Recent years have seen an increasing interest in the genetics and physiology of lactic acid bacteria because of their importance for major economic activities, primarily their use in a variety of dairy fermentations (Mierau & Kleerebezem, Reference Mierau and Kleerebezem2005).
Lactococcus lactis, the model lactic acid bacterium (LAB), is a food grade Gram-positive bacterium, which is well-characterised and ‘generally recognised as safe’ (GRAS). Many genetic tools have been developed and its complete genome was sequenced (Wessels et al. Reference Wessels, Axelsson, Bech Hansen, De Vuyst, Laulund, Lähteenmäki and Lindgren2004). Protein secretion by this GRAS bacterium would allow production directly in a food product and thus an interaction between the secreted protein (enzyme or antigen) and the environment (the food product itself or the digestive tract). Lc. lactis can be considered a good candidate for heterologous protein secretion for at least two reasons: (i) to date, relatively few proteins are known to be secreted by Lc. lactis (ii) secreted proteins are prone most of the time to extracellular degradation (Wu et al. Reference Wu, Ye, Wu, Ng and Wong1998; Lee et al. Reference Lee, Kim, Bae, Byun and Chung2000).
High-level production of heterologous proteins in Lc. lactis has been obtained using lactococcal constitutive or inducible promoters (Kuipers et al. Reference Kuipers, de Ruyter, Kleerebezem and de Vos1997; de Vos, Reference de Vos1999). One of the developed expression systems for Lc. lactis is the Nisin Controlled gene Expression system (NICE). This system has become one of the most successful and widely used tools for regulated gene expression in Gram-positive bacteria (Mierau & Kleerebezem, Reference Mierau and Kleerebezem2005).
The NICE system has been used to express genes of various different backgrounds (Gram-positive, Gram-negative and eukaryotic) to study metabolic and enzyme function and to produce larger amounts of an enzyme for food, medical or technical applications (de Ruyter et al. Reference de Ruyter, Kuipers and de Vos1996).
Lipases constitute the most important group of biocatalysts for biotechnological applications (Benjamin & Pandey, Reference Benjamin and Pandey1998). Lipases have been isolated from many species of plants, animals, bacteria, fungi and yeast. The enzymes from microorganisms are used in various industries such as dairy, food, detergents, textile, pharmaceutical, cosmetic and biodiesel industries, and in synthesis of fine chemicals, agrochemicals and new polymeric materials (Saxena et al. Reference Saxena, Ghosh, Gupta, Davidson, Bradoo and Gulati1999). Lipases are extensively used in the dairy industry for hydrolysis of milk fat. The dairy industry uses lipases in the modification of fatty acid chain lengths, to enhance the flavours of various cheeses. Current applications also include the acceleration of cheese ripening and the lipolysis of butter, fat and cream (Jaeger & Eggert, Reference Jaeger and Eggert2002). The free fatty acids generated by the action of lipases on milk fat endow many dairy products, particularly soft cheeses, with their specific flavour characteristics (Sharma et al. Reference Sharma, Chisti and Banerjee2001).
Burkholderia cepacia lipase formerly known as Pseudomonas cepacia lipase has been shown to be a useful enzyme which catalyses a broad range of different reactions in water and non-polar solvents under mild conditions. Bur. cepacia lipase is a highly selective catalyst for a broad range of substrates (Kazlauskas & Weber, Reference Kazlauskas and Weber1998), including the kinetic resolution of racemic mixtures of secondary alcohols by hydrolysis in water or esterification in organic solvents (Schmid & Verger, Reference Schmid and Verger1998; Bornscheuer & Kazlauskas, Reference Bornscheuer and Kazlauskas1999; Jaeger & Eggert, Reference Jaeger and Eggert2002).
Due to the importance of lipase enzyme and many advantages of Lc. lactis as a cell factory as well as unique characteristics of NICE system, this study was an attempt to clone and highly express the lipase from Bur. cepacia in Lc. lactis using NICE protein expression system.
Material and methods
Bacterial strains, media and growth condition
The bacterial strains and plasmids used are listed in Table 1. Lc. lactis was grown statically in M17medium (Oxoid, USA) at 30 °C in tightly capped flasks supplemented with 0·5% glucose. Escherichia coli was grown in Luria-Bertani (LB) medium (Oxoid, USA) at 37 °C. Chloramphenicol was used at a concentration of 10 μg/ml, for Lc. lactis and ampicillin was used at concentration of 100 μg/ml for Esch. coli when needed. Nisin powder (ICN; 2·5% nisin content) was used at concentrations of 1, 5, 10 and15 ng/ml.
