Introduction
It has been known for some time that eating disorders ‘run in families’, with a 7- to 12-fold increase in the prevalence of anorexia nervosa (AN) or bulimia nervosa (BN) in relatives of eating-disordered probands compared to families of controls (Klump et al. Reference Klump, Kaye and Strober2001). Historically, and in common with many other types of psychopathology, there has been a view that families cause eating disorders, a view reinforced by patients referred for treatment, who most commonly report the perceived cause of the eating problem to be a dysfunctional family (Tozzi et al. Reference Tozzi, Sullivan, Fear, McKenzie and Bulik2003). However, over the last 20 years a number of twin studies have shown that there is a clear and substantial genetic contribution to both BN and AN (Fairburn & Harrison, Reference Fairburn and Harrison2003), in conjunction with an influence of the non-shared environment. The role of the shared environment in disordered eating (DE) is less certain (Bulik et al. Reference Bulik, Sullivan, Wade and Kendler2000).
A more nuanced understanding of how genes and the environment impact on the aetiology of DE is slowly emerging. Two longitudinal studies suggest shifts in genetic and environmental risk factors between childhood and adolescence. In a study of female twins at ages 11, 14 and 18 years (Klump et al. Reference Klump, Burt, McGue and Iacono2007), the shared environment was found to be a significant contributor to DE in the pre-pubertal age group, with negligible impact at age 14. Conversely, the impact of heritability was negligible at 11 years, but increased significantly by age 14. The source of genetic variance active at 11 years of age continued to be a major contributor to variance at ages 14 and 18 years, and a new source of genetic variance appeared at age 14 which made only small contributions to the variance at ages 14 (1%) and 18 (18%), with no new sources of genetic influence appearing at 18 years. In contrast, new sources of non-shared environmental variance appeared at each age. Authors of this longitudinal study (and other cross-sectional investigations) suggest that puberty and associated ovarian hormones may contribute to an environmental trigger ‘switching on’ the main source of genetic risk for DE (Klump et al. Reference Klump, McGue and Iacono2003, Reference Klump, Culbert, Slane, Burt, Sisk and Nigg2012; Culbert et al. Reference Culbert, Burt, McGue, Iacono and Klump2009; Klump, Reference Klump2013). A second longitudinal study, this time assessing weight and shape concern in adolescent twins (Wade et al. Reference Wade, Hansell, Crosby, Bryant-Waugh, Treasure, Nixon, Byrne and Martin2013) over three occasions (12–13, 13–15, 14–16 years), showed a similar pattern of results, with non-shared environmental influences largely specific to each age cohort, and genetic influences influential at 12–13 years continuing to contribute to subsequent age cohorts, with independent sources of genetic influence emerging at ages 13–15, but not in later adolescence. This latter study omitted to examine older adolescents, where the transition from adolescence to adulthood represents an important risk period for the emergence of eating disorders, especially those related to binge and purge behaviours (Lewinsohn et al. Reference Lewinsohn, Striegel-Moore and Seeley2000; Hudson et al. Reference Hudson, Hiripi, Pope and Kessler2009).
While no specific genes have been identified for eating disorders yet, weight teasing has been identified as a potentially important environmental influence, predicting frequent dieting in girls, as well as binge eating and unhealthy weight control in boys (Haines et al. Reference Haines, Neumark-Sztainer, Eisenberg and Hannan2006). Relatedly, perceived pressure to be thin predicts growth in importance of weight and shape in adolescents (Wilksch & Wade, Reference Wilksch and Wade2010), a diagnostic criterion for eating disorders, and comments about amounts eaten or appearance by family members as children were growing up is a retrospective correlate for both AN and BN (Fairburn et al. Reference Fairburn, Cooper, Doll and Welch1999; Wade et al. Reference Wade, Gillespie and Martin2007). In addition, an increase in negative life events predicted onset of eating disorders in a female adolescent sample (McKnight Investigators, 2003), with retrospective recall of negative life events predicting onset of BN (Welch et al. Reference Welch, Doll and Fairburn1997).
