CHD is the most common birth malformation, occurring in 28% of all congenital birth defects. The prevalence of CHD at birth is estimated to be between 75 and 90/10,000 live births.Reference Kahr and Diller 1 CHD is the single largest cause of infant morbidity and mortality worldwide and represents a significant global economic burden.Reference Cowan and Ware 2 In the past few decades, several genetic and environmental factors have been shown to be involved in the aetiology of CHD, although the aetiology of most forms of CHD is still incompletely understood.Reference van der Bom, Zomer, Zwinderman, Meijboom, Bouma and Mulder 3 Many complex chromosomal abnormalities, submicroscopic rearrangements, whole-chromosome aneuploidies, and single-gene mutations have been demonstrated to be correlated with the occurrence of CHD. As a well-known example, 50% of trisomy 21 patients have atrioventricular canal defects and/or tetralogy of Fallot. A total of 80% of patients with 22q11.2 deletion syndrome are born with cardiovascular anomalies. The most prevalent anomalies among them are tetralogy of Fallot and tetralogy of Fallot with pulmonary atresia, as well as pulmonary atresia with ventricular septal defect.Reference Momma 4 Single-gene disorders such as NKX2.5, GATA4, TBX20, and growth/differentiation factor 1 (GDF1) mutations have been found in a wide spectrum of CHD, including ventricular septal defect, atrial septal defect, tetralogy of Fallot, and other kinds of cardiac malformations.
Pulmonary atresia is a rare kind of complex cyanotic CHD with a poor prognosis, occurring in 1.3–3.4% of all heart malformations.Reference Abid, Elloumi and Abid 5 , Reference Hoffman and Kaplan 6 It is characterised by an undeveloped pulmonary valve or pulmonary artery. The pulmonary valve is completely closed, thereby obstructing the outflow of blood from the right ventricle to the main pulmonary artery. Pulmonary atresia, including a wide spectrum of heart malformations with great morphological heterogeneity, is mainly divided into two categories: pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum. Although they are cousins in the pulmonary atresia family, the anatomical abnormalities, haemodynamics, surgery strategy, and prognosis are quite different. Table 1 shows the differences in anatomical abnormalities between pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum.
Table 1 Differences in anatomical abnormalities between PA/VSD and PA/IVS.

LPA=left pulmonary artery; MPA=main pulmonary artery; PA/IVS=pulmonary atresia with intact ventricular septum; PA/VSD=pulmonary atresia with ventricular septal defect; RPA=right pulmonary artery; RV=right ventricle; RVOT = right ventricular outflow tract; TV=tricuspid valve
Tetralogy of Fallot with pulmonary atresia, an extreme state of tetralogy of Fallot, has many similarities to pulmonary atresia with ventricular septal defect. For this reason, we included tetralogy of Fallot with pulmonary atresia under pulmonary atresia with ventricular septal defect in this study. In patients with this disease, no blood passes from the right ventricle to the lungs. Consequently, all pulmonary blood flow perfuse in a retrograde manner through the ductus arteriosus and/or the major aortopulmonary collateral arteries. Pulmonary artery dysplasia is common. The severity of cyanosis depends on the flow of collateral arteries and the distribution of pulmonary vasculature. Surgical treatments, palliative operations, or radical operations are determined by the development of the pulmonary vasculature.
In patients with pulmonary atresia with intact ventricular septum, all components of the right ventricle can be affected, including the atretic pulmonary valve and variable degrees of hypoplasia or other abnormalities in the right ventricular and tricuspid valve. Patients with adequate tricuspid valve, adequate right ventricle, and no evidence of right ventricle-dependent coronary circulation are candidates for biventricular repair, whereas others have to accept functional single-ventricle palliation. Overall, regardless of the anatomical subtype, the survival of pulmonary atresia with intact ventricular septum patients is reported to be as low as 50% at 5 years.Reference Zampi, Hirsch-Romano, Goldstein, Shaya and Armstrong 7
The incidence of pulmonary atresia has been reported to be 132–220/million live births.Reference Abid, Elloumi and Abid 5 , Reference Hoffman and Kaplan 6 The incidence of pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum is 0.18 and 0.04/1000 live births, occurring in 2.7 and 0.7% of all heart malformations, respectively. A study indicated that the recurrence rates of pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum were 5.3 and 14.3%, respectively, which are both higher than the total recurrence rate of CHD (3.98%).Reference Fesslova, Brankovic and Lalatta 8 In particular, the recurrence rate of pulmonary atresia with intact ventricular septum (14.3%) was the highest in various subgroups of CHD, indicating that genetic research in pulmonary atresia with intact ventricular septum patients may have great value.
