell parameter a [7.8211(6) Å]. The crystal structure of Pd3HgTe3 was described as being composed of layered modules (denoted as A through F) stacked along the c-axis. The modules B and F in temagamite structure show the same bond topology of Pd and Te atoms as Pd and Se atoms in Pd2HgSe3. Hg atoms occupy the anti-cubooctahedral voids in both compounds. The crystal structure of another Pd–Hg selenide, the mineral tischendorfite Pd8Hg3Se9 (Laufek et al., Reference Laufek, Vymazalová, Drábek, Navrátil and Drahokoupil2014), differs from Pd2HgSe3 to a certain extent. On one hand, Pd and Se atoms in Pd8Hg3Se9 show different bond topology. On the other hand, Hg atoms have essentially identical coordination in both compounds. The crystal structure of a third known Pd–Hg selenide, the phase Pd5HgSe (Laufek et al., Reference Laufek, Vymazalová, Drábek, Navrátil, Plecháček and Drahokoupil2012), is based on [HgPd12] cubooctahedra and [SePd8] rectangular prism, and hence deviates significantly from the structure of Pd3HgSe2.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0885715617001026.
ACKNOWLEDGEMENTS
This work was supported by the internal projects Nos. 332000 and 331400 from the Czech Geological Survey. The authors thank Jan Drahokoupil (Institute of Physics of the Czech Academy of Sciences) for assistance in XRD data collection.