Given the omission of an older adolescent group in a previous longitudinal examination of the cognitive component of DE (Wade et al. Reference Wade, Hansell, Crosby, Bryant-Waugh, Treasure, Nixon, Byrne and Martin2013), and lack of research addressing important environmental risk factors in the context of any shifts in genetic contribution to DE within this developmental period, the purpose of the current study was 2-fold. First, we wished to examine contributions of genetic and environmental variance contributing to DE in adolescents, measured using the global score of the Eating Disorder Examination (EDE: Fairburn & Cooper, Reference Fairburn, Cooper, Fairburn and Wilson1993), used as the primary indicator of outcome in treatment trials of eating disorders, both AN and BN, in children and adults (le Grange et al. Reference le Grange, Crosby, Rathouz and Leventhal2007; Fairburn et al. Reference Fairburn, Cooper, Doll, O'Connor, Bohn, Hawker, Wales and Palmer2009; Lock et al. Reference Lock, Le Grange, Agras, Moye, Bryson and Booil2010; Wade et al. Reference Wade, Treasure and Schmidt2011). It incorporates both cognitive and behavioural indicators of DE. In particular, our aim was to examine any changes in genetic and environmental influences between early (i.e. before age 16 years) and late adolescence (i.e. from 16 years of age) in order to revisit the question of whether new sources of heritability emerge later in adolescence, given that the only previous longitudinal study of DE suggested that this is not the case (Klump et al. Reference Klump, Burt, McGue and Iacono2007). Second, we examined the predictive role of two risk factors for DE, namely peer teasing about weight and negative life events. While puberty has been considered as an environmental influence in early adolescence, we wished to investigate the nature of any emerging environmental risk factors over late adolescence.
Method
Participants
The current study used outcome measures derived from three waves of data from adolescent female–female twin pairs, described previously (Wade et al. Reference Wade, Byrne and Bryant-Waugh2008, Reference Wade, Hansell, Crosby, Bryant-Waugh, Treasure, Nixon, Byrne and Martin2013; Wilksch & Wade, Reference Wilksch and Wade2009, Reference Wilksch and Wade2010). Given that we wished to examine DE outcomes in non-overlapping age groups that typified the peak risk periods of emergence of eating disorders (early and late adolescence), only the first and third wave of data collection were used as the age at the middle wave of data (wave 2; mean age 15.10 years, s.d. = 0.83, range 13.76–17.56 years) overlapped with age at the first and third waves. Data pertaining to environmental risk factors (i.e. weight-related peer teasing, negative life events) were derived from waves 1 and 2.
At wave 1, twin pairs who were between 12 and 15 years of age registered with the Australian Twin Registry (ATR) were approached, along with their parents, to participate in the present study by the ATR. Seven hundred and nineteen families were contacted and invited to participate, and of these 411 (57.2%) agreed, 237 (32.9%) declined, and 71 (9.9%) did not reply. Researchers approached those agreeing to take part and sent self-report questionnaires to both parents, including those families where the parents did not live together. After parents returned the questionnaires, the EDE (Fairburn & Cooper, Reference Fairburn, Cooper, Fairburn and Wilson1993) was conducted by telephone, such that 699 twins were interviewed at separate times with a different interviewer for each child in the family. The sample was Caucasian and the socioeconomic indexes for areas (SEIFA) – a standardized measure of socioeconomic status with a mean of 100 (s.d. = 15), using an amalgam of parental occupation, education (years of school), and income from 2006 census data related to the postcode of primary residence (Farish, Reference Farish2004) – was 101.14 (s.d. = 11.36). In order to ensure no overlap in ages between waves 1 and 3, eight pairs of twins where one or more was aged 16 at the time of first assessment were removed from the dataset, such that the mean age of the twins who participated was 13.90 years, (s.d. = 0.80, range 12.70–15.47)Footnote 1 Footnote †.
All twins were contacted again at wave 2 (including non-responders); the mean duration of time between waves 1 and 2 was 1.15 years (s.d. = 0.17). A total of 514 parents completed questionnaires (86% of wave 1) and 669 twins completed interviews (96% of wave 1). Mean age was 15.10 years (s.d. = 0.83, range 13.76–17.56).
