



The bicuspid aortic valve is one of the most common congenital heart defects and has a complex range of manifestations. At the benign end of this spectrum, the bicuspid aortic valve may be an incidental anomaly that remains subclinical over an entire lifetime (Fig 1),whereas at the severe end of the spectrum, the bicuspid aortic valve may be an outcome-determining factor owing to aortic valve dysfunction, left ventricular failure, and aortic disease, which can manifest at any point in life, from birth to adult life.Reference Fernandes, Khairy and Graham 1 , Reference Michelena, Desjardins and Avierinos 2 The bicuspid aortic valve may also occur in association with other congenital lesions – mainly left-sided obstructive lesions such as aortic coarctation and hypoplastic left heart syndrome.Reference Koenraadt, Bartelings and Bökenkamp 3
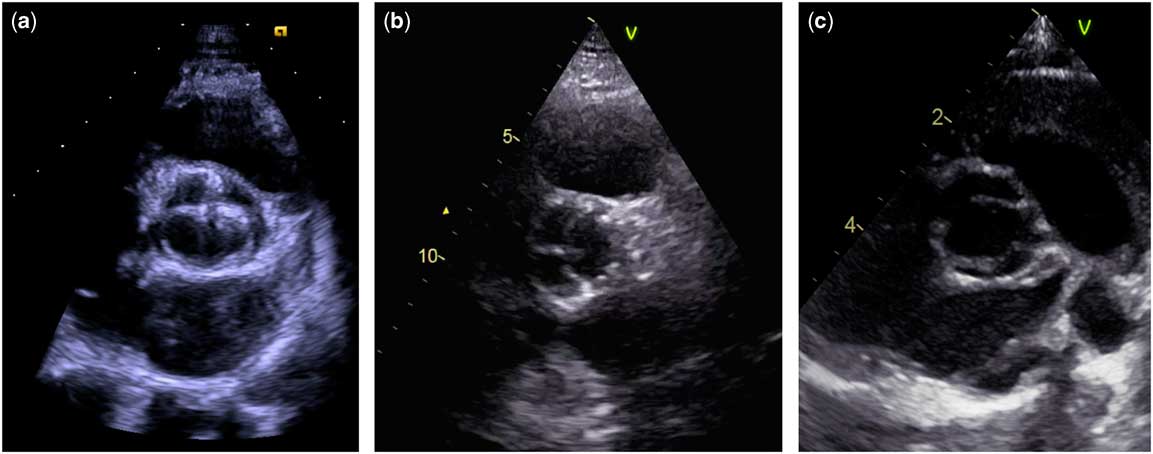
Figure 1 The three types of bicuspid aortic valve in three different clinical settings. ( a ) 17-year-old male with a type 1 bicuspid valve (left and right coronary cusp fusion) and moderate stenosis. ( b ) 78-year-old male with a normal functioning type 2 bicuspid valve (right and non-coronary cusp fusion). ( c ) Newborn boy with Edwards syndrome (trisomy 18) and a type 3 bicuspid valve (left and non-coronary cusp fusion). The bicuspid aortic valve classification is based on the paper by Schaefer et al.Reference Schaefer, Lewin and Stout 29
Over the last few decades, major advances in the field of embryology have improved our understanding of the multi-faceted development of both the normal tricuspid and abnormal bicuspid aortic valve.Reference Phillips, Mahendran, Singh, Anderson, Chaudhry and Henderson 4 – Reference Freeze, Landis, Ware and Helm 6 Further light has been shed on the aetiology of the bicuspid aortic valve, with the identification of several syndromes and chromosomal disorders associated with a bicuspid aortic valve and also numerous genes postulated to be involved in the process of cusp fusion.Reference Giusti, Sticchi, De Cario, Magi, Nistri and Pepe 7 , Reference Lin, Lin, Chen, Zhou and Chang 8
Furthermore, improved knowledge of clinical outcomes has charted an area of evidence-driven clinical diagnoses, surveillance, and intervention in asymptomatic and symptomatic bicuspid aortic valve disease.Reference Kerneis, Pasi and Arangalage 9 – Reference McKellar, Michelena, Li, Schaff and Sundt 11 Despite these major advances, considerable knowledge deficits prevail regarding the development, aetiology, and outcomes of the bicuspid aortic valve. For example, does the origin have bearings on the treatment of patients with a bicuspid valve and does it matter which cusps are fused?
The current clinical guidelines only leave a small space for specific recommendations on how to handle the bicuspid valve in regards to valve repair or replacement, aortic root surgery, pregnancy issues, and so on, and the relevance of population screening programmes also remains undetermined. Do we save lives by identifying persons with a bicuspid aortic valve or do we over-diagnose persons with well-functioning bicuspid aortic valves who will never need treatment?Reference Brodersen, Schwartz, Heneghan, O’Sullivan, Aronson and Woloshin 12
Considering the above questions, the aim of this review is to present an overview of the current understanding of the embryological development and genes associated with the development of the bicuspid aortic valve. The review also seeks to map the current state-of-the-art clinical care in valve disease in children and adults and aortic disease in patients with a bicuspid aortic valve, along with pregnancy-related challenges and considerations related to population screening.