The aetiology of the differences between pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum may be attributed to the different genetic patterns present in patients with each condition. This is supported by some studies, which have shown that there are statistical differences in gene expression patterns in patients with pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum.Reference Sun, Meng and You 9 , Reference Lee, Su and Cheng 10 Understanding the molecular mechanisms that influence the development of these diseases is essential for future diagnosis, prevention, therapeutic approaches, and genetic consulting; however, complex manifestations of CHD such as pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum cannot be simply attributed to monogenetic mutations. It has become increasingly accepted that the genetic pathogenesis of sporadic CHD arises from the accumulation of rare mutations, copy number variants, and common variants in cardiac development genes.Reference Bentham and Bhattacharya 11 In this article, we briefly introduce relevant genes in pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum and review the recent advances in genetic understanding.
Copy number variants
Copy number variants, a form of structural variants, refer to gains or losses of contiguous DNA ranging in size from 1 kb to several MBs. They contribute to the gene heterogeneity of various complex human diseases.Reference Gelb and Chung 12 , Reference Silversides, Lionel and Costain 13 As genetic testing has evolved to offer higher resolutions and greater diagnostic yields than chromosomal analyses, copy number variants have emerged as important causes of both syndromic and non-syndromic CHD.Reference Lander and Ware 14 – Reference Tomita-Mitchell, Mahnke and Struble 17 It is estimated that copy number variants contribute to 3–25% of CHD with extracardiac abnormalities and 3–10% of isolated CHD.Reference Lander and Ware 14 Serra-Juhe et al selected 95 fetuses with congenital malformations from medically terminated pregnancies at 17–22 weeks of gestation, including CHD, congenital nervous system malformations, and renal malformations. After karyotyping by chromosome microarray analysis and multiplex ligation-dependent probe amplification, 22 rare or very uncommon copy number variants were identified. The proportion of copy number variants with rearrangements was different between the different groups of malformations, being higher in fetuses with CHD (10/33 samples, 30.30%) and even higher when considering heart hypoplasia diseases alone (8/17, 47.06%).Reference Serra-Juhe, Rodriguez-Santiago and Cusco 18 These results illustrate the significance of copy number variants in the pathogenesis of CHD, particularly in complex CHD such as heart hypoplasia diseases.
As one kind of complex CHD, the role of copy number variants in pulmonary atresia was investigated by Xie et alReference Xie, Chen and Zhang 19 in 2014. A total of 82 pulmonary atresia, including five pulmonary atresia with intact ventricular septum, patient–parent trios and 189 controls were assayed using high-resolution genome-wide microarrays for copy number variants with an Illumina single nucleotide polymorphisms array platform (Illumina Inc., San Diego, United States of America). Out of 82 patients, 17 (20.7%) had identified genomic lesions, and eight of these copy number variants are considered pathogenic or likely pathogenic, including five de novo copy number variants occurring at two known CHD loci – 16p13.1 and 22q11.2 – and three rare copy number variants that were previously undiscovered – duplication of 5q14.1 (dihydrofolate reductase, DHFR), duplication of 10p13 (CUBN), and deletion of 17p13.2 (CAMTA2). Table 2 lists pathogenic and likely pathogenic copy number variants found in pulmonary atresia patients. Being widely considered as pathogenic, a 16p13.1 duplication was also reported in other kinds of CHD such as aortic dissections, tetralogy of Fallot, transposition of the great arteries, and hypoplastic left heart syndrome,Reference Warburton, Ronemus and Kline 16 , Reference Serra-Juhe, Rodriguez-Santiago and Cusco 18 – Reference Prakash, LeMaire and Guo 21 as well as other extracardiac clinical features such as behavioural abnormalities, cognitive impairment, and skeletal manifestations.Reference Nagamani, Erez and Bader 20 This region contains 12 genes, including myosin heavy chain 11 (MYH11), whose mutations may cause non-syndromic thoracic aortic aneurysms and patent ductus arteriosus.Reference Pannu, Tran-Fadulu and Papke 22 The 22q11.2 deletion is a well-known pathogenic variant discovered in a large spectrum of CHD such as DiGeorge syndrome, tetralogy of Fallot, pulmonary atresia with ventricular septal defect, double outflow of the right ventricle, ventricular septal defect, atrial septal defect, and interrupted aortic arch.Reference Momma 4 , Reference Silversides, Lionel and Costain 13 , Reference Warburton, Ronemus and Kline 16 Conotruncal heart defects, especially tetralogy of Fallot and pulmonary atresia with