At wave 3 all twins, responders and non-responders, were again approached, of which 499 (71% of wave 1) completed interviews, with a mean duration of time between waves 1 and 3 of 2.96 years (s.d. = 0.27), ranging from 1.91 to 4.65. The mean age of the twins was 16.90 years, s.d. = 0.70 (range 15.49–19.84), and age was significantly different from wave 1 [t(df = 488) 261.18, p < 0.001]. Attrition at wave 3 was unrelated to EDE scores at wave 1 (OR 1.15, 95% CI 0.88–1.51); this was also the case for the environmental risk factors at both waves 1 and 2 for life events (W1: OR 1.04, 95% CI 0.89–1.21; W2: OR 0.96, 95% CI 0.87–1.06) and weight-related peer teasing (W1: OR 1.00, 95% CI 0.76–1.34; W2: OR 1.01, 95% CI 0.72–1.42). While not significant, dizygotic (DZ) twins showed a tendency to be less likely to participate at wave 3 than monozygotic (MZ) twins (OR 0.72, 95% CI 0.51–1.00).
Zygosity assignment used parental responses to standard questions about physical similarity and confusion of twins by parents, teachers, and strangers; this method gives better than 95% agreement with genotyping (Eaves et al. Reference Eaves, Eysenck, Martin, Jardine, Heath, Feingold, Young and Kendler1989). Where there was uncertainty (N = 46 pairs), DNA testing was used to assign zygosity for 39 pairs (DNA was not available for seven pairs and these pairs were therefore not included in the analyses). At wave 1 there were 367 MZ twins (182 complete pairs and three incomplete pairs), 302 DZ twins (151 complete pairs), and seven pairs where zygosity was unknown, i.e. 340 complete pairs and three incomplete pairs. Wave 2 constituted 330 complete pairs (178 MZ, 146 DZ, 6 without zygosity) and eight incomplete pairs (5 MZ, 3 DZ). At wave 3, there were 243 complete pairs (136 MZ, 106 DZ, 1 without zygosity) and five incomplete pairs (4 MZ, 1 DZ). The number of twin pairs completing the EDE at wave 1 was 340, with 243 complete pairs (72% of wave 1) available across both waves. The Flinders University Clinical Ethics Committee approved the data collection process; parents gave written informed consent and twins gave written assent.
Disordered eating
The twin interview consisted of two parts for waves 1 and 2; the EDE (Fairburn & Cooper, Reference Fairburn, Cooper, Fairburn and Wilson1993) and questions from self-report questionnaires assessing a range of variables including weight-related peer teasing and life events (Wilksch & Wade, Reference Wilksch and Wade2009, Reference Wilksch and Wade2010). Parents also completed self-report questionnaires at waves 1 and 2, including twin weight, height, and life events. The interview at wave 3 comprised only the EDE interview. Sixteen postgraduate Clinical Psychology trainees competent in the use of the EDE conducted all interviews.
The EDE interview generates a global measure of eating psychopathology (22 items) that includes four subscales weight concern, shape concern, eating concern and dietary restraint. It was adapted for the younger age of the sample simply by encouraging participants to verbalize the list related to the following question: ‘Imagine the things that influence how you feel about (judge, think, evaluate) yourself as a person – such as how you are doing at school, what sort of friend you are, how you get along with other people – and put these things in order of importance, where does your weight/shape fit in?’ The EDE is generally considered the ‘pre-eminent’ eating disorder interview assessment tool with sound validity and reliability (Berg et al. Reference Berg, Peterson, Frazier and Crow2011, Reference Berg, Peterson, Frazier and Crow2012a , Reference Berg, Stiles-Shields, Swanson, Peterson, Leobow and Le Grange b ).
The global EDE was found to perform validly in our sample. First, construct validity was demonstrated by global EDE scores among those meeting diagnostic thresholds for key DE symptoms (e.g. fear of weight gain, purging) being significantly higher than those who did not meet criteria (data available from authors on request). Second, convergent validity was confirmed with medium-large correlations with Eating Disorder Inventory (EDI: Garner et al. Reference Garner, Olmstead and Polivy1983) interoceptive awareness and drive for thinness measures. Third, factorial invariance suggested that the global EDE remained invariant across waves 1 and 3. The χ2 difference test showed the metric invariance model was not a significantly poorer fit to these data (χ2 test for difference = 30.997, df = 21, p = 0.07), nor the full threshold invariance model (χ2 test for difference = 166.52, df = 140, p = 0.06). Medium-large cross-wave correlations (range 0.37–0.42) suggest the global EDE score remains quite stable over time. Fourth, the internal reliability of the global EDE was 0.93 at both waves 1 and 3.