Table 1. Bacterial strains and plasmids used in this study

DNA manipulation
Chromosomal DNA of Bur. cepacia was obtained using miniprep DNA extraction Kit (GenAll, Korea) according to the manufacturer's instructions. The presence and quality of genomic DNA was confirmed by agarose gel electrophoresis. The optical density (OD) ratio at 260 and 280 nm (OD260/OD280) was calculated to estimate the purity of extracted DNA. Plasmid DNA was isolated using plasmid extraction miniprep spin kit (Fermentas) according to the manufacturer's instructions.
Synthesis and sequencing lipase gene
Lipase gene (LipA) from Bur. cepacia was amplified by PCR using a pair of primers designed according to the lipase nucleotide sequences deposited in GeneBank under accession number (M58494). The PCR product was cloned into Esch. coli TOP10 cells using the pCR Blunt vector (Fermentas).
The recombinant plasmid was sequenced by Sigma Company (Singapore). The results of DNA sequencing were then run in Chromas Lite program to analyse the similarity of sequenced gene in GenBank library. The comparison of the DNA sequence was carried out using the database of the National Centre of Biotechnology (http://www.ncbi.nih.gov). Meanwhile, the analysis of the lipase gene was achieved with Biology Workbench (http://www.biology.ncsa.sdsc.edu) and ExPASy (Expert Protein Analysis System) (http://www.expasy.org.tools).
Cloning and expression of lipase gene
A set of primers were designed based on the sequence of Bur. cepacia (Forward: 5′-ATGGCCAAATCGATGCGTTCC-3′ and Reverse: 5′-TTACACGCCCGCGAGCTTCA-3′). Amplification process was carried out in a reaction mixture containing 50–100 ng DNA template, 30 pmol each forward and reverse primers, 0·2 mm dNTP mix, 2 mm MgCl2, 2 U Taq DNA polymerase (Fermentas; Lithuania), 10×PCR buffer with the following PCR conditions: an initial denaturation step at 94 °C for 4 min, 35 cycles at 94 °C for 1 min, annealing at 64 °C for 50 s, and extension at 72 °C for 2 min, except for the final extension of 10 min, and preservation at 4 °C.
The PCR product was directly cloned into the pCR-Blunt vector (Ferments) according to the manufacturer's instructions and transformed into Esch. coli Top 10 competent cells. The positive clones were obtained on the LB agar plate contains 100 μg/ml ampicillin after 24 h at 37 °C. The positive clones were confirmed using colony PCR as well as plasmid extraction following to the run on gel electrophoresis.
Expression of lipase gene in Lactococcus lactis
The PCR-Blunt vector used as template to amplify the lipase gene using following primers; Forward 5′- GCCTGCAGCATCACCATCACCATCACGGGATGGCCAAATCGATGCGTTCC-3′ and Reverse 5′- AAGCTTTTACACGCCCGCGAGCTTCA-3′, to generate a product that would encode a LipA recombinant protein with a six His-tag at the N-terminus. In the forward primer, nucleotides representing the six histidine residues (italicised) are located downstream of the incorporated PstI restriction site (underlined); in the reverse primer, a HindIII restriction site (underlined) was incorporated downstream of the LipA sequence.
PCR was optimised and performed with initial denaturation at 94 °C for 4 min, followed by 35 cycles of 94 °C for 1 min, 62 °C for 50 s, and 72 °C for 2 min. Amplification was ended at 72 °C for 10 min. The corresponding 1092 bp PCR product was purified and digested with both PstI and HindIII and ligated into the corresponding digested sites in pNZ8148, producing the recombinant plasmid pNZ-LipA.
Competent Lc. Lactis NZ9000 cells were then electro-transformed with the recombinant plasmids (Eppendorf, USA) at 2000 V for 5 ms, spread on M17 agar contains 0·5% glucose and 10 μg/ml Chloramphenicol incubated at 30 °C for 24 h (Holo & Nes, Reference Holo and Nes1989). Transformants harbouring the recombinant plasmids were verified through enzymatic digestion; PCR and sequencing verified the correct lipase gene sequence, orientation, and frame.
