INTRODUCTION
The Eimeria species are apicomplexan parasites that cause the life-threatening disease coccidiosis in all livestock animals, most notably poultry. The annual cost incurred by the Eimeria species has been estimated to exceed £2 billion per annum (Shirley et al. Reference Shirley, Ivens, Gruber, Madeira, Wan, Dear and Tomley2004). Of the Eimeria species that infect the chicken, Eimeria tenella is considered to be one of the most economically important based upon a combination of prevalence and pathogenicity. Control of the Eimeria species is largely achieved through prophylactic chemotherapy, although drug resistance is rife and the portfolio of effective drugs is limited. Live wild-type and attenuated vaccines for the Eimeria species that infect chickens are effective but relatively expensive and their use is limited to a small part of the global poultry industry. New, cost-effective anti-coccidial control strategies are required but the high cost of developing novel chemotherapeutics has proven to be limiting. Importantly, the Eimeria species have been adept at rapidly evolving to escape deleterious selective pressures such as those imposed by drug selection. For example, genetic resistance was described within the first year after the introduction of drugs including buquinolate, lasalocid and salinomycin (Chapman, Reference Chapman1997). Nonetheless, chemotherapy remains a valid anti-coccidial approach. A key component in the development of new anti-coccidial drugs will be knowledge defining the rapidity at which resistance can be selected. An assessment of relevant pre-existing genetic diversity will be integral to this process.
The increasing availability of transcriptomic, proteomic and genomic data for pathogens such as the Eimeria species now renders rational drug development increasingly feasible (Wan et al. Reference Wan, Chong, Ng, Shirley, Tomley and Jangi1999; Ng et al. Reference Ng, Jangi, Shirley, Tomley and Wan2002; Bromley et al. Reference Bromley, Leeds, Clark, McGregor, Ward, Dunn and Tomley2003; Shirley et al. Reference Shirley, Ivens, Gruber, Madeira, Wan, Dear and Tomley2004). Studies with related protozoan parasites including Toxoplasma gondii and the Kinetoplastida have highlighted glucose-6-phosphate isomerase (G6-PI) as a novel chemotherapeutic target (Dzierszinski et al. Reference Dzierszinski, Popescu, Toursel, Slomianny, Yahiaoui and Tomavo1999; Hardré et al. Reference Hardré, Salmon and Opperdoes2000). Three-dimensional protein structure modelling and phylogenetics based upon T. gondii G6-PI have implied that it is a plant homologue, while studies using Trypanosoma brucei G6-PI revealed the possibility of inhibiting this enzyme using novel competitive inhibitors. Thus G6-PI, an enzyme that catalyses the reversible isomerization of D-glucose 6-phosphate to D-fructose 6-phosphate in the glycolysis pathway, could prove valuable as a target for the development of novel anti-coccidial drugs for use in poultry. G6-PI has previously been used in electrophoretic enzyme mobility studies with Eimeria as a means of differentiating between strains of species including Eimeria acervulina, Eimeria mitis and E. tenella (Sutton et al. Reference Sutton, Shirley and McDonald1986; Shirley et al. Reference Shirley, Chapman, Kucera, Jeffers and Bedrnir1989). Although the basis of the observed electrophoretic diversity remains unknown, knowledge that G6-PI is characterized by naturally occurring phenotypic diversity within Eimeria species may limit its attraction as a drug target. The occurrence and extent of genetic polymorphism is as yet unknown for Eimeria G6-PI prior to drug selection, although the distribution of coding polymorphism across sites which may be considered ‘relevant’ (e.g. predicted active site) would temper enthusiasm for further development given the elevated possibility of pre-existing inherently resistant genotypes. Thus, identification of the E. tenella G6-PI (EtG6-PI) orthologous coding sequence and determination of any putative genetic basis for the observed electrophoretic polymorphism between E. tenella strains can inform on the suitability of G6-PI for inclusion in the future development of anti-coccidial drug strategies.
MATERIALS AND METHODS
Parasites
The E. tenella Houghton (H), Weybridge (Wey) and Wisconsin (Wis) strains were kindly provided by Professor Martin W. Shirley of the Institute for Animal Health, Compton, UK. The H and Wey strains were isolated in the UK and the Wis strain was isolated in the USA (Joyner and Norton, Reference Joyner and Norton1969; Jeffers, Reference Jeffers1975; Chapman and Shirley, Reference Chapman and Shirley2003). E. tenella parasites were propagated and isolated in accordance with previously outlined procedures (Shirley, Reference Shirley, Eckert, Braun, Shirley and Coudert1995).