Body mass index (BMI) centile
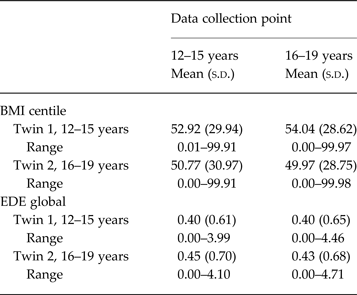
As data relating to parental report of the twins’ weight and height were highly correlated, the mothers’ report was used, aside from instances where these data were missing, when the fathers’ report was used. At wave 3, twins reported their own weight and height. We adopted the Center for Disease Control recommendation to use BMI-for-age (or BMI centile) in this sample. Mean (standard deviation) BMI centile and range for both twins 1 and 2 with analogous details for the global EDE score are reported (Table 1).
Table 1. BMI centile and global EDE means (s.d.) and range at wave 1 (12–15 years) and wave 3 (16–19 years)

BMI, Body mass index; EDE, eating disorder examination.
Measures of specific riskFootnote 2
At waves 1 and 2, a measure of weight-related peer teasing (McKnight Investigators, 2003) was undertaken by twins. The scale consisted of eight items (e.g. In the past year, how often have girls – including sisters – made fun of you because of your weight?) where responses were made on a 3-point scale (1 = never, 2 = sometimes, 3 = a lot) from which a mean score was calculated. Cronbach's alpha was 0.87 and 0.89, respectively. Parents were asked to report if the twin had encountered negative life events in the preceding 12 months (yes/no) to waves 1 and 2, listing 13 items including death of a family member, moving home, starting at a new school, parental separation, or a breakdown of a close friendship. A count variable of those life events was created which summed the aforementioned items.
Statistical analysis
Twin correlations
All subsequent analyses treated data as continuous, and a full information maximum likelihood (FIML) approach was used with the statistical package Mx (Neale, Reference Neale1994) designed to employ structural equation modelling approaches to twin data with complete and incomplete pairs of twins across the waves automatically creating respective mean vector and covariance matrix for each data point (Neale, Reference Neale1994). The twin pair correlations for MZ and dizygotic DZ twins for each phenotype were calculated. As MZ twins share 100% of their genes, but DZ twins share only 50% (on average), additive genetic effects on a phenotype can be deduced from MZ twin correlations being approximately double DZ twin correlations (Plomin et al. Reference Plomin, DeFries and McClearn1990).