ventricular septal defect, are the most common disorders associated with the 22q11.2 deletion syndrome.Reference Xu, Wang and Xu 23 Of three rare copy number variants, all were considered likely pathogenic. Both the DHFR 5q14.1 duplication and CUBN 10p13 duplication are linked with folate-mediated one-carbon metabolism. It has been reported that genetic disturbances in folate metabolism may increase the risk of CHD.Reference Christensen, Zada and Rohlicek 24 DHFR is a key folate-metabolising enzyme that catalyses the reduction of dihydrofolate to tetrahydrofolate and folic acid to dihydrofolate.Reference Cario, Smith and Blom 25 Encoded by CUBN, the cubilin receptor is expressed in the apical pole of the absorptive epithelia, and it plays an important role in the absorption of vitamin B12.Reference Kozyraki 26 , Reference Tsaroucha, Chatzaki and Lambropoulou 27 An interesting fact is that another important gene in folate and vitamin B12 metabolism, 5, 10-methylenetetrahydrofolate reductase (MTHFR), which will be discussed below, may contribute to the pathogenesis of pulmonary atresia with intact ventricular septum.Reference Lee, Su and Cheng 10 All these former studies implied an intimate connection, which might be a dosage-sensitive association, between folate-related genes and a specific subtype of CHD (pulmonary atresia) in humans. The last rare copy number variant is the 17q13.2 deletion, which affects CAMTA2. CAMTA2 has been shown to interact with NKX2.5 and promote cardiac hypertrophy in mice.Reference Song, Backs and McAnally 28 In this article, 9.8% of pulmonary atresia patients were identified with potentially pathogenic copy number variants, suggesting a contribution of copy number variants in the pathogenesis of pulmonary atresia; however, owing to the relatively low number of pulmonary atresia with intact ventricular septum cases, the author did not separate pulmonary atresia with intact ventricular septum from pulmonary atresia with ventricular septal defect. The contributions of copy number variants to pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum, respectively, still require further study.
Table 2 Pathogenic and likely pathogenic CNVs reported in PA patients.

ASD=atrial septal defect; CN=copy number; CNV=copy number variant; COA=coarctation of the aorta; DORV=double outflow of right ventricle; HLHS=hypoplastic left heart syndrome; IAA=interrupted aortic arch; LP=likely pathogenic; P=pathogenic; PA=pulmonary atresia (without knowing PA/VSD or PA/intact ventricular septum); TGA=transposition of the great arteries; TOF=tetralogy of Fallot; VSD=ventricular septal defect
* TBX1 is not the only gene in this interval. It is listed alone because it has been identified as necessary for CHD
Moreover, in some studies on tetralogy of Fallot chromosome rearrangements, a few pulmonary atresia patients were discovered with similar gene variants. In 2012, Soemedi et alReference Soemedi, Topf and Wilson 29 reported a phenotype-specific effect of chromosome 1q21.1 rearrangements in CHD. After examining 948 patients with tetralogy of Fallot, 1488 patients with other kinds of CHD, and 6760 controls, they found that duplication of 1q21.1 was more common in cases of tetralogy of Fallot than in controls, whereas deletion of 1q21.1 was more common in cases of non-tetralogy of Fallot than in controls. They also reported a 1q21.1 duplication in a patient with tetralogy of Fallot and pulmonary atresia and a 1q21.1 triplication in a patient with pulmonary atresia, without knowing about the presence of pulmonary atresia with intact ventricular septum or pulmonary atresia with ventricular septal defect. The 1q21.1 region contains many important genes including GJA5, GJA8, and BCL9. Owing to its critical function and mutations associated with pulmonary atresia, the GJA5 gene is notable and will be discussed below. Duplication of 1q21.1 was also found in other types of CHD, such as coarctation of the aorta, transposition of the great arteries, double outflow of the right ventricle, hypoplastic left heart syndrome, and interrupted aortic arch.Reference Silversides, Lionel and Costain 13 , Reference Soemedi, Wilson and Bentham 15 , Reference Warburton, Ronemus and Kline 16 , Reference Christiansen, Dyck and Elyas 30 , Reference Mefford, Sharp and Baker 31
Although some copy number variants were reported in pulmonary atresia cases, there is still not enough information to evaluate the contribution of these mutations in pulmonary atresia, especially in pulmonary atresia with intact ventricular septum. Low number of pulmonary atresia with intact ventricular septum patients was the main limitation in estimating the role of copy number variants in pulmonary atresia and distinguishing the different genetic mechanisms of pulmonary atresia with ventricular septal defect versus pulmonary atresia with intact ventricular septum. Future studies could detect copy number variants in pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum and better estimate the contribution of these mutations to each disease variant.