Generation of the E. tenella G6-PI cDNA sequence
Total RNA was isolated from sporulated oocysts using TRI Reagent (Molecular Research Center Inc., USA) for use as a template in 5′ and 3′ Rapid Amplification of cDNA Ends (RACE) reactions performed with the SMART RACE cDNA Amplification kit (Clontech, USA). The forward (GSP2: 5′ GAT TTG CGT GGG GAT TGG GGG AAG TTA 3′) and reverse (GSP1: 5′ CCA TTG GTC CCT GGT TCT CCG AAG TAA 3′) RACE primers were designed based on the predicted EtG6-PI coding sequence (GenBank Accession no. AM269894). The PCR temperature and cycling conditions were as follows: 5 cycles of 94°C for 30 s and 72°C for 3 min; 5 cycles of 94°C for 30 s, 70°C for 30 s and 72°C for 3 min; and 25 cycles of 94°C for 30 s, 68°C for 30 s and 72°C for 3 min. PCR products were then purified, cloned into the pTZ57R vector (Fermentas, Lithuania), and sequenced using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, USA) on the ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, USA). The most probable coding sequence for EtG6-PI was predicted using NCBI ORF Finder, while the similarity search was performed against the GenBank non-redundant database using blastx (Altschul et al. Reference Altschul, Gish, Miller, Myers and Lipman1990).
Phylogenetic analysis
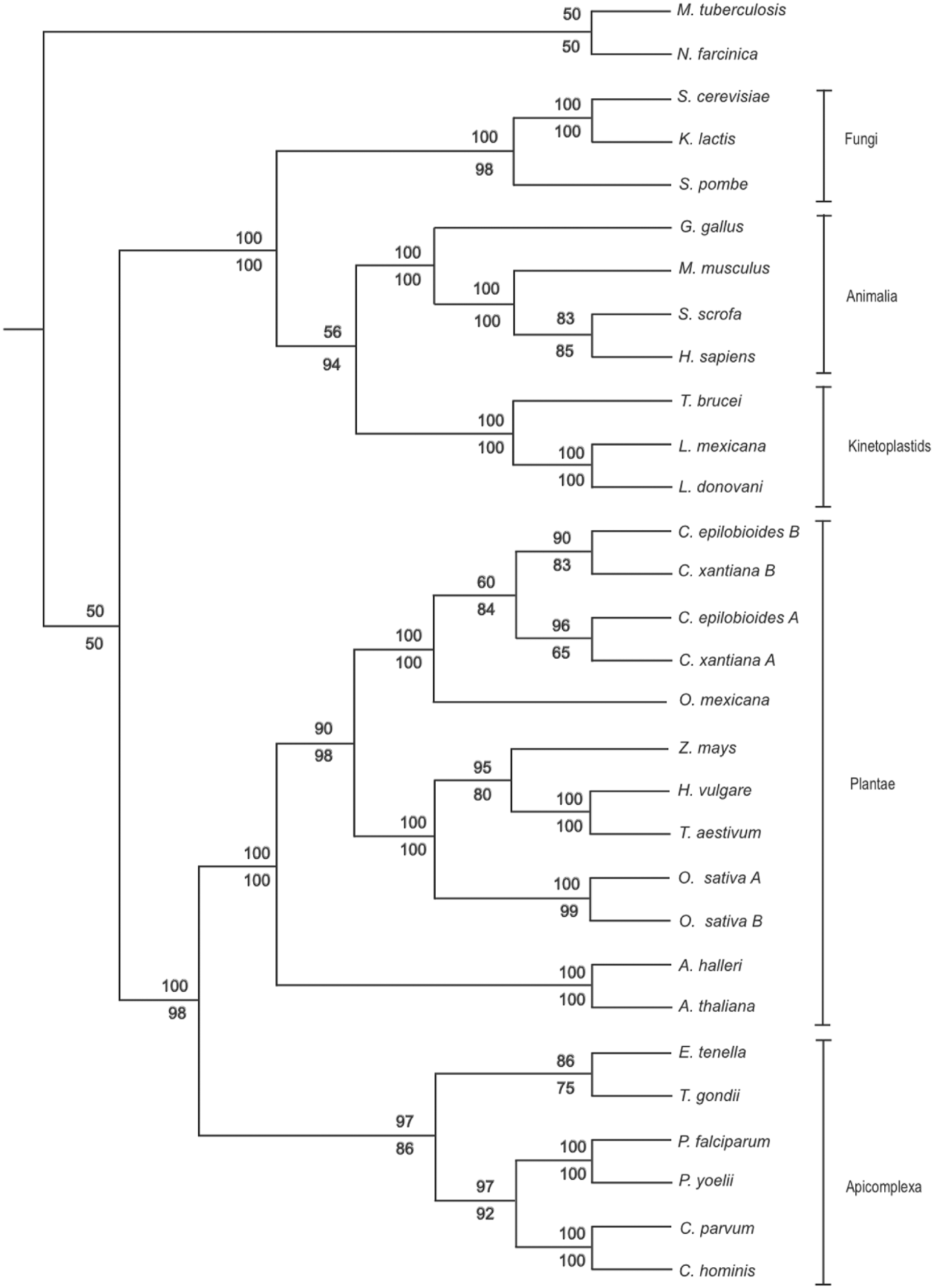
Multiple orthologous G6-PI amino acid sequences were retrieved by PSI-BLAST (Altschul et al. Reference Altschul, Madden, Schäffer, Zhang, Zhang, Miller and Lipman1997) with a minimum match cut-off value of E ⩽e−100. A total of 32 unique taxa containing 682 characters were selected for further analyses. Sequences were aligned using MUSCLE (Edgar, Reference Edgar2004a, Reference Edgarb) followed by visual inspection and manual adjustment. Phylogenetic analyses were conducted using PHYLIP (Felsenstein, Reference Felsenstein1989). Phylogenetic trees were constructed using the Neighbor-Joining (NJ) method (as implemented in NEIGHBOR) from distance matrixes calculated by the Jones-Taylor-Thornton model (using PROTDIST). The Maximum Parsimony (MP) method was also used (using PROTPARS). The parsimony settings for these analyses were ordinary parsimony, with non-weighted sites. The robustness of the trees was evaluated by bootstrap analysis of 1000 random iterations (using SEQBOOT). All trees were then viewed and edited using TREEVIEW 1.6.6 (Page, Reference Page1996) with Mycobacterium tuberculosis and Nocardia farcinica being rooted as outgroups.