Multivariate model fitting
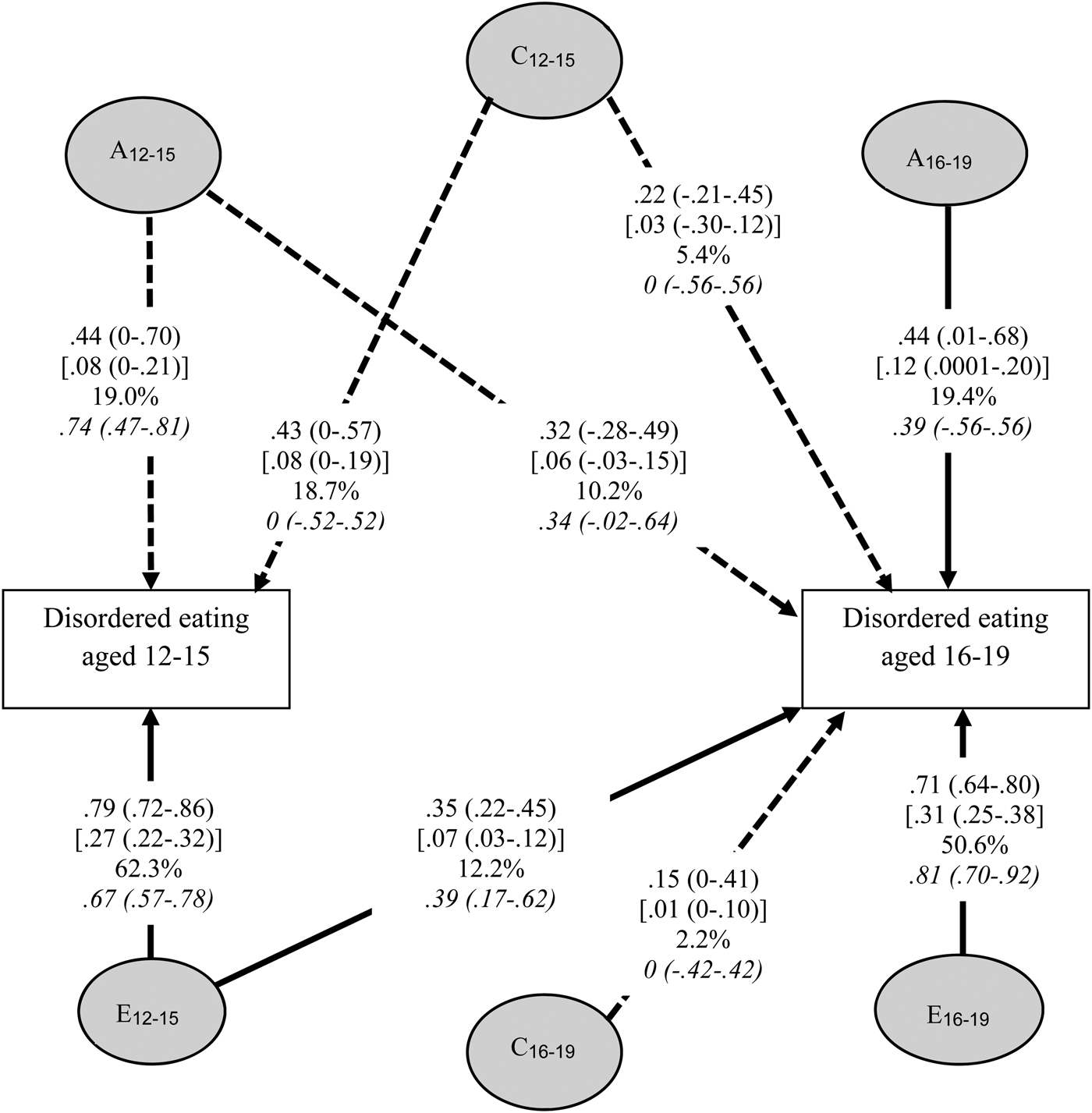
A bivariate Cholesky decomposition model was used to analyse DE at waves 1 and 3; models were fitted with and without adjustment for BMI centile. The structure of the model can be seen in Fig. 1. As bivariate models use both variances of individual variables and covariances between the different variables to estimate parameters they are more powerful than their univariate counterparts (Neale et al. Reference Neale, Mazzeo and Bulik2003). Further, use of repeated measures enables correction of any ascertainment bias due to differential attrition (Little & Rubin, Reference Little and Rubin1987), also diminishing proportion of measurement error attributed to the non-shared environment (Bulik et al. Reference Bulik, Sullivan and Kendler1998).

Fig. 1. Bivariate Cholesky decomposition path diagram for disordered eating at 12–15 and 16–19 years of age showing the standardized [unstandardized] parameter estimates for A, C and E, proportion of total heritability (%), and body mass index adjusted standardized estimates (italics) with respective 95% confidence intervals (A, additive genetic influences; C, shared environment; E, non-shared environmental influences). Non-significant pathways are dashed, significant pathways are solid.
Initially, models were fitted in which the magnitude of the parameter estimates was allowed to vary across assessment periods (i.e. waves 1 and 3), starting with a full model [i.e. containing the additive genetic variance (A), shared environment (C) and non-shared environment (E) sources of variance]. Subsequently, a series of nested models examined whether all sources of variance were necessary, where 95% confidence intervals (CI) for all estimates assisted identifying the significance of the models. Twice the difference in the log likelihood (–2LL) between a higher order and submodel yields a statistic that is asymptotically distributed as χ2, with the degrees of freedom (df) equal to the difference in their number of parameters, and can be used to determine if the submodel is significantly worse fitting than the full model. Typically, where models do not differ significantly, Akaike's Information Criterion (AIC) determines the selection of the submodel as the best fitting model, where the lowest value represents the best combination between explanatory power and parsimony.