Reported single-gene mutations in patients with pulmonary atresia
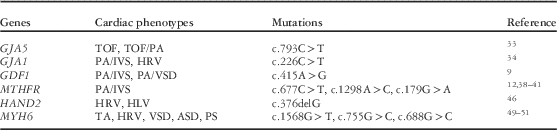
The cumulative effect of single-gene mutations is considered as a possible cause of CHD. In recent years, some studies reported that single-gene mutations such as GAJ5, GDF1, and MTHFR were found in pulmonary atresia patients. Table 3 lists reported single-gene mutations in patients with pulmonary atresia or similar phenotypes.
Table 3 Reported single-gene mutations in patients with pulmonary atresia or other similar phenotypes.

ASD=atrial septal defect; HLV=hypoplasia left ventricular; HRV=hypoplasia right ventricular; PA=pulmonary artery; PA/IVS=pulmonary atresia with intact ventricular septum; PA/VSD=pulmonary atresia with ventricular septal defect; PS=pulmonary stenosis; TA=tricuspid atresia; TOF=tetralogy of Fallot; VSD=ventricular septal defect
GAJ5
The GJA5 gene, located at 1q21.1, encodes the cardiac gap junction protein connexin 40 (Cx40). Cx40 has key functions in cell adhesion and cell–cell communication. Deletion of Cx40 in a mouse model caused various structural heart abnormalities;Reference Christiansen, Dyck and Elyas 30 however, as of yet, there are no data from animal models for GJA5 overexpression.Reference Silversides, Lionel and Costain 13 GJA5 was recently shown to be expressed in cells derived from the second heart field during outflow tract development, where it is regulated by the key cardiac transcription factor HAND2.Reference Holler, Hendershot, Troy, Vincentz, Firulli and Howard 32 Thus, GJA5 is an important candidate for CHD.
As previously mentioned, duplications of GJA1 in region 1q21.1 were detected in many CHD. Soemedi et alReference Soemedi, Topf and Wilson 29 detected one GJA5 duplication in a tetralogy of Fallot with pulmonary atresia patient and one GJA5 triplication in a pulmonary atresia patient. Similarly, in 2013, Guida et alReference Guida, Ferese and Rocchetti 33 found a heterozygous nucleotide change c.793C>T in exon 2 of the GJA5 gene, leading to the p.Pro265Ser variant at the carboxyl terminus of the protein in two unrelated sporadic patients, one with tetralogy of Fallot and one with tetralogy of Fallot with pulmonary atresia. Mutations in the GJA1 gene, related to GJA5, can cause oculodentodigital dysplasia. Izumi et alReference Izumi, Lippa, Wilkens, Feret, McDonald-McGinn and Zackai 34 reported an oculodentodigital dysplasia patient with a c.226C>T (p.Arg76Cys) mutation in the GJA1 gene presenting with pulmonary atresia with intact ventricular septum, right ventricular hypoplasia, and tricuspid stenosis.