Prevalence
A bicuspid aortic valve occurs in 1–2%.Reference Hoffman and Kaplan 13 In newborns, the prevalence has been described to be as low as 4.6 per 1000 live-born children and is far more common in boys (7.1 per 1000 boys) than that in girls (1.9 per 1000 girls).Reference Tutar, Ekici, Atalay and Nacar 14 These gender differences suggest that the unopposed X chromosome may play a major role because males only have one X chromosome similar to women with Turner syndrome in whom the incidence of a bicuspid aortic valve is as high as 15–30%.Reference Mortensen, Andersen and Gravholt 15 A bicuspid aortic valve is also common in chromosomal diseases, such as Down’s syndrome (trisomy 21),Reference Niaz, Poterucha and Olson 16 DiGeorge (22q11),Reference Niaz, Poterucha and Olson 16 , Reference Pierpont, Basson and Benson 17 and Edwards syndrome (trisomy 18),Reference Pierpont, Basson and Benson 17 and also in genetic syndromes, such as Williams syndrome, Holt-Oram syndrome,Reference Niaz, Poterucha and Olson 16 , Reference Pierpont, Basson and Benson 17 Marfan (4.7%),Reference Nistri, Porciani, Attanasio, Abbate, Gensini and Pepe 18 and Loeys-Dietz syndromes (8.8%).Reference Van Hemelrijk, Renard and Loeys 19 This mix of chromosomal and genetic disorders implies that a “common trigger” is unlikely; instead, the bicuspid aortic valve may be the final consequence of numerous complex mechanisms influencing the creation of the semilunar valves (Fig 2).
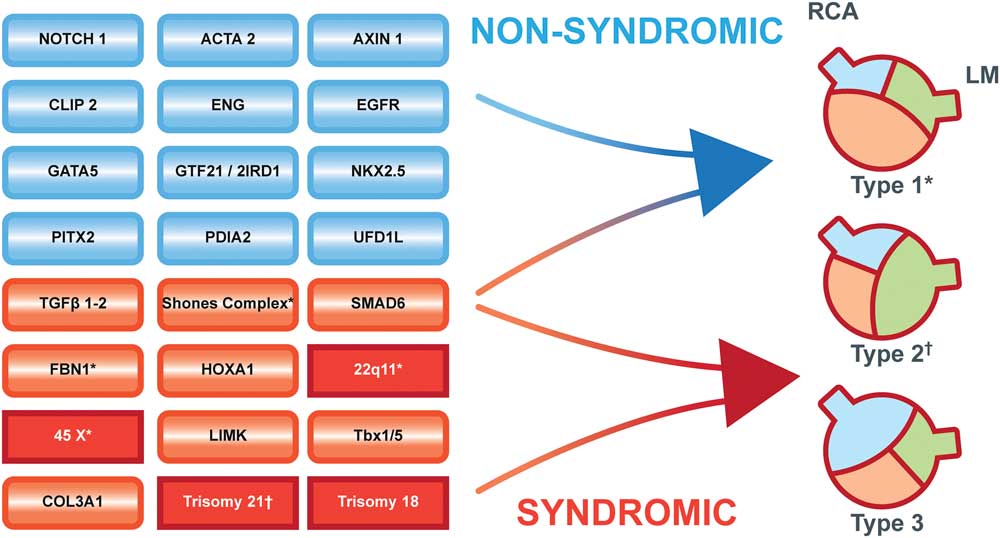
Figure 2 Multiple pathways to the development of a bicuspid aortic valve. Blue boxes indicate genes associated with non-syndromic bicuspid valves; red boxes indicate genetic syndromes; dark red squared boxes are chromosomal disorders. Type 1 bicuspid valve with fusion of the left and right coronary cusps. Type 2 bicuspid valve with fusion of the right and non-coronary cusps. Type 3 bicuspid valve with fusion of the left and non-coronary cusps. (*) Diseases associated with a type 1 valve. (†) Diseases associated with a type 2 valve. The bicuspid aortic valve classification is based on the paper by Schaefer et al.Reference Schaefer, Lewin and Stout 29 LM: Left main coronary artery. RCA: Right coronary artery.
Foetal development
The aortic and pulmonary valves develop within the very first weeks in the embryo. Cardiac mesodermal cells from the primitive streak give origin to the first and second heart fields in the embryo.Reference Yassine, Shahram and Body 20 The first heart field cells give origin to the early heart tube. Then, cells from the second heart field contribute to the development of the conotruncal tissue that gives origin to the outflow tracts.Reference Martin, Kloesel, Norris, Lindsay, Milan and Body 21 This tissue also forms the semilunar valves.Reference Martin, Kloesel, Norris, Lindsay, Milan and Body 21 Cells from the neural crest migrate into the conotruncal tissue and septate the common vessel during the spiralling of the outflow tracts. The final development of the semilunar valves is a complex interplay between the second heart field cells and the neural crest cells, where the rudimentary aortic valve leaflets are shaped by neural crest cells that create “cavities” in the rudimentary valve.Reference Phillips, Mahendran, Singh, Anderson, Chaudhry and Henderson 4 , Reference Freeze, Landis, Ware and Helm 6 If the cavities are not created, the cusps will be fused.Reference Phillips, Mahendran, Singh, Anderson, Chaudhry and Henderson 4 , Reference Freeze, Landis, Ware and Helm 6
It is hypothesised that right coronary and non-coronary cusp fusion is caused by defective morphogenesis of the outflow tract tissue before “valve cavitation”, whereas perturbations in the neural crest cell behaviour result in the lack of “cavitation” and thereby right and left coronary cusp fusion occurs.Reference Fernandez, Duran and Fernandez-Gallego 22 These hypotheses are supported by the observation that patients with right and left coronary cusp fusion are more prone to anomalies of the coronary arteries, and because neural crest cells appear to play a major part in the positioning of the coronary arteries, the combination of a bicuspid valve and coronary anomalies can have neural crest cell dysfunction as a common denominator.Reference Phillips, Mahendran, Singh, Anderson, Chaudhry and Henderson 4 The mechanism behind the rare fusion of the left and non-coronary cusp is unresolved.