Protein analysis
Overnight culture of recombinant Lc. lactis (pNZ-lipA) was diluted 100-folds in fresh M17-glucose-chloramphenicol and grown at 30 °C until an OD600 nm value of 0·5–1. Expression of lipase was induced with different concentration of nisin 1, 5, 10 and 15 ng/ml at 30 °C. Preparation of protein was carried out according to Schmidtke et al. (Reference Schmidtke, Schmidt and Kloetzel1997) with some modifications. Bacterial cells were collected by two-times centrifugation at 4000 g for 5 min followed by washing and resuspension in phosphate buffered saline (PBS) containing 1% (w/v) sterile, acid-washed glass-beads (Merck, Germany). The cells were disrupted by vortexing for 1 min at maximum speed, and then chilled on ice for 2 min, repeating this process 8 times. The suspension was centrifuged at 4000 g for 20 min at 4 °C and the resulting supernatant was transferred into a separate tube. Both cellular supernatant and cell pellet were treated with 2% SDS detergent at 37 °C for 30 min before being centrifuged at 20 000 g for 30 min at 4 °C.
SDS-PAGE and western blotting
SDS-PAGE was carried out according to the protocol of Laemmli (Reference Laemmli1970). The protein preparations were further subjected to boiling at 100 °C for 5 min in SDS and dithiothreitol (DTT) before being analysed on a 12% (w/v) denaturing polyacrylamide gel. SDS-PAGE resolved polypeptides were transferred onto a polyvinyllidene difluoride (PVDF) membrane using a semi-dry electroblotter (BioRad, USA) at 64 mA for 45 min.
Lipase activity assay
Liberated free fatty acids were determined by calorimetric method using olive oil as a substrate (Rahman et al. Reference Rahman, Kamarudin, Yunus, Salleh and Basri2010). An equal volume of olive oil (Merck, Germany) and 50 mm phosphate buffer (pH 7) was mixed to prepare the emulsion. One millilitre of enzyme was added to 2·5 ml of the emulsion plus 20 μl 0·02 m CaCl2 and was shaken at 200 rpm for 30 min at 37 °C. One millilitre of 6 m HCl and 5 ml isooctane were then added to stop the enzyme reaction. This was followed by a vigorous mixing for 30 s with a vortex mixer. Four millilitres of the upper isooctane layer was transferred to a test tube containing 1 ml copper reagent. The copper reagent was prepared by 5% (w/v) copper (II) acetate-1-hydrate and the pH was adjusted to 6·1 with pyridine. The absorbance of the upper layer which contained the liberated fatty acids was read at 715 nm. Lipase activity was determined by measuring the amount of free fatty acid released by referring to the standard curves of free fatty acids. One unit of lipase activity was defined as the rate of 1 μmol of fatty acid released per minute.