Relationship between specific risk variables and wave 3 disordered eating
To investigate putative influences on DE at 16–19 years, linear mixed model analyses were conducted with five models: (a) weight-related peer teasing at wave 1; (b) weight-related peer teasing at wave 2; (c) change in level of peer teasing between waves 1 and 2; (d) total life events at wave 1; and, (e) total life events at wave 2. All models adjusted for baseline (wave 1) EDE global scores and BMI centile by including them as covariates. Effect sizes (ES) were computed using
 ${\rm (2}\sqrt {\rm F} )/\sqrt {{\rm df}({\rm error})} $
.
${\rm (2}\sqrt {\rm F} )/\sqrt {{\rm df}({\rm error})} $
.
Results
Twin correlations
Table 2 reports the cross-twin, cross-trait correlations of the global EDE score at waves 1 and 3 for twins 1 and 2. Bolded correlations highlight correlations within each twin pair, and show MZ (r age 12–15: 0.43; r age 16–19: 0.36) are observed to be greater than DZ (r age 12–15: 0.20, r age 16–19: 0.01) correlations, such that it is probable additive genetic factors contribute to the association between EDE levels in both early and late adolescence.
Table 2. Cross-twin and cross-wave FIML correlations (95% confidence intervals) for twins 1 and 2; within-twin correlations are shown in bold

FIML, Full information maximum likelihood.
MZ above diagonal and DZ below diagonal.
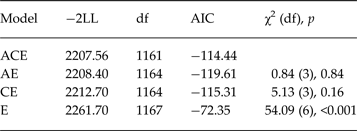
Multivariate model fitting
The results of the Cholesky model fitting are shown in Table 3. The ACE model fit was retained as recommended (Sullivan & Eaves, Reference Sullivan and Eaves2002) although the AE submodel achieved greater parsimony, without decrement in model fit (as determined by the lowest AIC value). The standardized and unstandardized parameters (and 95% CIs), percentage variance, and standardized estimates adjusting for the influence of BMI centile for the ACE model are shown in Fig. 1. The unstandardized parameter estimates of genetic influence more than doubled over time (0.08–0.18) accompanied by a 30% increase in non-shared environmental influence (0.27–0.38). Importantly, late adolescence includes new sources of both genetic and non-shared environmental influences that predominate the risk period in late adolescence, with two-thirds of the heritable influence contributing to DE in late adolescence being unique to this age group. There is a moderate correlation between the latent genetic risk variables (r A = 0.63, 95% CI −1.0 to 1.0), shared environment (r C = 1.0, 95% CI 1.0–1.0), and between the two latent non-shared environment variables (r E = 0.26, 95% CI 0.19–0.45) contributing to DE.
Table 3. Model comparisons of the bivariate Cholesky model of disordered eating between 12–15 years and 16–19 years of age

A, Additive genetic influences; C, shared environmental influences; E, non-shared environmental influences; AIC, Akaike's Information Criterion; preferred model (using AIC) in bold; −2LL, two times the log likelihood using ACE model as the comparison; χ2 (df) p indicates AE and CE have no significant decrement in fit, but ACE model is preferred based on recommendations by Sullivan & Eaves (Reference Sullivan and Eaves2002).
Standardized estimates indicated that, in early adolescence, latent sources of genetic (19.0%), shared (18.7%) and non-shared environmental (62.3%) variance impact DE, with 29.6% of the sources acting on late adolescence being genetic, contrasting with shared (7.6%) and non-shared sources (62.7%) that decrease. When adjusting for the influence of BMI centile these estimates were similar despite CIs being marginally broader, a probable consequence of reduced power as inferred by one further path becoming non-significant.
Relationship between sources of specific risk variables and disordered eating at wave 3
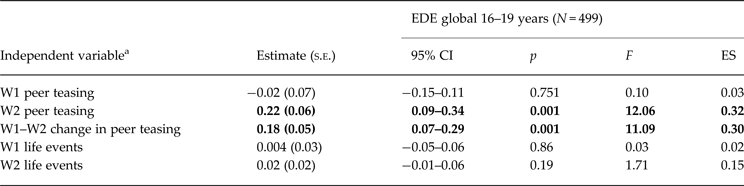
Table 4 shows two of the five independent specific risk variables predicted growth in DE. This included higher levels of weight-related peer teasing at wave 2 and an increase in peer teasing between waves 1 and 2. There was no relationship with life events in the previous 12-month period.