Genetics
The temporal interplay of the genes necessary to develop a normal heart and outflow tracts is unclear. Several genes are involved in the normal development of the semilunar valves, and abnormal development into a bicuspid aortic valve is part of what is called left ventricular outflow tract obstructive lesions. The spectrum of left ventricular outflow tract obstructive lesions includes a number of malformations other than a bicuspid aortic valve, including mitral valve abnormalities, subaortic stenosis, and coarctation of the aorta.Reference Koenraadt, Bartelings and Bökenkamp 3 These congenital defects may appear in isolation or as a constellation of abnormalities in the complete Shone’s complex and hypoplastic left heart syndrome, conditions that can be seen in females with Turner syndrome.Reference Freeze, Landis, Ware and Helm 6 The fact that females with Turner syndrome have a very frequent occurrence of a bicuspid valve (15–30%),Reference Mortensen, Andersen and Gravholt 15 particularly in the 45, X karyotype, points towards the importance of genes on the X chromosome for development of the bicuspid valve.Reference Phillips, Mahendran, Singh, Anderson, Chaudhry and Henderson 4 However, only a minority of females with 45, X will develop a bicuspid valve, indicating that more complex genetic mechanisms including more than only genes on the X chromosomes are involved.Reference Mortensen, Andersen and Gravholt 15
Normal cardiac morphogenesis involves several key signalling pathways, including the tumour growth factor-β superfamily, nitric oxide synthase 3, Wnt/β-catenin, vascular endothelial growth factor, Tbx20, and the GATA family.Reference Freeze, Landis, Ware and Helm 6 It is clear that different mutations in the transcriptional regulatory NOTCH1 gene can lead to the development of a bicuspid aortic valve.Reference Garg, Muth and Ransom 5 , Reference McKellar, Tester, Yagubyan, Majumdar, Ackerman and Sundt 23 Tumour growth factor-β signalling is critical for the differentiation and survival of smooth muscle cells of neural crest origin and for maintaining the contractile phenotype of these smooth muscle cells.Reference Li, Li and Jiao 24 , Reference Jiao, Xiong and Wang 25 In many familial cases, the isolated bicuspid aortic valve has an autosomal dominant inheritance with reduced penetrance and variable expressivity.Reference Freeze, Landis, Ware and Helm 6 , Reference Laforest and Nemer 26 However, in other cases, notably in many syndromes, this valve morphology appears as part of a more serious cardiovascular phenotype, such as FBN1 (Marfan syndrome), TGFBR1/2 (Loeys-Dietz syndrome), and ACTA2 (thoracic aortic aneurysm and dissection syndrome).Reference Giusti, Sticchi, De Cario, Magi, Nistri and Pepe 7 , Reference Papagiannis 27
Variations in a number of other genes, such as HOXA1 (Bosley–Salih–Alorainy syndrome, Athabaskan brainstem dysgenesis syndrome), COL3A1 (vascular Ehlers-Danlos syndrome), Tbx1/5 (Holt-Oram syndrome), NKX2.5, UFD1L, GATA4/5, SMAD6, PITX2, CLIP2, GTF2I, GTF2IRD1, AXIN1, EGFR, ENG, PDIA2, and LIMK (Williams–Beuren syndrome) and MEF2c,Reference Giusti, Sticchi, De Cario, Magi, Nistri and Pepe 7 , Reference Lin, Lin, Chen, Zhou and Chang 8 have been found to be associated with bicuspid aortic valve formation (Fig 2). Variations in yet other genes have been identified that may be involved in the development of the bicuspid aortic valve and associated malformations.Reference Freeze, Landis, Ware and Helm 6 These findings indicate a genetic diversity behind the occurrence of bicuspid aortic valves, highlighting the need for a complete and comprehensive understanding of the genetic machinery behind normal cardiovascular embryogenesis.
We recently associated haploinsufficiency of the TIMP1 gene with bicuspid aortic valves in Turner syndrome in the presence of two common single-nucleotide variants in the TIMP3 gene (submitted data). Therefore, we proposed that the combination of X chromosome TIMP1 haploinsufficiency and variants of the chromosome 22 paralogue TIMP3 increases the risk of bicuspid aortic valve formation in Turner syndrome. The TIMP1 gene is situated on the X chromosome and shows variable and tissue-dependent escape status and is normally expressed from both X chromosomes in normal females.Reference Tukiainen, Villani and Yen 28 Therefore, such a mechanism only increases the genetic complexity behind normal cardiac development and should also spur new developments within the field and allow for new comparative bioinformatic approaches to the study of the bicuspid aortic valve and associated disorders.