Amplification and sequencing of the EtG6-PI genomic locus
The EtG6-PI genomic locus was PCR amplified using the enhanced proof-reading hybrid Expand Long Template PCR System kit (Roche Applied Science, Germany). Primers (5′UTR-F: 5′ ACA CAC CTT TGC GTT TCG AGC CCC T 3′ and 3′UTR-R: 5′ CTG GCG CCC CGA ATC AAT TGT CTA A 3′) were designed in the untranslated regions flanking EtG6-PI identified through comparison of the EtG6-PI cDNA with the E. tenella chromosome 1 sequence (GenBank Accession no. NC008685; Ling et al. Reference Ling, Rajandream, Rivailler, Ivens, Yap, Madeira, Mungall, Billington, Yee, Bankier, Carroll, Durham, Peters, Loo, Mat-Isa, Novaes, Quail, Rosli, Mariana, Sobreira, Tivey, Wai, White, Wu, Kerhornou, Blake, Mohamed, Shirley, Gruber, Berriman, Tomley, Dear and Wan2007b). Genomic DNA was extracted from sporulated oocysts as previously described (Shirley, Reference Shirley, Eckert, Braun, Shirley and Coudert1995). The PCR temperature and cycling conditions were 94°C for 2 min, followed by 10 cycles of 94°C for 10 s, 63°C for 30 s and 68°C for 8 min; 25 cycles of 94°C for 15 s, 63°C for 30 s and 68°C for 8 min+5 s per cycle; and a final step of 68°C for 7 min. PCR products were subsequently purified, cloned into the pCR-XL-TOPO vector (Invitrogen Corporation, USA) and sequenced as described above.
EtG6-PI structure homology modelling
The EtG6-PI H strain coding sequence was used to identify the equivalent Wey and Wis strain coding sequences from the sequenced genomic loci. All 3 coding sequences were translated and subjected to automated protein structure homology modelling using the SWISS-MODEL workspace (Arnold et al. Reference Arnold, Bordoli, Kopp and Schwede2006) via the ExPASy web server and studied using Invitrogen VectorNTI Advance 11.0 3D Molecule Viewer.
RESULTS
Identification and characterization of the EtG6-PI coding sequence
EtG6-PI is predicted to be encoded in 1647 bp, yielding a polypeptide of 549 amino acids (GenBank Accession no. GU117732). Blastx analysis of the coding sequence revealed the highest degree of similarity to the T. gondii G6-PI sequence (E value 0.0, 61% identity). All 100 of the top hits were annotated as, or similar to, G6-PI (E values ⩽e-166). The EtG6-PI predicted amino protein sequence was of a comparable size to each of these hits.