GDF1
GDF1 is a secreted glycoprotein and a member of the transforming growth factor β (TGF-β) superfamily, which is widely known as a necessary factor in left–right axes formation and asymmetric organ morphogenesis. Working as a coligand of nodal signalling and a member of the TGF-β family, GDF1 plays an essential role in the establishment of left–right axes and regulates the growth and differentiation of cells in both embryonic and adult tissues. A previous study indicated that individuals with NODAL mutations had a significantly higher occurrence of pulmonary valve atresia compared with cases without a detectable NODAL mutation.Reference Mohapatra, Casey and Li 35 Evidence from human and animal studies suggests that GDF1 may play an important role in cardiac physiology and pathology.Reference Zhang, Zhang and Gao 36
Mutations in the GDF1 gene have been found in 2% of a large group of patients with a wide spectrum of CHD, including transposition of the great arteries, double outflow of the right ventricle, tetralogy of Fallot, and interrupted aortic arch.Reference Wessels and Willems 37 Sun et alReference Sun, Meng and You 9 reported four target single nucleotide polymorphisms of GDF1 in non-syndromic CHD patients by using polymerase chain reaction (PCR)-based denaturing high-performance liquid chromatography and restriction fragment length polymorphism. According to these results, the AA genotype and A allele of GDF1 rs4808870 (NM_001290265.1:c.415A>G) showed statistically significant differences between pulmonary atresia patients and the control group. In the pulmonary atresia with intact ventricular septum subgroup, these statistically significant differences were especially pronounced. This implies that a distinguishable genetic pattern occurs between pulmonary atresia with intact ventricular septum and pulmonary atresia with ventricular septal defect, and alterations in the GDF1 gene may be more common in pulmonary atresia with intact ventricular septum. Future studies will determine the causality between the mutation in GDF1 and downstream signalling. These results need to be confirmed in a larger population of pulmonary atresia patients.
MTHFR
Different enzymes including solute carrier family 19 (folate transporter), member 1 (SLC19A1), DHFR, MTHFR, and methionine synthase reductase (MTRR) influence DNA synthesis through the methylation cycle by converting homocysteine into methionine.Reference Wessels and Willems 37 Among these, MTHFR, catalyses the reduction of methylenetetrahydrofolate to 5-methyltetrahydrofolate. During the past decade, mutations in the MTHFR gene were found to be related to many diseases including different kinds of cancer, Parkinson’s disease, and venous thrombosis. Recent studies propose that hyperhomocysteinemia is a high risk for the development of heart defects.Reference Verkleij-Hagoort, Bliek, Sayed-Tabatabaei, Ursem, Steegers and Steegers-Theunissen 38 Therefore, polymorphisms in MTHFR may be closely related to the risk of CHD.Reference Wang, Hou, Wang, Wei, Li and Jiang 39
The relationship between MTHFR mutations and CHD is debatable, however. Verkleij-Hagoort et alReference Verkleij-Hagoort, Bliek, Sayed-Tabatabaei, Ursem, Steegers and Steegers-Theunissen 38 published a meta-analysis in 2007, concluding that the MTHFR variant c.677C>T on patient or maternal genotypes was not significantly associated with CHD. Wang et alReference Wang, Hou, Wang, Wei, Li and Jiang 39 also published a meta-analysis in 2013, indicating that the MTHFR c.677C>T polymorphism was significantly associated with CHD in Asian but not Caucasian persons. The results of Xu et alReference Xu, Xu and Xue 40 showed that MTHFR c.179G>A is associated with CHD in Chinese populations. These results were also supported by the observations that there is a relatively greater incidence of right-sided over left-sided defects among Asians.Reference Gelb and Chung 12 In 2014, Sayin Kocakap et alReference Sayin Kocakap, Sanli, Cabuk, Koc and Kutsal 41 suggested that the MTHFR c.1298A>C is a risk factor for conotruncal heart disease; however, whether pulmonary atresia patients have this variant has not been determined. In 2006, Lee et alReference Lee, Su and Cheng 10 conducted a study involving 213 CHD patients and 195 healthy controls. The results showed no significant differences between CHD patients and healthy controls as far as the overall genotype was concerned. Taking into account the variation in subgroups of all CHD patients, however, they noticed a significantly increased proportion of homozygous TT genotypes of MTHFR c.677C>T in pulmonary stenosis and pulmonary atresia with intact ventricular septum patients. These discrepant results depict the heterogeneity in the developmental mechanism of CHD and remind us that different forms of CHD may have different associated gene mutations. MTHFR is more closely related to the occurrence of pulmonary atresia with intact ventricular septum than pulmonary atresia with ventricular septal defect. A notable fact is that MTHFR, DHFR, and CUBN, which are mentioned in copy number variants, are important enzymes in folate and vitamin B12 metabolism. The relevance between MTHFR and other genes active in folate and vitamin B12 metabolism has caused researchers to speculate that MTHFR plays a role in the molecular mechanism of pulmonary atresia. It is reasonable to assume that there is an association between folate and vitamin B12 metabolism and CHD, or more specifically pulmonary atresia. Future studies should narrow the diagnosis to focus on one subtype of CHD – such as pulmonary atresia with intact ventricular septum – and explore the pathophysiology from susceptibility genes to expressed proteins and cardiac development.