Valve morphology
As previously mentioned, there are three morphological types of the bicuspid aortic valve. Type 1 is the most common, where the left and right coronary cusps are fused (79%). In type 2, the second most common, the right and non-coronary cusps are fused (20.5%). Type 3 is a fusion of the left and non-coronary and is quite rare (0.5%)Reference Schaefer, Lewin and Stout 29 (Fig 2). In the majority of cases, the bicuspid aortic valve is associated with a raphe, where the cusps are fused and some even have two raphes.Reference Sievers and Schmidtke 30 Few cases have no visible raphe.Reference Sievers and Schmidtke 30 The foetal origin of the raphe is unresolved, but abnormal foetal haemodynamics during cusp fusion or variations in foetal cellular migration may be involved. The presence of a raphe appears to have some relevance since valve dysfunction and aortic dilation are more likely when a raphe is present.Reference Della Corte, Body and Booher 31 There is a trend towards more type 1 valve morphology in patients from Asia,Reference Kong, Regeer and Poh 32 Turner syndrome,Reference Mortensen, Hjerrild and Stochholm 33 DiGeorge and Marfan syndrome, and Shone’s complex,Reference Niaz, Poterucha and Olson 16 whereas type 2 is predominate in trisomy 21Reference Niaz, Poterucha and Olson 16 (Fig 2).
Aortic dilatation and dissection
The development of aortic dilatation in patients with a bicuspid aortic valve is enigmatic. Several theses have been proposed, spanning from detrimental haemodynamic effects of the aortic flow because of cusp fusion, to aortopathy because of an underlying genetic disease that also caused cusp fusion.
Overall, there are three different phenotypes of aortic dilatation. The most common type involves dilatation of the tubular part of the ascending aorta with mild to moderate involvement of the aortic root.Reference Verma and Siu 34 This type of dilatation appears to be associated with older age, aortic valve stenosis, and right and left cusp fusion.Reference Schaefer, Lewin and Stout 29 , Reference Verma and Siu 34 , Reference Mahadevia, Barker and Schnell 35 Isolated involvement of the tubular part of the ascending aorta is the second most common. There is usually relative sparing of the aortic root, but the dilatation frequently extends into the transverse aortic arch.Reference Jiao, Xiong and Wang 25 Patients with solitary dilatation of the aortic sinus are not very common, and in these cases, a syndromic (genetic) cause should be considered.Reference Nistri, Porciani, Attanasio, Abbate, Gensini and Pepe 18 , Reference Van Hemelrijk, Renard and Loeys 19 , Reference Papagiannis 27 , Reference Verma and Siu 34
In children, it is not uncommon to see dilatation of the aorta, but dissections in non-syndromic children are extremely rare.Reference Fernandes, Khairy and Graham 1 , Reference Mahle, Sutherland and Frias 36 However, in syndromic diseases such as Marfan and Loeys-Dietz syndrome (dilated sinuses), dissections can appear during childhood or adolescence (Fig 3).Reference Van Hemelrijk, Renard and Loeys 19 , Reference Groth, Stochholm and Hove 37 The true mechanisms behind these different types of dilatation of the aorta are most likely complex and probably involve a combination of abnormal tissue quality and changes in flow and wall stress.

Figure 3 Aortic aneurysm (41 mm) in a 6-year-old child with Loeys-Dietz syndrome and a bicuspid valve.
When a patient with a bicuspid aortic valve has a normal aorta size, the aortic tissue also appears histologically normal.Reference Yassine, Shahram and Body 20 However, in bicuspid aortic valve patients with aortic dilation, there is usually normal fibre architecture but non-inflammatory loss of smooth muscle cells, with multi-focal apoptosis and media degeneration with lower fibrillin content and increased tissue tumour growth factor-β1 levels.Reference Yassine, Shahram and Body 20
The foetal development of the outflow tracts is primarily derived from the second heart field cells,Reference Ramsbottom, Sharma and Rhee 38 whereas the smooth muscle cells in the ascending aorta are mostly derived from the cardiac neural crest cells.Reference Phillips, Mahendran, Singh, Anderson, Chaudhry and Henderson 4 Whether this different source has any bearings on the dilatation of the aorta is not completely clear, but some studies indicate that the presence of a bicuspid aortic valve is part of an aortopathy in the ascending part of the vessel.Reference Abdulkareem, Smelt and Jahangiri 39
The other major theory is the formation of unfavourable flow patterns owing to specific cusp fusion.Reference Meierhofer, Schneider and Lyko 40 Several studies have shown flow abnormalities in patients with a bicuspid aortic valve depending on which cusps are fused.Reference Vergara, Viscardi, Antiga and Luciani 41 , Reference Viscardi, Vergara and Antiga 42 The typical flow through the ascending aorta is usually with a slight helical spiral, whereas patients with a bicuspid aortic valve have accentuated helical flow. Dependent on cusp fusion, wall stress is increased in different areas in the aortic root.Reference Viscardi, Vergara and Antiga 42 Right helical flow and fusion of the right and non-coronary cusp (type 2) have been shown to create the most inappropriate flow patterns and the most unfavourable wall shear stress.Reference Mahadevia, Barker and Schnell 35 , Reference Bissell, Hess and Biasiolli 43 However, left and right cusp fusion (type 1) also creates abnormal flow patterns and increased wall stress.Reference Mahadevia, Barker and Schnell 35 Irrespective of the bicuspid valve type, valve dysfunction can accentuate the aortic dilatation.Reference Verma and Siu 34 Aortic regurgitation, in particular, will result in abnormal wall stress and subsequently causes aortic dilation.Reference Thanassoulis, Yip and Filion 44 , Reference Novaro, Tiong, Pearce, Grimm, Smedira and Griffin 45
Despite these observations, it is not uncommon to see elderly patients with normal aortic diameters even in the presence of a bicuspid aortic valve, so it appears that there must be an extra factor present beyond cusp fusion to make the aorta dilate to a significant extent.