Alignment of the predicted EtG6-PI amino acid sequence with G6-PI amino acid sequences identified from multiple diverse taxonomic ranks revealed conserved G6-PI superfamily residue patterns (Fig. 1). Among the most notable residue patterns were [DENSA]-x-[LIVM]-[GP]-G-R-[FY]-[ST]-[LIVMFSTAP]-x-[GSTA]-[PSTACM]-[LIVMSA]-[GSAN] (PROSITE Accession no. PS00765) at residues 289-302 and [GSA]-x-[LIVMCAYQS]-[LIVMFYWN]-x-(4)-[FY]-[DNTH]-Q-x-[GA]-[IV][EQST]-x-(2)-K (PROSITE Accession no. PS00174) at residues 528-545. Consideration of the proposed EtG6-PI translation start site with consensus sequence from T. gondii translational start sites revealed conservation of the 3 most favoured nucleotides (C−4, A−3, A−3; Seeber, Reference Seeber1997). Identification of serine as the second amino acid was also consistent with previous reports from T. gondii (Matrajt et al. Reference Matrajt, Nishi, Fraunholz, Peter and Roos2002).

Fig. 1. Multiple sequence alignment of the EtG6-PI coding sequence with G6-PI sequences of other organisms. Black-shaded areas show conserved amino acid residues while grey-shaded areas show