Candidate single-gene mutations in other similar phenotypes
Owing to the great heterogeneity of CHD, studies on single-gene mutations with pulmonary atresia are not sufficient. Similar phenotypes of CHD may carry candidate gene mutations, which are also involved in the pathogenesis of pulmonary atresia. Right heart hypoplasia and tricuspid atresia have haemodynamics and anatomical abnormalities similar to pulmonary atresia with intact ventricular septum. In this portion, we review some genes that may relate to right ventricular hypoplasia, tricuspid atresia, and pulmonary valve formation. These genes are listed in Table 3. Although not detected in pulmonary atresia patients earlier, these genes are potential single-gene mutations involved in similar diseases and provide new directions for research into the genetic mechanism of pulmonary atresia.
HAND1 and HAND2
The HAND1 and HAND2 genes encode heart and neural crest derivative-expressed protein 1 and 2, respectively, which belong to the basic helix-loop-helix family of transcription factors. HAND proteins are expressed within the developing ventricular chambers, cardiac neural crest, endocardium (HAND2 only), and epicardium (HAND2 only). They perform critical roles in cardiomyocyte growth in the ventricle, proliferation of cardioblasts in the distal outflow tract, and the balance between proliferation and differentiation in the developing heart.Reference Vincentz, Barnes and Firulli 42 , Reference Risebro, Smart, Dupays, Breckenridge, Mohun and Riley 43
In early-stage studies, mice lacking both HAND genes and NKX2.5 failed to form any ventricular chamber.Reference Srivastava, Gottlieb and Olson 44 VanDusen et alReference VanDusen, Casanovas and Vincentz 45 demonstrated that endocardial ablation of HAND2 resulted in the failure to develop a patent tricuspid valve, intraventricular septal defects, and hypotrabeculated ventricles, which collectively resembled human congenital defect tricuspid atresia. Reamon-Buettner et alReference Reamon-Buettner, Ciribilli, Inga and Borlak 46 sequenced the HAND1 gene in 31 hypoplastic hearts, including seven right hypoplastic hearts. Of these 31 hearts, 24 had a c.376delG (p.Ala126fs) frameshift mutation, which led to the inability of HAND1 to modulate transcription either alone or in conjunction with basic helix-loop-helix binding partners. The results of this study, however, are questionable, as the mutations were found in formalin-fixed malformed hearts. Owing to the damage caused by the fixation artefact, it may be possible to attribute these mutations to the acquired factors, rather than congenital factors. Therefore, the association between HAND1 and human cardiac hypoplasia, or other CHD, requires further study.