Table 4. Associations (unstandardized estimate and standard error) between disordered eating at wave 3 and specified risk factors at wave 1 (W1) and wave2 (W2)

EDE, Eating Disorder Examination; CI, confidence interval; ES, effect size.
a
All models adjust for EDE global and body mass index centile at wave 1. Effect sizes were computed using
 ${\rm (2}\sqrt {\rm F} )/\sqrt {{\rm df}({\rm error})} $
.
${\rm (2}\sqrt {\rm F} )/\sqrt {{\rm df}({\rm error})} $
.
Discussion
The role of genetic and specific risk factors on the development of DE remains unclear, given few existing longitudinal studies (Klump et al. Reference Klump, Burt, McGue and Iacono2007, Wade et al. Reference Wade, Hansell, Crosby, Bryant-Waugh, Treasure, Nixon, Byrne and Martin2013), especially those that examine a critical risk period for the emergence of DE: the transition from adolescence to adulthood.
We therefore used longitudinal adolescent twin data to determine whether significant change in heritable variance of the DE phenotype occurred between early (before 16 years) and late adolescence (after 16 years). While non-shared environmental influence was observed to occur early and to persist across adolescence, we also found evidence for a new source of genetic influence contributing to expression of DE symptomatology in late adolescence. Importantly, this is the first study to report the presence of an independent heritable source – putatively, a late adolescence genetic diathesis. Of note, two-thirds of the heritable influence contributing to DE in late adolescence is unique to this age group. This time-frame accords with the later emergence of binge and purge symptomatology, well documented to have peak age of onset in late adolescence and early adulthood (Favaro et al. Reference Favaro, Caregaro, Tenconi, Bosello and Santonastaso2009; Kessler et al. Reference Kessler, Berglund, Chiu, Deitz, Hudson, Shahly, Aguilar-Gaxiola, Alonso, Angermeyer, Benjet, Bruffaerts, de Girolamo, de Graaf, Maria Haro, Kovess-Masfety, O'Neill, Posada-Villa, Sasu, Scott, Viana and Xavier2013). A new source of non-shared environmental risk was also identified in late adolescence, accounting for 80% of the environmental contribution at this time. In accordance with the recommendation by Sullivan & Eaves (Reference Sullivan and Eaves2002) we modelled influence derived from shared environmental sources, but as with other studies utilising Cholesky decomposition modelling with such data, we found non-significant influence – a likely consequence of insufficient power. Certainly, by late adolescence the variance accounted for by the shared environment is very small, estimated at 7.6%.
Our second aim explored the nature of specific risk factors for DE that occurred in early- to mid-adolescence, preceding the increase in genetic influence in later adolescence for DE. Despite negative life events having been shown to associate with DE in adolescence (e.g. McKnight Investigators, 2003), in the current sample it was not predictive of DE in late adolescence. There are a number of reasons for this: 12-month life events preceding the first two waves of data collection may not have provided sufficient numbers of traumatic experiences to adequately produce a detectable effect; earlier sources of trauma may be more influential in the later development of DE, e.g. parental loss in the form of separation prior to age 17 years was associated with increased risk for major depression as an adult (Kendler et al. Reference Kendler, Neale, Kessler, Heath and Eaves1992); the number of life events may be a blunt instrument in this context as opposed to a weighting based on self-report of impact; as the traumatic life events were reported by parents, it may be that the most traumatic events were kept secret from the parents by the twin, such as sexual assault or bullying at school, and thus were not assessed in the current study. This latter suggestion is somewhat supported by the finding that the life events measure was heavily influenced by the shared environment.
By contrast, both increased levels of weight-related peer teasing from early- to mid-adolescence, as well as greater weight-related peer teasing measured during mid-adolescence, were related to development of DE in late adolescence. These results underscore experience of weight-related peer teasing as being exceptionally challenging during adolescence, consistent with previous research that has highlighted the role of factors related to peer group acceptance in the emergence of DE. This is a developmental period combining significant hormonal changes usually functioning to redistribute or increase body adiposity (e.g. Garn et al. Reference Garn, LaVelle, Rosenberg and Hawthorne1986) and transitions from relationships predominantly family-focused to greater independence and peer-oriented connectivity associated with increased investment in seeking acceptance by peers (Brown et al. Reference Brown, Eicher and Petri1986; Larsen et al. Reference Larsen, Maryse, Moneta, Holmbeck and Duckett1996).