Results
Cloning and sequencing of the lipase gene
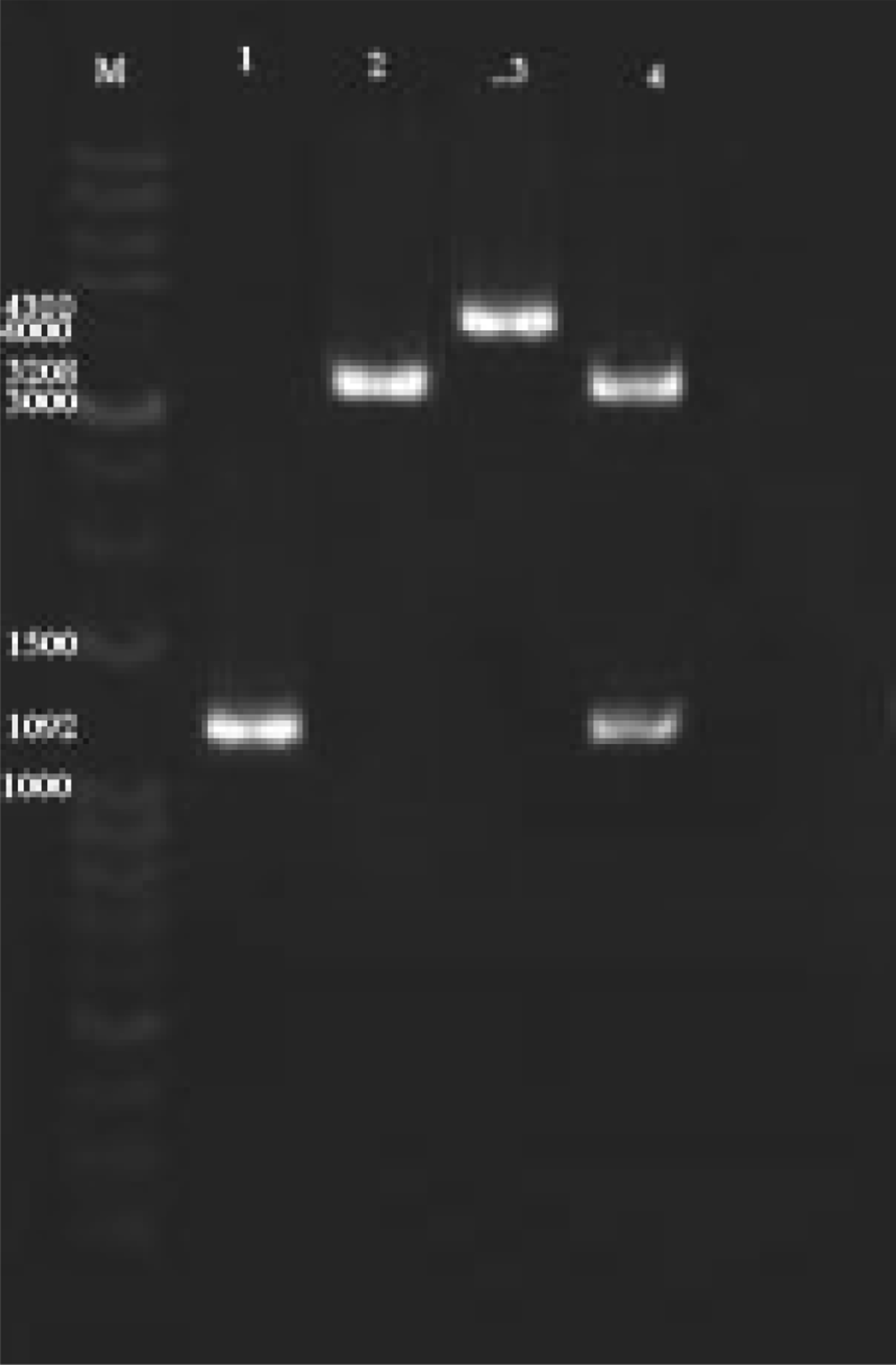
PCR fragments of the expected size (1092 bp) were obtained using degenerated primers, cloned and sequenced (Fig. 1). Nucleotide sequence analysis revealed an open reading frame (ORF) starting at bp 1 with a ATG codon and ending at AAT at bp 1092, thereby encoding a protein of 364 amino acids. Lipases are known to have an active-site consensus sequence Gly-X-Ser-X-Gly and to form a catalytic triad consisting of Ser, Asp and His residues. The lipase had the Gly-X-Ser-X-Gly sequence at positions 106–110, and Ser, Asp and His residues at 208, 315 and 278, respectively. In addition, two Asp residues, known Ca+2-binding sites, were found at positions 240 and 279, and two Cys residues forming a disulphide bond at positions 210 and 261 were also conserved.

Fig. 1. Agarose gel electrophoresis results Gene amplification and cloned. (M) Marker, (1) PCR Amplification of lipase gene, (2) single digested pNZ8148, (3) single digested cloned pNZ-LipA, (4) Double digested pNZ-LipA.
Expression of lipase in Lc. lactis
The lipase gene was successfully cloned and expressed in pNZ8148, an expression vector carrying nisin inducible nisA promoter, by Lc. lactis NZ9000 as host. To achieve this the lipase gene was inserted into pNZ8148 between PstI and HindIII restriction sites, then the recombinant vector (pNZ-lipA) was transformed into Lc. lactis NZ9000 by electroporation method. The recombinant Lc. lactis was cultured on M17-Glu-Cm and incubated at 30 °C for 24 h.
To detect the lipase expression of recombinant Lc. lactis containing pNZ8148, Lc. lactis containing pNZ-lipA were cultured on the Nile Blue Sulphate agar, and incubated at 30 °C for 24 h. Screening the results showed no lipase expression in cultured plates with Lc. lactis-pNZ8148 (no blue colour colonies) while the dark blue colonies was observed in plate containing Lc. lactis-pNZ-lipA (results not shown). These primary screenings indicate the expression of lipase by recombinant Lc. lactis.