similar residues. Gaps (−) formed for optimum alignment. A and B denote conserved residue patterns of the G6-PI superfamily. (Et) Eimeria tenella; (Tg) Toxoplasma gondii; (Pi) Phytophthora infestans; (Om) Oenothera mexicana; (At) Arabidopsis thaliana; (Zm) Zea mays; (Hs) Homo sapiens; (Gg) Gallus gallus; (Kl) Kluyveromyces lactis; (Sc) Saccharomyces cerevisiae.
Phylogenetic analysis
The E. tenella G6-PI amino acid sequence was aligned with previously published orthologous sequences from other apicomplexan species and other major lineages including Plantae, Fungi, Animalia and Kinetoplastida. The topology of the NJ and MP trees generated from these sequences supported previously described taxonomic relationships associating the Plantae with the Apicomplexa in a distinct cluster more distant to sequences derived from fungi, animals and kinetoplastids (Fig. 2; Dzierszinski et al. Reference Dzierszinski, Popescu, Toursel, Slomianny, Yahiaoui and Tomavo1999). Similarly, within the apicomplexan clade, E. tenella G6-PI was more closely related to the T. gondii G6-PI orthologue than those of Cryptosporidium parvum, Cryptosporidium hominis, Plasmodium falciparum and Plasmodium yoelii (>75% bootstrap confidence support). The separation of the Cryptosporidium species from the other coccidian genera was also statistically reliable (bootstrap value, >85%).
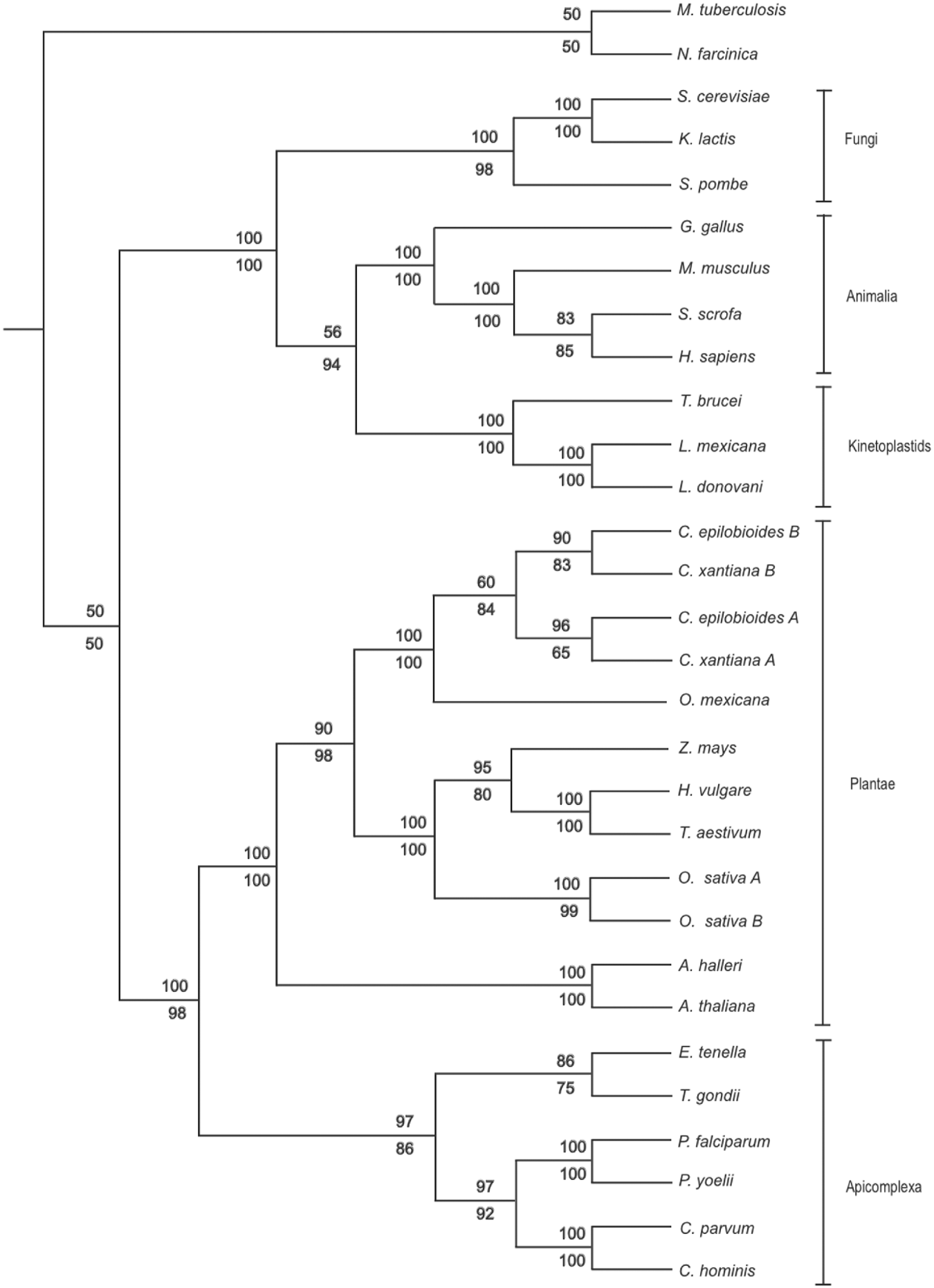
Fig. 2. Neighbor-joining (NJ, upper figure) and maximum parsimony (MP, lower figure) results illustrating the phylogenetic relationships between complete G6-PI amino acid sequences.
Structural organization of the EtG6-PI locus
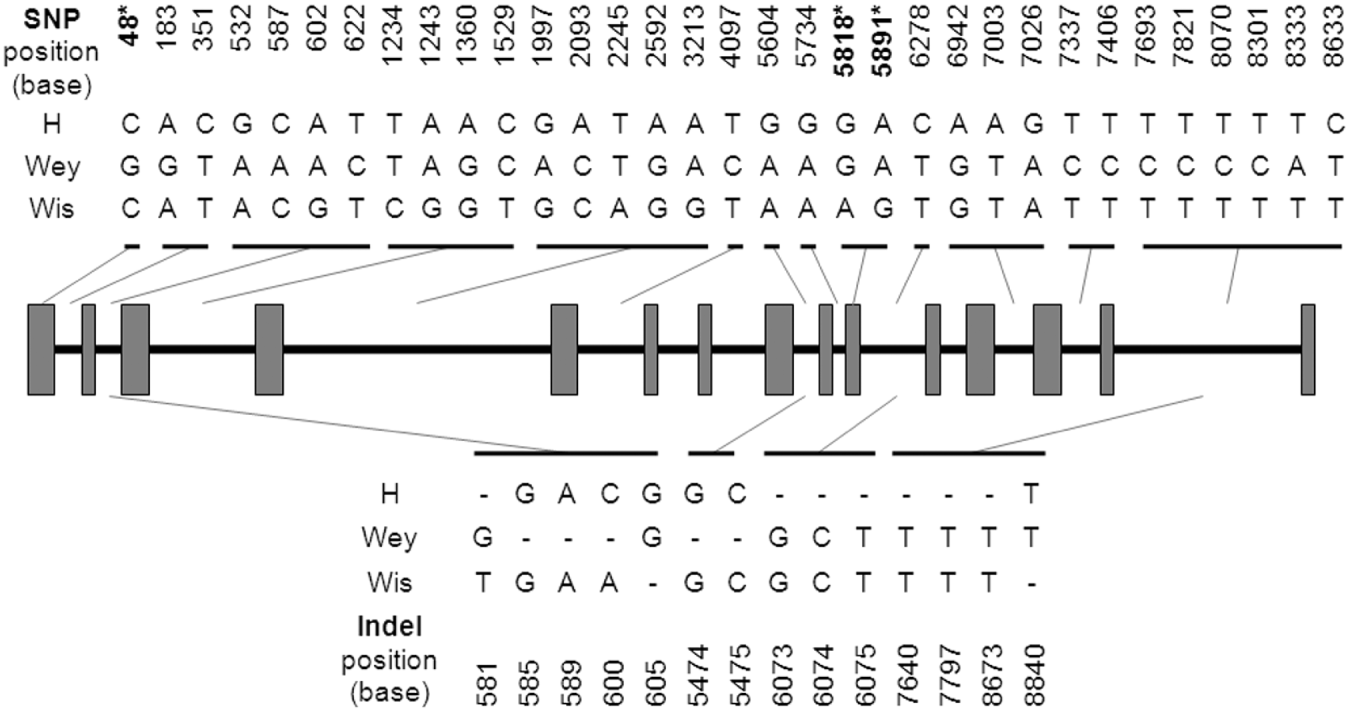
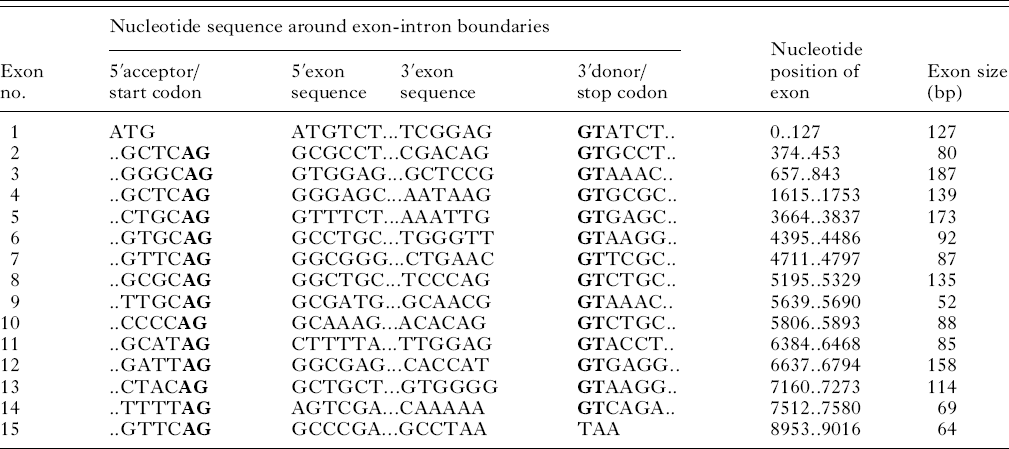
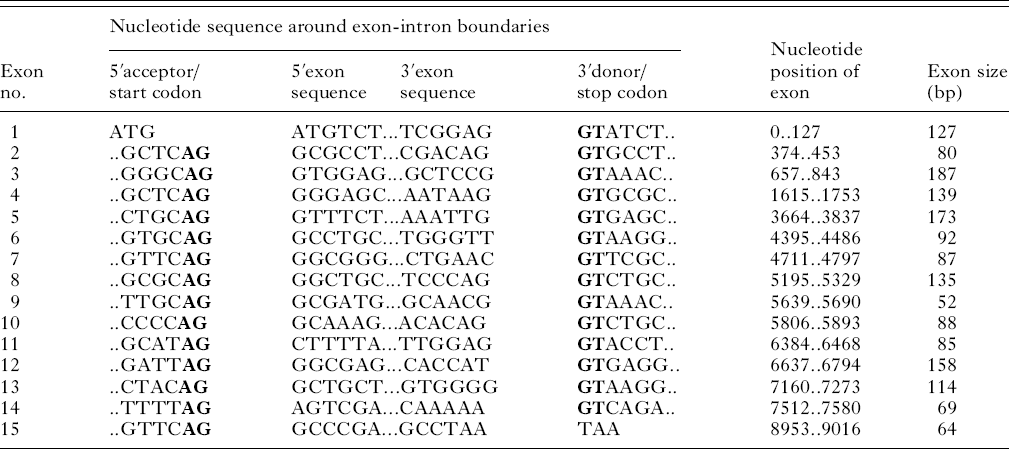
Amplification and sequencing of the EtG6-PI genomic locus yielded a contig of approximately 9 kb in length (GenBank Accession no. GU117733). Sequence comparison between the genomic and cDNA EtG6-PI sequences demonstrated that the gene consisted of 15 exons and 14 introns (Table 1, Fig. 3). All exons were found to obey the adopted intron-AG-/exon/-GT-intron splicing rule (Mount, Reference Mount1982). The average length of exons and introns was 110 bp and 527 bp respectively.