Clinical follow-up of the dilated aorta
Basically, the only reason to do regular check-ups in a patient with a normal functioning bicuspid aortic valve is the risk of aortic dilatation and subsequent dissection. When to see the patients and how often depends on the size of the aorta, age of the patient, and doctor’s state of mind. Despite the previously mentioned flow abnormalities, the annual dilatation rate is very slow. It is reported in several different publications to be between 0.1 and 0.5 mm per year.Reference Page, Mongeon, Stevens, Souliere, Khairy and El-Hamamsy 46 , Reference Avadhani, Martin-Doyle, Shaikh and Pape 47 There is nothing that indicates the dilatation rate is faster if there is concomitant aortic valve stenosis.Reference Kerneis, Pasi and Arangalage 9
The dilatation rate is much slower than in patients with Marfan syndrome but significantly faster than in non-syndromic patients with degenerative aortopathy.Reference Detaint, Michelena, Nkomo, Vahanian, Jondeau and Sarano 48 In the study by Detaint et al,Reference Detaint, Michelena, Nkomo, Vahanian, Jondeau and Sarano 48 43% of the patients did not progress at all during 3.6 years of follow-up.
The dilatation rate is also likely to be dependent on the presence of hypertension,Reference de Simone, Roman and De Marco 49 whether there are additional congenital defect such as coarctation of the aortaReference Quenot, Boichot and Petit 50 or presence of a syndrome, although there are no studies to date that have followed different groups of syndromic patients with bicuspid valves.
Aortic dissection
In the general population, patients with a bicuspid aortic valve experience aorta dissections at a significantly younger age (46.7 years) compared with their tricuspid peers (61.6 years)Reference Etz, von Aspern and Hoyer 51 but at an older age than Marfan patients (41.2 years).Reference Groth, Stochholm and Hove 52 However, the question is whether this potential risk can be translated to any patient with a bicuspid aortic valve. In the GENTAC register, there were only two dissections in patients with bicuspid aortic valves. These patients had aortic sizes of 42 and 49 mm, but there were no data on the indexed aortic size, cusp morphology, or whether these patients showed rapid progression of the aortic diameter before the dissection.Reference Weinsaft, Devereux and Preiss 53 The phenotype of aortic root dilatation may be a marker of a more severe aortopathy, necessitating closer follow-up and earlier root replacement. The solitary dilatation of the aortic sinus appears to progress faster than the phenotype with even dilatation of the ascending aorta.Reference Della Corte, Bancone and Buonocore 54
Nothing appears to indicate that certain types of cusp fusion should be followed more closely than others,Reference Detaint, Michelena, Nkomo, Vahanian, Jondeau and Sarano 48 and the recommendation to use β-blockers for prophylaxis is not supported by evidence.Reference Erbel, Aboyans and Boileau 55 However, an ongoing study – “The Beta Blockers and Angiotensin Receptor Blockers in Bicuspid Aortic Valve Disease Aortopathy (BAV) study” (ClinicalTrials.gov number, NCT01202721) – will likely provide new insights on this subject. It is important to identify syndromic patients, such as Turner, Marfan, and Loeys-Dietz, with bicuspid aortic valves because they have a much higher risk of dissection and sudden death than non-syndromic cases with a bicuspid aortic valve.Reference Papagiannis 27 This may seem trivial, but it is a fact that many patients with syndromes such as Turner syndrome or Marfan syndrome are not diagnosed until adulthood.Reference Groth, Hove and Kyhl 56 , Reference Stochholm, Juul, Juel, Naeraa and Gravholt 57 For these patient categories, close follow-up and timely aortic surgery is of upmost importance.
Currently, there is consensus that annual check-ups should be recommended if the aortic size is >45 mm,Reference Erbel, Aboyans and Boileau 55 and aortic root surgery is warranted if the size exceeds 55 mm.Reference Erbel, Aboyans and Boileau 55 , Reference Hiratzka, Creager and Isselbacher 58
Previously, there was an indication for aortic surgery at 50 mm in the United States, but a recently updated guideline from the American Heart Association/American College of Cardiology changed the indication to 55 mmReference Hiratzka, Creager and Isselbacher 58 on the basis of two retrospective studies that actually showed patients with a bicuspid aortic valve had an aortic size >60 mm at the time of dissection compared with patients with a tricuspid valves who had an aortic size ~53 mm at the time of dissection.Reference Etz, von Aspern and Hoyer 51 , Reference Eleid, Forde and Edwards 59
However, in some patients with an aortic diameter ~50 mm, the risk of dissection is already considerably increased. In an unselected surgical cohort including 380 patients followed for a median of 3 years (0–17 years), the dissection incidence was as high as 5.3%.Reference Wojnarski, Svensson and Roselli 60
For this reason, it is still recommended that surgeons be careful and offer surgery if the aortic diameter gets >50 mm and there is either a family history of dissection, syndromic disease, or rapid dilatation, with a dilatation rate of >5 mm per year.Reference Hiratzka, Creager and Isselbacher 58 In the European guidelines, surgeons are advised to offer surgery at a diameter of 50 mm if the dilatation rate exceeds 3 mm per year.Reference Erbel, Aboyans and Boileau 55 It is most certain that future inclusion of genetic status with presence or absence of a known mutation that leads to development of a bicuspid aortic valve will improve risk prediction and provide a more tailored surgical course.Reference Erbel, Aboyans and Boileau 55 , Reference Hiratzka, Creager and Isselbacher 58 , Reference Gravholt, Andersen and Conway 61
Key aortic sizes are shown in Table 1.