Expression of lipase in Lc. lactis was analysed using SDS-PAGE and western blotting techniques. Expression optimisations were carried out with 1, 5, 10 and 20 ng/ml nisin while, the highest protein expression was detected for10 ng/ml nisin. In the protein extracts comparing induced and uninduced cultures for pNZ-lipA as well as the negative control (clones harbouring empty pNZ8148 plasmid), the SDS-PAGE results showed distinct bands corresponding to an expected protein size of 37·5 kDa which was predicted to be exclusively visible in the induced cultures (Fig. 2). Also, subsequent analysis by western blotting showed a clear expected band on 37·5 kDa which was only present in the induced cultures but not in the uninduced cultures or the negative control (Fig. 3). The results confirm successful expression of recombinant protein in Lc. lactis. Optimisation of the induction conditions showed that lipase expression can be induced using 1–15 ng/ml nisin with highest expression at 10 ng/ml nisin.
Fig. 2. SDS-PAGE results of lipase produced by Lc. lactis cells harbuoring LipA. The highest protein expression was detected after 4 h of induction with 10 ng/ml nisin.
Fig. 3. Western Blotting results of lipase produced by Lc. lactis cells harbuoring LipA. The highest protein expression was detected after 4 h of induction with 10 ng/ml nisin.
Functionality of the recombinant lipase of purified recombinant protein extracts was analysed by enzymatic assay. The protein assay showed high lipase production by recombinant Lc. lactis around 152·2 μg/ml.h. The specific lipolytic activity of the recombinant lipase was found 180 U/ml which was 34 fold higher than the native enzyme.
Discussion
To date, many expression systems have been developed to produce recombinant proteins for various biotechnological applications. Among prokaryotic systems, the highest protein levels are obtained using Esch. coli as the cell factory (Jana & Deb, Reference Jana and Deb2005; Raftari et al. Reference Raftari, Ghafourian, Sadeghifard, Sekawi, Saari and Abu Bakar2012). However, in Esch. coli, the most commonly used production strategies are intracellular (in the periplasm or even in the cytoplasm), and involve expensive and often problematic downstream purification processes. In contrast, heterologous proteins produced in Gram positive bacterial hosts can be easily secreted into the medium, thus facilitating their purification (Morello et al. Reference Morello, Bermúdez-Humarán, Llull, Solé, Miraglio, Langella and Poquet2008; Raftari et al. Reference Raftari, Ghafourian and Abu Bakar2013).
Gram-positive bacterium having a GRAS status, Lc. lactis, is becoming an attractive alternative for heterologous protein secretion because of a number of properties which make it suitable for large scale protein expression; no inclusion bodies and endotoxin formation (Miyoshi et al. Reference Miyoshi, Poquet, Azevedo, Commissaire, Bermudez-Humaran, Domakova, Leloir, Oliveira, Gruss and Langella2002; Nouaille et al. Reference Nouaille, Ribeiro, Miyoshi, Pontes, Le Loir, Oliveira, Langella and Azevedo2003; Peterbauer et al. Reference Peterbauer, Masischberger and Haltrich2011), simple fermentation procedure which make direct scale up from 1 to 1000 litres possible, self-cloning and food grade plasmid selection mechanism (Mierau et al. Reference Mierau, Leij, van Swam, Blommestein, Floris, Mond and Smid2005a; Pinto et al. Reference Pinto, Kuipers, Marreddy, Poolman and Kok2011), and finally being food grade (Konings et al. 2000; Nouaille et al. Reference Nouaille, Ribeiro, Miyoshi, Pontes, Le Loir, Oliveira, Langella and Azevedo2003; Peterbauer et al. Reference Peterbauer, Masischberger and Haltrich2011). Lc. lactis has been studied for the last 2 decades: its metabolism is relatively simple and well known, and the genome information of at least five strains of Lc. lactis were described and available (Bolotin et al. Reference Bolotin, Wincker, Mauger, Jaillon, Malarme, Weissenbach, Ehrlich and Sorokin2001; Klaenhammer et al. Reference Klaenhammer, Altermann, Arigoni, Bolotin, Breidt, Broadbent, Cano, Chaillou, Deutscher, Gasson, van de Guchte, Guzzo, Hartke, Hawkins, Hols, Hutkins, Kleerebezem, Kok, Kuipers, Lubbers, Maguin, McKay, Mills, Nauta, Overbeek, Pel, Pridmore, Saier, van Sinderen, Sorokin, Steele, O'Sullivan, de Vos, Weimer, Zagorec and Siezen2002). The comprehensive experience and knowledge gained has led to the application of Lc. lactis in areas other than in food production. It is used as a cell factory for pilot production of pharmaceutical products, production of human cytokines and other therapeutic agents for in situ treatments, expression of bacterial and viral antigens for safe vaccination via mucosal immunisation (Hols et al. Reference Hols, Kleerebezem, Schanck, Ferain, Hugenholtz, Delcour and de Vos1999; Hugenholtz et al. Reference Hugenholtz, Kleerebezem, Starrenburg, Delcour, de Vos and Hols2002; Nouaille et al. Reference Nouaille, Ribeiro, Miyoshi, Pontes, Le Loir, Oliveira, Langella and Azevedo2003; Hanniffy et al. Reference Hanniffy, Wiedermann, Repa, Mercenier, Daniel, Fioramonti, Tlaskolova, Kozakova, Israelsen, Madsen, Vrang, Hols, Delcour, Bron, Kleerebezem and Wells2004; Leroy & Devuyst, Reference Leroy and Devuyst2004; Mierau et al. Reference Mierau, Leij, van Swam, Blommestein, Floris, Mond and Smid2005a).