Fig. 3. SNP and indel positions identified within the EtG6-PI locus of the Eimeria tenella H, Wey and Wis strains. SNPs are shown in the upper panel, indels are shown in the lower panel. Asterisk (*) denotes positions located within coding (exonic) sequence.
Table 1. Features of exon-intron structure of EtG6-PI (H strain)
Intra-specific EtG6-PI coding sequence comparison
Multiple sequence alignment analysis of the predicted EtG6-PI genomic sequence from the E. tenella H, Wey (GenBank Accession no. GU117735) and Wis (GenBank Accession no. GU117734) strains revealed a panel of insertions/deletions (indels) and SNPs. From a total of 14 indels, 7 specifically characterized the H strain while 5 and 2 defined the Wey and Wis strains respectively (Fig. 3). No deletion was shared among the strains investigated here. In all but 2 examples, a base deleted from one strain was non-polymorphic in the other strains. The exceptions occurred at base positions 581 and 600 (581: H[−], Wey[G], Wis[T]; 600: H[C], Wey[−], Wis[A]).
In addition to the indels, 33 SNPs were detected. Among these SNPs, 14 were found to involve purine substitutions (A and G bases) and 13 involved pyrimidine substitutions (C and T bases) (Fig. 3). Base substitutions between the purine and pyrimidine groups occurred at 6 sites. No single site was found to be polymorphic between all 3 strains. Only 3 SNPs were located within exonic regions, 1 within exon 1 and 2 within exon 10 (Fig. 3). All 3 exonic SNPs incurred non-synonymous substitutions.
EtG6-PI structure homology modelling
SWISS-MODEL protein structure homology modelling revealed a best hit to PDB Chain 1t10A for all 3 EtG6-PI proteins (E value 0.0). Based upon this model intra-specific EtG6-PI secondary structure polymorphism was only predicted to be associated with coding SNP 1 (Fig. 4), suggesting an extended α-helix associated with the Wey strain compared to the H and Wis strains (residues 7-19 compared to residues 6-13).

Fig. 4. Influence of exonic SNPs on EtG6-PI secondary structure predicted in the Swiss Model workspace (Arnold et al. Reference Arnold, Bordoli, Kopp and Schwede2006). Non-polymorphic amino acids are not shown (−), polymorphic amino acids and surrounding regions are shown in boxes 1–3. Only box 1 contains a predicted structural variation. h=α helix, s=β sheet.
DISCUSSION
G6-PI has previously been identified as a candidate drug target for novel strategies to control protozoan parasites including T. gondii and the Kinetoplastida (Dzierszinski et al. Reference Dzierszinski, Popescu, Toursel, Slomianny, Yahiaoui and Tomavo1999; Hardré et al. Reference Hardré, Salmon and Opperdoes2000). By comparison, it can be considered a valid target for anti-coccidial drug development. Historically, resistance has been observed soon after the introduction of every new anti-coccidial drug arising through a combination of apparent genomic plasticity and inadequate application against manufacturers' guidelines (Chapman, Reference Chapman1997). Thus, defining the potential for resistance has become a key criterion in rational drug development. Previous studies with eimerian G6-PI have revealed polymorphic intra-specific electrophoretic mobility (Sutton et al. Reference Sutton, Shirley and McDonald1986; Shirley et al. Reference Shirley, Chapman, Kucera, Jeffers and Bedrnir1989), although deficiencies in our knowledge of this molecule have hindered understanding the cause of the observed polymorphism.
The putative EtG6-PI described here has been shown to have a high percentage identity and similar sequence length to previously identified G6-PI coding sequences, supporting annotation as a genuine G6-PI orthologue. The identification of conserved G6-PI superfamily residue patterns, in common with Bacillus stearothermophilus and Entamoeba histolytica, provided further supporting evidence (Sun et al. Reference Sun, Chou, Chen, Wu, Meng and Hsiao1999; Razmjou et al. Reference Razmjou, Haghighi, Rezaian, Kobayashi and Nozaki2006). Comparison with the sequenced EtG6-PI genomic locus revealed adherence to the intron-AG-/exon/-GT-intron splicing rule with similar predicted intron and exon sizes to those described previously for the E. tenella phosphatidylinositol 4-phosphate 5-kinase gene (PIP5K; Ling et al. Reference Ling, Loo, Rosli, Mariana, Mohamed and Wan2007a).
