


Introduction
Association mapping via linkage disequilibrium or LD is a useful technique to identify quantitative trait loci (QTLs) (Oraguzie et al., Reference Oraguzie, Wilcox, Rikkerink, Oraguzie, Rikkerink, De Silva and Gardiner2007). The physical association between a marker locus and the DNA sequence responsible for a desirable trait is the basic concept in the search for genes controlling the trait. Although any association found will be generally referred to as LD (Weir, Reference Weir1996), only those caused by physical linkage are significant for association mapping (Stich et al., Reference Stich, Mohring, Piepho, Heckenberger, Buckler and Melchinger2008).
The choice of germplasm is critical to the success of association analysis (Yu et al., Reference Yu, Pressoir, Briggs, Bi, Yamasaki, Doebley, McMullen, Gaut, Nielsen, Holland, Kresovich and Buckler2006). The extent of LD, the genetic diversity, the structure and the relatedness within the population condition the mapping resolution (Remington et al., Reference Remington, Thornsberry, Matsuoka, Wilson, Whitt, Doebley, Kresovich, Goodman and Buckler2001; Yu et al., Reference Yu, Pressoir, Briggs, Bi, Yamasaki, Doebley, McMullen, Gaut, Nielsen, Holland, Kresovich and Buckler2006; Zhu et al., Reference Zhu, Gore, Buckler and Yu2008). The extent of LD is a major determinant of the resolution, marker density and sample size (Ball, Reference Ball, Oraguzie, Rikkerink, De Silva and Gardiner2007). If the extent of LD is low, the resolution is high but a greater marker density is required (Rafalski, Reference Rafalski2002). Knowledge on the extent of LD and LD patterns in plants is currently limited; however, it is clear that the extent of LD varies widely between species, populations, genomic regions and allelic frequencies (Cockram et al., Reference Cockram, White, Leigh, Lea, Chiapparino, Laurie, Mackay, Powell and O'Sullivan2008; Slatkin, Reference Slatkin2008; Yan et al., Reference Yan, Shah, Warburton, Buckler, McMullen and Crouch2009; Mandel et al., Reference Mandel, Nambeesan, Bowers, Marek, Ebert, Rieseberg, Knapp and Burke2013).
Model-based clustering methods have been used in several species, including maize, to correct for population structure in association mapping studies (Liu et al., Reference Liu, Goodman, Muse, Smith, Buckler and Doebley2003; Camus-Kulandaivelu et al., Reference Camus-Kulandaivelu, Veyrieras, Madur, Combes, Fourmann, Nordborg, Bergelson, Cuguen and Roux2006) and to contrast the conformation of heterotic groups in maize (Liu et al., Reference Liu, Goodman, Muse, Smith, Buckler and Doebley2003). Besides, model-based clustering methods allow estimating population genetic parameters to reduce redundancy and select core sets from association mapping populations. Reducing the population size for mapping purposes may lead to spurious results in association studies (Ball, Reference Ball, Oraguzie, Rikkerink, De Silva and Gardiner2007). However, core sets can be envisaged as useful tools to deepen phenotypic variation analysis by using top-down approaches (Andrade et al., Reference Andrade, Sala, Pontaroli, León, Sadras and Calderini2015) or as valuable reservoirs for rapid allele mining (Ashkani et al., Reference Ashkani, Yusop, Shabanimofrad, Azadi, Ghasemzadeh, Azizi and Latif2015).
Core sets of crop germplasm can be developed by drawing a weighted number of individuals within each group of a structured population (Frankel and Brown, Reference Frankel, Brown, Holden and Williams1984; Brown and Schoen, Reference Brown, Schoen, Loeschcke, Tomiuk and Jain1994). In maize, a simulated annealing (SA) algorithm and the model-based clustering method have been proposed to choose a core set that maximizes allelic richness (Liu and Muse, Reference Liu and Muse2005).
In a previous work, we assessed the genetic diversity of 103 maize inbreds with 50 single sequence repeats (SSRs) (Olmos et al., Reference Olmos, Delucchi, Ravera, Negri, Mandolino and Eyhérabide2014a). In the present study, we extended the genotyping by adding 24 SSRs and eight historic flint inbreds.
The objectives of this study were to assess: (1) the relative genetic relationships, diversity and extent of LD within the Argentine public maize inbred line collection after the addition of new germplasm and SSRs; and (2) the genetic diversity and the extent of LD within mini-core sets defined with the model-based clustering method and the SA algorithm.