The NICE system (de Ruyter et al. Reference de Ruyter, Kuipers and de Vos1996; Kuipers et al. Reference Kuipers, de Ruyter, Kleerebezem and de Vos1997, Reference Kuipers, de Ruyter, Kleerebezem and de Vos1998) derives from the nis (nisABTCIPRKEFG) operon present in some Lc. lactis strains, which is involved in the biosynthesis of the antimicrobial peptide nisin (Kuipers et al. Reference Kuipers, Rollema, Siezen and de Vos1995). The NICE comprises the regulatory elements of the nis operon: PnisA, the nisin-inducible promoter (cloned into several expression vectors), and nisRK, the regulator-sensor 2-component system (either carried by compatible plasmids or integrated in the chromosome). The NICE system has been extensively used to produce proteins in Lc. lactis (Kunji et al. Reference Kunji, Slotboom and Poolman2003; Le Loir et al. Reference Le Loir, Azevedo, Oliveira, Freitas, Miyoshi, Bermudez-Humaran, Nouaille, Ribeiro, Leclercq, Gabriel, Guimaraes, Oliveira, Charlier, Gautier and Langella2005; Mierau & Kleerebezem, Reference Mierau and Kleerebezem2005). It offers numerous advantages: (1) easy use, (2) tightly controlled and efficiently induced expression leading to high protein yields (Mierau & Kleerebezem, Reference Mierau and Kleerebezem2005) and (3) large-scale production process, as shown for Staphylococcus simulans lysostaphin (Mierau et al. Reference Mierau, Leij, van Swam, Blommestein, Floris, Mond and Smid2005a, Reference Mierau, Olieman, Mond and Smidb).
In this study, we succeeded in overproduction of lipase from Bur. cepacia in Lc. lactis using NICE system. For achieving that the complete lipase gene according to sequence deposited in GeneBank, was cloned and expressed in Lc. lactis NZ9000 using pNZ8148 inducible vector. The lipase gene encoding an extracellular lipase cloned from the meta-genomic library was composed of 1092 bp in length. The predicted protein consisted of 364 amino acids with a calculated molecular mass of 37·5-kDa. Nucleotide sequence alignment showed that lipase gene has 100% identity with Bur. cepaciae lipase and the lowest identity was observed with Vibrio vulnificus lipase. In previous study (Raftari et al. Reference Raftari, Ghafourian, Sadeghifard, Sekawi, Saari and Abu Bakar2012) we sub-cloned and expressed the lipase in Esch. coli. The results indicated expression of recombinant lipase by Esch. coli but comparing the results showed that Lc. lactis has ability to produce higher level of recombinant lipase in shorter period, while extracellular expression by Lc. lactis could also reduce the purification cost and time as well as being safe.
Sequence analysis also showed that there is only one cysteine in the lipase protein, which is similar to other reported lipase (Steele & Streit, Reference Steele and Streit2005). Cysteines are often involved in the formation of disulphide bonds in proteins, and proteins without cysteines are generally more flexible because of the lack of disulphide bonds (Li et al. 1995). This characteristic is important for protein secretion of extracellular bacterial proteins, which also readily allows the conformational change that accompanies interfacial activation.
In conclusion the NICE system was found to be a strong and practical protein expression system in Lc. lactis for production recombinant lipase enzyme that can be used for commercial purposes.
The authors are thankful to Ministry of Higher Education, Malaysia for financial support.