Table 1 Key aortic sizes in bicuspid aortic valve disease.

Valve dysfunction
In rare cases, severe aortic stenosis develops in foetal life.Reference Hochstrasser, Ruchat, Sekarski, Hurni and von Segesser 62 This development can cause hypoplasia of the left ventricle or heart failure in the infant. However, in most cases, people with a bicuspid aortic valve are asymptomatic until adult life, and an unknown number of people remain undiagnosed.
In selected paediatric cohorts with bicuspid aortic valves but without severe stenosis or concomitant CHD, <5% require valve interventions before adult life.Reference Mahle, Sutherland and Frias 36 , Reference Spaziani, Ballo and Favilli 63 The majority of children who need intervention have valve stenosis.Reference Spaziani, Ballo and Favilli 63
If a patient enters adult life with either mild dysfunction or normal function of the bicuspid aortic valve, then the number of patients needing aortic valve replacement increases, but the numbers remain small.Reference Michelena, Desjardins and Avierinos 2 In 212 individuals followed for 20 years, 18% needed aortic valve replacement at a mean age of 49±20 years. This cohort also included an 89-year-old person with normal function of the bicuspid aortic valve. In a cohort including a broader spectrum of valve dysfunction, 22% needed valve intervention during 10 years of follow-up.Reference Tzemos, Therrien and Yip 64 In adult patients, the presence of a raphe considerably increases the risk of valve dysfunction, and the treatment of aortic stenosis is more common than regurgitation, although the difference is small.Reference Della Corte, Body and Booher 31 , Reference Kong, Delgado and Poh 65
What about the aorta in patients who need valve replacement?
When the patient develops severe valve dysfunction and needs valve replacement, a crucial question is raised: Should only the valve or the aortic root be replaced? Moreover, what about patients with aortic stenosis and only dilatation of the tubular part of the ascending aorta? Can these patients avoid root replacement with coronary artery reimplantation and simply undergo aortoplasty and valve replacement? Obviously, the goal is to prevent aortic dissection over time; however, by choosing root replacement we include reimplantation of the coronary arteries, which is a more complex operation with increased risk. Therefore, the first question is whether the future risk of dissection can justify the increased surgical risk of embarking on root surgery.
It is not uncommon to see aortic dilatation late after aortic root replacement in bicuspid valve patients (Fig 4), but is it more common than in patients with tricuspid valves? A few studies have addressed this question. A cohort study of 1286 patients, who had valve replacement because of valve dysfunction, found that a preoperative aortic size >40 mm was not associated with a higher occurrence of dissection than a preoperative size <40 mm. However, there was a higher occurrence of new surgery owing to aortic dilatation in the group with an enlarged aorta (0.3% versus 1.8%, p=0.01).Reference McKellar, Michelena, Li, Schaff and Sundt 11 A recent study showed similar trends; patients with a bicuspid aortic valve – 2079 patients and 11,053 control patients with acquired valve disease – had the same risk of dissection during a follow-up period of 6.6 years.Reference Itagaki, Chikwe, Chiang, Egorova and Adams 10

Figure 4 Aortic dilatation (58 mm) 20 years after aortic root replacement owing to aortic valve stenosis at the age of 24. The aortic size at the time of surgery was 39 mm.
Consequently, the decision to choose aortic surgery and not just valve replacement in patients with bicuspid aortic valve dysfunction must be based on several factors as follows. The size of the aortic root is, of course, important. There is international consensus that aortic root replacement should be offered if the diameter is ⩾45 mm,Reference Erbel, Aboyans and Boileau 55 , Reference Hiratzka, Creager and Isselbacher 58 but the evidence is poor.Reference Borger, Preston and Ivanov 66 Age is also important. The increased surgical risk of aortic surgery must be justified by a reduction in the future risk of aortic dissection, but since increased risk cannot be found in the two large follow-up studies mentioned earlier, it is worth considering whether adult patients with a moderate life expectancy should be offered aortic surgery, as they will probably not live long enough to experience aortic dissection.