Taken together, our two main study outcomes are suggestive of the possibility of an interaction between the emergence of a period of genetic risk for DE and weight-related peer teasing in mid to late adolescence. In other words, this period in adolescence conveys heritable risk for DE at a time when non-shared environmental influences increase in importance as greater emphasis is placed on independence and tighter peer-focused relationships. Our results suggest approaches that strategically targeting peer relationships and skills for standing up to teasing and other weight related pressures within preventive interventions are likely to present key opportunities to protect older adolescents from the emergence of heritable risk factors for the development of DE, especially binge and purge behaviours. This is consistent with the success of approaches for young adolescents that prevent the growth of weight and shape concern by targeting internalization of cultural appearance ideals promoted by media and peers (Richardson & Paxton, Reference Richardson and Paxton2010; Bird et al. Reference Bird, Halliwell, Diedrichs and Harcourts2013; Wilksch et al. Reference Wilksch, Paxton, Byrne, Austin, McLean, Thompson, Dorairaj and Wade2015).
It is of interest to note in the current study that the profile of risk factors for DE change even while the mean levels of DE have remained stable. One recent study using the self-report version of the EDE showed mean scores were elevated slightly at 17 years, while symptoms prior (14 years old) and post (20 years old) were of similar magnitudes (Allen et al. Reference Allen, Crosby, Oddy and Byrne2013). It is possible that our groups have bisected Allen et al.'s peak symptom group, resulting in an ‘averaging’ of their profile. Studies using other measures to track DE symptomatology over time show no coherent common trajectory across adolescence (e.g. Kotler et al. Reference Kotler, Cohen, Davies, Pine and Walsh2001; Klump et al. Reference Klump, Burt, McGue and Iacono2007; Neumark-Sztainer et al. Reference Neumark-Sztainer, Wall, Larson, Eisenberg and Loth2011; Abebe et al. Reference Abebe, Lien and von Soest2012).
The current study needs to be considered within a number of limitations, the first of which relates to no formal measure of puberty being available in the current study. However, the population-based age ranges suggests our sample is highly likely not to be pre-pubertal; rather, participants are already on a trajectory to sexual maturity. Further work is required to assess how stages within puberty progression impact on the results. Second, the low mean of the EDE global score, although the range was considerable, which may have limited error variance estimations. Third, while the impact of life events and weight-related peer teasing were found to be predominantly environmental origin, the small heritable influences contributed by these variables may conflate the additive genetic and non-shared environmental variance in our analysis. Fourth, the initial response rate was 49%, akin to equivalently large epidemiological studies of twins incorporating multiple time points (Wade et al. Reference Wade, Bergin, Tiggemann, Bulik and Fairburn2006; Stice et al. Reference Stice, Marti and Rohde2013). Fifth, while our primary analyses did not control for BMI centile, we did run additional models adjusting for BMI. Minor differences were observable, but CIs were broadened, indicating a lack of power which argues for the use of larger samples in the investigation of the complexity of genetic and environmental risk factors on DE over adolescent development. Finally, participants reporting DE at wave 1 were offered referrals, which may have influenced DE at wave 3. Unfortunately no treatment data are available to test this suggestion.
Future research should more explicitly aim to test the presence of gene and environment interactions occurring at later adolescence in terms of increasing risk for later onset DE in early adulthood. This is not a trivial problem, given previous research has found that 23% of women aged between 22 and 27 years experience DE in the 12-month period before assessment, which results in a sustained negative impact on quality of life (Wade et al. Reference Wade, Wilksch and Lee2012). A better understanding of how to protect young women from the onset of DE can be expected to result in a greater participation of young women in society, given the impact of DE on social, vocational and educational functioning, and resultant reduced productivity (Simon et al. Reference Simon, Schmidt and Pilling2005).
Acknowledgements
Grants 324715 and 480420 from the National Health and Medical Research Council (NHMRC) to T.D.W. supported this work. Administrative support for data collection was received from the Australian Twin Registry, which is supported by an Enabling Grant (ID 310667) from the NHMRC administered by the University of Melbourne. The authors thank the twins and their families for their participation in this research, and Ms. Judith Slater for co-ordinating the data collection.
Declaration of Interest
None.