Myosin heavy chain 6 (MYH6)
MYH6, encoded by the MYH6 gene, is a conventional myosin consisting of head, neck, and tail domains; two MYH6 proteins in the tail domain form a coiled coil that stabilises the molecule so that the head domain is able to generate force through its interaction with actin.Reference England and Loughna 47 Thus, MYH6 plays a critical role in myofibril assembly and proper heart development. Several human genetics studies have reported dominant heterozygous mutations in MYH6 linked with hypertrophic cardiomyopathy, dilated cardiomyopathy, or CHD primarily consisting of a secundum atrial septal defect.Reference England and Loughna 47 Furthermore, MYH6 has been described as the predominant sarcomeric disease gene for familial atrial septal defects, and, particularly, perturbations in the MYH6 head domains are regarded as the major genetic cause of familial atrial septal defects.Reference Posch, Waldmuller and Muller 48 On the other hand, MHY6 has also been identified in patients with a variety of CHD phenotypes including transposition of the great arteries, tricuspid atresia, and patent foramen ovale, revealing the genetic heterogeneity.Reference Granados-Riveron, Ghosh and Pope 49
Granados-Riveron et alReference Granados-Riveron, Ghosh and Pope 49 used denaturing high-performance liquid chromatography and sequence analysis to detect point mutations in MYH6 in 470 cases of isolated CHD. In a patient with tricuspid atresia, restrictive ventricular septal defect, and hypoplastic right ventricle, a nonsense mutation within exon 14, c.1568G>T, was discovered. This nonsense mutation is predicted to encode a truncated peptide of 500 residues containing most of the segments required for nucleotide and actin binding but lacking most of the lower 50 kDa domain and the entire 20 kDa domain, as well as the neck and rod regions. In another patient with tricuspid atresia, hypoplastic right ventricle, large secundum atrial septal defect, and valvular and supravalvular pulmonary stenosis, a missense mutation in the MYH6 gene, c.755G>C (p.Ala230Pro), was identified. The alanine residue found in the non-mutant protein was conserved in every muscle myosin sequence available and was predicted to disrupt the helical configuration of the segment contiguous to the switch-1 loop N-terminus. From another independent study, a similar variant, MYH6 c.688G>C (p.Ala290Pro), was also identified by exon sequencing technology in all but one family member in a highly penetrant pleiotropic CHD family with a preponderance of secundum atrial septal defects.Reference Arrington, Bleyl and Matsunami 50 , Reference Arrington, Bleyl, Brunelli and Bowles 51 This suggested that the MYH6 p.Ala230Pro variant was likely playing a major role in the development of CHD in this family. Recently, a whole-genome sequencing analysis was performed in 21 individuals from five families.Reference Theis, Zimmermann and Evans 52 The result revealed that molecular genetic defects in MYH6 were associated with a phenotype characterised by left heart underdevelopment and impaired systemic right ventricular performance. These findings implicated a shared molecular basis for developmental arrest and latent myopathy of left and right ventricles, respectively. Another study based on intracardiac flow dynamics determined that atrial contractility defects caused by mutations in MYH6 in zebrafish could affect ventricular morphogenesis and lead to a more compact ventricular myocardium.Reference Kalogirou, Malissovas, Moro, Argenton, Stainier and Beis 53 The influence of intracardiac flow dynamics on valve and ventricular morphogenesis may provide a different aspect on heart hypoplastic disease studies.
HEY gene family
The HEY gene family including HEY1, HEY2, and HEYL encodes hairy/enhancer-of-split, which is related to the YRPW motif protein. It is a basic helix-loop-helix transcription factor, working as a repressor through the basic helix-loop-helix domain. On the basis of previous studies, it is an important determinant of mammalian heart development.Reference Reamon-Buettner and Borlak 54 It also works as a direct target of the NOTCH signalling pathway, which recently has been demonstrated to be associated with some forms of CHD – for example, bicuspid aortic valve and left ventricular outflow tract obstructions.Reference Wessels and Willems 37
The relationship between the HEY gene family and CHD is unclear. The Hey gene family is considered to play a role in valve formation diseases, such as pulmonary atresia or tricuspid atresia. In animal studies, homozygotes of the HEY2 mutant allele display a spectrum of cardiac malformations including ventricular septal defect, tetralogy of Fallot, and tricuspid atresia.Reference Donovan, Kordylewska, Jan and Utset 55 In patient studies, the results of Fischer et alReference Fischer, Klamt and Schumacher 56 in 2004 suggested that the mutation in HEY2 was not a major contributing factor to CHD or Alagille syndrome. In a study involving 40 tricuspid atresia patients in 2005, the results also failed to demonstrate a major role for HEY2 mutations in the pathogenesis of human tricuspid atresia.Reference Sarkozy, Conti and D’Agostino 57 In another study from Fischer et alReference Fischer, Steidl and Wagner 58 in 2007, combined inactivation of HEY1 and HEYL caused severe heart malformations including membranous ventricular septal defect and dysplastic atrioventricular and pulmonary valves, which suggested that the HEY gene family is involved in a process critical for valve and septum formation. Thus, further studies of HEY gene mutations may help reveal the genetic mechanism of pulmonary atresia.