The second question is how to choose the best solution when addressing the different types of aortic dilatation if the choice is to replace both the valve and the dilated aorta. Radical resection of the aorta including the root and the entire ascending aorta (Bentall procedure) will, of course, eliminate possible potential for late dilatation and dissection.Reference Hagl, Strauch and Spielvogel 67 However, reimplantation of the coronary arteries adds to the surgical risk, particularly if they have an abnormal origin.Reference Naito, Petersen, Reichenspurner and Girdauskas 68 Since coronary anomalies are quite common in bicuspid aortic valve patients, of whom ~5% have a separate origin of the left anterior descending artery or circumflex arteryReference Koenraadt, Bartelings and Bökenkamp 3 and a high tubular origin is seen in almost a third (Fig 5),Reference Koenraadt, Bartelings and Bökenkamp 3 the option to perform root sparing surgery becomes relevant. The choice to perform a separate valve replacement and an ascending aortic graft avoids the risks associated with coronary reimplantation but leaves a potential risk of sinus dilatation over time. Some studies have examined the long-term results of this less extensive surgical technique, but it appears that this could be a good surgical solution in many patients with bicuspid valve dysfunction and ascending aortic dilatation, provided the aortic sinus is not significantly enlarged.Reference Park, Greason, Suri, Michelena, Schaff and Sundt 69 , Reference Nazer, Elhenawy, Fazel, Garrido-Olivares, Armstrong and David 70 On the contrary, significant dilatation of the aortic sinus and a bicuspid aortic valve should always be suspected to be part of a connective tissue disorder, and in these cases, extensive aortic surgery with replacement of the root and the entire ascending aorta must always be advised owing to the increased risk of dissection later in life.Reference Groth, Stochholm and Hove 52

Figure 5 High tubular origin of the right coronary artery (arrow) in a patient with aortic dilatation and right and left coronary cusp fusion.
Screening
As there appears to be a genetic component in the development of non-syndromic bicuspid aortic valve disease, it has been discussed whether screening of family members is relevant and important. Huntington et al, 20 years ago, found a prevalence of 9.1% in first-degree relatives,Reference Huntington, Hunter and Chan 71 and since then, several other studies of first-degree relatives have provided numbers between 4.6 and 22%.Reference Cozijnsen, van der Zaag-Loonen and Braam 72 , Reference Kerstjens-Frederikse, Du Marchie Sarvaas and Ruiter 73 Some studies strongly recommend screening of siblings,Reference Hales and Mahle 74 but does the potential beneficial effect of screening actually outweigh the unintended harms?
If we screen siblings with echocardiography, we want to find individuals with normal functioning bicuspid aortic valves with a potentially increased risk of dissection over time. We must assume that children or adults with valve dysfunction already get diagnosed because of a murmur or symptoms, similarly to any other patient. The other patients will follow the large population studies in adults where <1% will dissect over 10–20 years.Reference Michelena, Desjardins and Avierinos 2 , Reference Tzemos, Therrien and Yip 64 , Reference Rodrigues, Agapito and de Sousa 75 If we assume that 1% of the general population has a bicuspid aortic valve and 10% of these individuals have an undiagnosed relative with a bicuspid aortic valve, we will identify 1000 persons per 1 million citizens and probably prevent 10 dissections over 10–20 years if the identified patients adhere to the follow-up programme. The remaining 990 individuals can be regarded as over-diagnosed with a bicuspid aortic valve and many unnecessary echocardiograms will be done on a population scale.Reference Brodersen, Schwartz, Heneghan, O’Sullivan, Aronson and Woloshin 12 The over-diagnosed persons will be offered regular checks-ups throughout their entire life and have no beneficial effects in relation to longer life with less morbidity. In contrast, these individuals will be labelled with a potentially life-threatening condition, which could have negative psychosocial consequences.Reference Cotter, Vuong and Mustelin 76 Moreover, for those living in countries with privately run healthcare systems, overdiagnosis of bicuspid aortic valves can be costly for the individual person.Reference Harris, Sheridan and Lewis 77 In countries with public run healthcare systems, overdiagnosis will result in societal costs.Reference Harris, Sheridan and Lewis 77 Finally, an overdiagnosis can have an impact on a person’s possibilities to get life insurance or to have a professional carrier in sports, and so on. For this reason, the effort is probably better spent on other structural health preventive interventions.
As genetic unravelling of the underlying propensity to develop a bicuspid aortic valve is evolving, the above-mentioned deliberations may be changed to focus screening of relatives of patients with high-risk bicuspid valve disease owing to a specific genetic change. This might be a better approach, where benefits will outweigh the harms of screening.