Phylogenetic comparison of the E. tenella G6-PI amino acid sequence supported the previously reported relationship between the Apicomplexa and the Plantae (Köhler et al. Reference Köhler, Delwiche, Denny, Tilney, Webster, Wilson, Palmer and Roos1997; Dzierszinski et al. Reference Dzierszinski, Popescu, Toursel, Slomianny, Yahiaoui and Tomavo1999; Funes et al. Reference Funes, Davidson, Reyes-Prieto, Magallón, Herion, King and González-Halphen2002; Grauvogel et al. Reference Grauvogel, Brinkmann and Peterson2007). Despite the relatively low support for the distinction between G6-PI sequences of apicomplexan parasites and their higher order hosts described here, these findings have been supported by others (e.g. Dzierszinski et al. Reference Dzierszinski, Popescu, Toursel, Slomianny, Yahiaoui and Tomavo1999), demonstrating their candidacy as specific drug targets. The NJ and MP phylogenetic trees illustrated the taxonomic relationship between E. tenella and T. gondii, both of which are categorized under the order Eucoccidiorida. Although Cryptosporidium spp. are also classified under the order Eucoccidiorida together with E. tenella and T. gondii, this and previous phylogenetic studies have revealed a greater distance relationship from E. tenella to the Cryptosporidium spp. than T. gondii. The Cryptosporidium spp. are now thought to have a closer relationship with apicomplexan parasites under the order Eugregarinorida than to other members of the order Eucoccidiorida (Carreno et al. Reference Carreno, Martin and Barta1999; Zhu et al. Reference Zhu, Keithly and Philippe2000; Leander et al. Reference Leander, Clopton and Keeling2003).
A panel of 33 SNPs and 14 indels was detected across the EtG6-PI locus but only 3 SNPs, at base positions 48, 5818 and 5891, were detected in coding regions. Translation of exon 1 revealed that the H and Wis strains encode the histidine (H) residue while the Wey strain encoded the glutamine (Q) residue. The polar H residue is positively charged while the Q residue, an analogue of the acidic glutamic acid (E), is neutral. Thus, the SNP at base position 48 on exon 1 conferred an overall loss of charge. Two SNPs were detected in exon 10, both of which differentiated the Wis strain from the H and Wey strains. The first SNP in exon 10 resulted in a change from aspartic acid (D; H and Wey strains) to asparagine (N; Wis). The D residue is negatively charged while the N residue is a derivative without charge. The substitution incurs a loss of charge to the amino acid residue. The second SNP in exon 10 resulted in a change from an H residue (H and Wey strains) to arginine (R) residue, both positively charged, incurring no change in charge. Preliminary structural predictions using the SWISS-MODEL workspace (Arnold et al. Reference Arnold, Bordoli, Kopp and Schwede2006) suggest an extended α-helix associated with the Wey strain-specific exon 1 SNP. Previous study has shown that E. tenella H and Wis strain isoenzyme profiles are characterized by the EtG6-PI electrophoretic variant GPI-1, while the Wey strain is characterized by the electrophoretic variant GPI-9 (Shirley et al. Reference Shirley, Chapman, Kucera, Jeffers and Bedrnir1989), a shift possibly induced by this α-helix polymorphism. The SNPs in exon 10 are not predicted to influence secondary protein structure although the charge polymorphism associated with one of the SNPs could affect tertiary protein structure. Similarly, post-translational modifications such as glycosylation could impact upon electrophoretic mobility. The functional significance of the predicted structural polymorphism is not known. However, although detailed crystallography studies have yet to be conducted on EtG6-PI, in silico comparison with Leishmania mexicana G6-PI suggests that none of the changes are closely associated with predicted catalytic residues or residues thought to be involved in the coordination of the substrate's phosphate group (Cordeiro et al. Reference Cordeiro, Michels, Delboni and Thiemann2004). A more complete description of the EtG6-PI protein structure is ongoing alongside these studies (manuscript in preparation).
Consideration of the limited EtG6-PI coding and predicted structural polymorphism detected here between E. tenella strains isolated on 2 continents, combined with the phylogenetic divergence between the parasite and its host G6-PI, suggest that EtG6-PI remains a valid candidate anti-coccidial drug target. Nevertheless, the finding of only 2 EtG6-PI isoenzyme types to date suggests that it would be prudent to test the efficacy of any candidate anti-EtG6-PI drug on representatives of both types.
FINANCIAL SUPPORT
This work was supported by the IRPA Top Down Grant awarded by the Ministry of Science, Technology and Innovation (MOSTI), Malaysia (R. M., grant number 09-02-02-002 [BTK/TD/003]); the Genomics and Molecular Biology Initiatives Programme of the Malaysia Genome Institute, MOSTI, Malaysia (K.-L. W., grant number 07-05-16-MGI-GMB10); and the Biotechnology and Biological Sciences Research Council (BBSRC), UK (D. P. B., grant number BBE01089X1).