Advances in research methods regarding the genetic mechanism of CHD
Traditional methods to detect susceptible gene mutations for CHD use karyotype and pedigree analysis to locate mutations in related chromosomal regions, and then sequence the susceptible genes; however, insufficient resolutions of Sanger sequencing can hardly resolve the heterogeneity of CHD. With the completion of the Human Genome Project, genetic testing technologies have made considerable progress in the past few decades. With high sensitivity, high resolutions, and low average cost, chromosomal microarray assays are a first-line tool to detect genome abnormalities. Not only can these technologies detect microscopic and submicroscopic variants across the whole-genome structure, but they also provide information regarding copy number variants and uniparental disomy. With the help of chromosomal microarray assays, many microdeletions and duplications of chromosomal abnormalities, which were too small to be discovered by traditional methods, have been recently identified as a significant factor in CHD pathogenesis. Thus, chromosomal microarray assays are the recommended first-line genetic diagnostic tool for CHD patients.Reference Cowan and Ware 2 , Reference Geng, Picker and Zheng 59 , Reference Breckpot, Thienpont and Peeters 60 This technology was used in pulmonary atresia patients by Xie et al in 2014, which was reviewed above. If a large number of pulmonary atresia with intact ventricular septum patients are tested by this method, it will be helpful to explain the genetic mechanism of pulmonary atresia with intact ventricular septum and distinguish the genetic pattern between pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum.
Next-generation sequencing technologies are undoubtedly the most significant progresses in the genetic testing field in recent years. These techniques facilitate considerably greater depths of coverage, faster turnaround times, and a greater degree of cost-effectiveness compared with traditional capillary-based sequencing methods.Reference Cowan and Ware 2 , Reference Mardis 61 They provide a greater diagnostic utility for CHD and cover all suspected genes. On the basis of next-generation sequencing technologies, whole exome sequencing gives us a more comprehensive and efficient method to identify candidate genetic defects. The powerful function of whole exome sequencing has been widely recognised in single-gene diseases; however, because of the heterogeneity of complex diseases such as CHD, interpretation of detected mutations from whole exome sequencing remains a great challenge. Pathological variants should be screened out from most benign variants to estimate the potential relevancy between the variants and the disease.
With the progress of sequencing technology and the decline in its cost, it is possible to use whole-genome sequencing technology in many pulmonary atresia patients, including a suitable proportion of pulmonary atresia with intact ventricular septum cases. These results may be helpful in further exploring the molecular pathogenesis of pulmonary atresia and demonstrating the influence of gene modifications. Comparing susceptibility genes between pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum may be useful, because the differences in genetic pattern between these populations may help explain the difference in phenotypes, and further elucidate the pathogenesis of pulmonary atresia and CHD. We believe this work is helpful and necessary to clarify the genetic mechanism of pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum in the future.
Conclusions
Over the past decade, a number of copy number variants and single-gene mutations have been identified in CHD patients. It has been widely considered that the accumulation of copy number variants, single-gene mutations such as GDF1 and MTHFR, and environmental factors may affect pulmonary valve, pulmonary artery, and right ventricular morphogenesis, further leading to this complex cyanotic CHD; however, because of the great heterogeneity, the genetic molecular mechanism of pulmonary atresia is still unclear. More information is required to illustrate the relationship between genotype and phenotype. Narrowing the diagnosis and aiming at one subtype of pulmonary atresia – such as pulmonary atresia with intact ventricular septum – may be helpful to increase the specificity of pathological genes analysis. Furthermore, with the progress in genetic testing technologies and the use of next-generation sequencing, additional gene factors involved in pulmonary atresia with ventricular septal defect and pulmonary atresia with intact ventricular septum and the interaction between these should be revealed.
Acknowledgements
The authors gratefully acknowledge the valuable cooperation of Dr Zhaojing Zheng and Dr Juan Geng in providing the professional guidance of genetic knowledge.
Financial Support
This study was supported by National Natural Science Fund of China (grant nos 31370982 and 81501604). The authors have no other relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Conflicts of Interest
None.