Pregnancy
Pregnancy is not a problem in the absence of valvular dysfunction or dilatation of the aortic root. However, if the future mother has a bicuspid aortic valve because of a syndrome such as Turner or Marfan syndrome, the pregnancy is a condition that should be treated by a specialist.Reference Gravholt, Andersen and Conway 61 , Reference Regitz-Zagrosek, Blomstrom Lundqvist and Borghi 78
Aortic valve regurgitation is well tolerated in the otherwise healthy woman, whereas a stenotic aortic valve can cause problems with heart failure. Rescue balloon dilatation may be necessary during pregnancy and is a situation that absolutely should be avoided. Nevertheless, a recent study based on data from the The Registry on Pregnancy and Cardiac Disease register showed that women with even moderate to severe aortic stenosis were able to deliver without fatalities and with a low number of valve interventions, despite a relative high rate of hospitalisations because of heart failure symptoms (20.8%).Reference Orwat, Diller and van Hagen 79
Pregnancy and aortic dilatation is also quite safe.Reference McKellar, MacDonald, Michelena, Connolly and Sundt 80 There are specific recommendations for women with Turner syndrome, in which pregnancy is not recommended in women with an ascending aortic size of an ascending aortic size of 20–25 mm/m2 and a bicuspid aortic valve.Reference Gravholt, Andersen and Conway 61 In Marfan syndrome, there are no specific recommendations for women with a bicuspid aortic valve, but an extra safe approach must be recommended.Reference Groth, Greisen, Nielsen and Andersen 81
In non-syndromic women, it is recommended to advise against pregnancy if the aortic diameter is >50 mm;Reference Regitz-Zagrosek, Blomstrom Lundqvist and Borghi 78 however, there is not any clear evidence behind this statement. Pregnancy-related dissections are very rare,Reference McKellar, MacDonald, Michelena, Connolly and Sundt 80 and larger aortic sizes could probably be tolerated. Serial echocardiograms during the pregnancy are warranted if the aortic size is >40 mm, even though pregnancy rarely makes the aorta dilate.Reference McKellar, MacDonald, Michelena, Connolly and Sundt 80
The worst scenario is the opposite situation, where pre-pregnancy aortic root surgery is recommended at a very early stage “just in case”.Reference Freeze, Landis, Ware and Helm 6 If this surgery includes aortic valve replacement with a mechanical valve, the woman is put at a much higher risk of valve thrombosis or bleeding than she had for aortic dissection and also an increased mortality of up to 1.2%.Reference Lawley, Lain, Algert, Ford, Figtree and Roberts 82 If the woman is offered a solution with a bioprosthetic aortic valve, then the risk is lower, but she will be committed to numerous interventions over an entire lifetime.Reference Kaza and Pigula 83
There is also an option for a pulmonary autograft procedure, such as Ross procedure, where the major advantage is the avoidance of anticoagulation. In experienced centres, the Ross procedure has a very good long-term prognosis.Reference Mazine, David and Rao 84 With continuous refinements of the surgical technique to avoid late root dilatationReference Brown, Ruzmetov, Shahriari, Rodefeld, Mahomed and Turrentine 85 and the introduction of transcatheter treatment of pulmonary homograft stenosis,Reference Gillespie, McElhinney and Kreutzer 86 it appears that this patient category can live even longer before another surgery is needed.
What is new within the area of bicuspid aortic valve disease?
We still do not understand the genetics behind the development of bicuspid semilunar valves. A major breakthrough will probably not be found in the imminent future, but we are getting closer to a more thorough understanding of the mechanisms behind the development of the heart.Reference Chen and VanBuren 87
Initially, transcatheter aortic valve replacement for bicuspid aortic valves was controversial, but improved preprocedural imaging and optimised techniques have made it a safe and effective valve replacement option for patients with a stenotic bicuspid valve.Reference Sannino, Cedars, Stoler, Szerlip, Mack and Grayburn 88 , Reference Popma and Ramadan 89 With the new generation of transcatheter aortic valves, there also appears to be fewer paravalvular leaks and a higher device success rate.Reference Yoon, Lefevre and Ahn 90 This treatment option is still reserved for the elderly (77±8 years),Reference Yoon, Bleiziffer and De Backer 91 and time will tell whether transcatheter aortic valve replacement can be used with good long-term outcomes in patients in their 50s or 60s.
Reconstructive surgery of the bicuspid valve is also an option that has become more common in many centres.Reference Luciani, De Rita and Lucchese 92 , Reference Schneider, Feldner and Hofmann 93 This is a brilliant option for young individuals with aortic valve regurgitation as they can avoid warfarin treatment. Until now, this treatment is not offered worldwide, but the results appear promising.Reference Thudt, Papadopoulos and Monsefi 94
Recently, the Ozaki procedureReference Ozaki, Kawase and Yamashita 95 with neocuspidisation using glutaraldehyde-treated autologous pericardium has been applied to both regurgitant and stenotic bicuspid valves.Reference Ozaki, Kawase and Yamashita 96 This valve replacement technique seems very promising, but there are only a few studiesReference Reuthebuch, Koechlin, Schurr, Grapow, Fassl and Eckstein 97 , Reference Ozaki, Kawase, Yamashita, Uchida, Takatoh and Kiyohara 98 with relatively short follow-up and varying results among the cases of stenotic bicuspid valves.Reference Ozaki, Kawase, Yamashita, Uchida, Takatoh and Kiyohara 98 At present, this technique is only available for the elderly population, but hopefully this method will be an established treatment option, either for life or as a bridge to transcatheter treatment in adult life.
Conclusion
The pathophysiological mechanisms behind a bicuspid valve are complex and multi-faceted and focus on syndromic and non-syndromic cases is of significant clinical value. A bicuspid aortic valve is related to a low risk of valve interventions in childhood, but this risk incidence increases in adult life.
If a person enters adulthood with a normal functioning bicuspid valve, the prognosis is excellent, with a low risk of dissection; there is also a good prognosis if aortic valve replacement is needed. Mass screening for bicuspid aortic valve is not recommended because the likely benefits do not appear to outweigh the potential harms. Pregnant patients with a bicuspid aortic valve have a very low complication risk if the aorta is <50 mm, and pre-pregnancy aortic valve or root interventions should therefore be reserved for high-risk patients.
Acknowledgements
None.
Financial Support
This research received no specific grant either from any funding agency or from commercial or not-for-profit sectors.
Conflicts of Interest
None.