Material and methods
Plant material
A set of 111 inbred lines representing the most important public lines from Argentina and including some reference lines from the USA were chosen to represent the diversity available at the maize breeding programme of the National Institute of Agricultural Technology (INTA, Argentina). These included public inbred lines mostly adapted to temperate environments obtained from intermated open-pollinated populations (composites and synthetics) and in a lesser extent from commercial hybrids. The high genetic diversity within the Argentine public maize inbred lines can also be observed at the phenotypic level. This collection currently harbours 17 inbreds that are parents of several recombinant inbred line (RIL) populations that are used to conduct conventional QTL studies for different agronomic traits (Sampietro et al., Reference Sampietro, Vattuone, Presello, Fauguel and Catalán2009; Campos-Bermudez et al., Reference Campos-Bermudez, Fauguel, Tronconi, Casati, Presello and Andreo2013; D'Andrea et al., Reference D'Andrea, Piedra, Mandolino, Bender, Cerri, Cirilo and Otegui2016) as part of the maize breeding program at INTA Pergamino (Argentina). In addition, these lines also show a high variation in colour and grain texture, yield, maturity and common rust resistance.
Out of the 111 inbred lines examined here, a subset of 103 inbred lines was previously genotyped with 50 SSRs (Olmos et al., Reference Olmos, Delucchi, Ravera, Negri, Mandolino and Eyhérabide2014a). The eight newly incorporated historic flint lines have Cuarentín and Argentine × Caribbean background; however, the precise origin and pedigree of these materials remain unknown. Coding numbers and pedigrees of the lines (when records were available) are listed in online Supplementary Table S1.
SSR genotyping
For SSR genotyping, we used 74 SSR loci evenly distributed throughout the maize genome. These SSR loci included a set of 50 SSRs previously selected based on their high polymorphism information content (online Supplementary Table S2). No prior information about the genomic location of loci in coding or non-coding regions or about locus proximity to genes was used for the selection of loci. Forty-nine out of the 50 previously used SSRs were used in this work. The exception was bnlg540 because it was difficult to score. In addition, we mapped 25 SSRs, seven of which were from chromosomes 1, 3, 4, 8, 9 and 10 and the remaining 18 from chromosome 6. The denser coverage of chromosome 6 was carried out to conduct further studies on candidate QTLs for the high oleic acid content located in that position. Almost all SSRs matched the genomic regions of the AGPv2, 2009-03-20 assembly version (online Supplementary Table S2). The exceptions were bnlg1429 (1.02), bnlg420 (3.05) and phi034 (7.02), for which primer sequences were not found in the B73 RefGen_v2 sequence. One of the mapped SSRs (umc1583) was assigned by the MaizeGDB into bin 7.00; however, the query of primer pairs with Blastn (Altschul et al., Reference Altschul, Gish, Miller, Myers and Lipman1990) positioned this locus at coordinate 59,866,172 bp (B73 RefGen_v2 assembly). This coordinate matched bin 7.02, between the Chr7:13,852,673..128,140,577 interval; consequently, this genomic position was then used to arrange the loci for LD assessment.
The PCR marker called p2526 was developed to amplify a predicted HapMap1 INDEL allele (PZE06104824646,----/ACCC) at bin 6.04. The INDEL sequence context was retrieved from the Panzea database www.panzea.org and primers were designed with Primer3plus interface (http://primer3plus.com/) to amplify a 233-bp fragment in the B73 reference sequence (Forward: GGCAACCGTTGAAGAGAGTC, Reverse: AGGATCGTCTGGGGAACTTT). Unexpectedly, the PCR amplification of this locus revealed a multi-allelic pattern of variation. Further Sanger Sequencing showed that this locus also included a microsatellite and transposon-like variation. Because this locus resulted in easily scorable allelic differences in the entire inbred panel, it was finally included for genotyping purposes. All SSR primer sequences are available at MaizeGDB (http://www.maizegdb.org/).
DNA extraction and PCR conditions have been previously described in Olmos et al., (Reference Olmos, Delucchi, Ravera, Negri, Mandolino and Eyhérabide2014a). Gels were silver-stained and alleles were identified by comparison with products of known size from the B73 inbred line accession B73-05-6081 (Olmos et al., Reference Olmos, Lorea and Eyhérabide2014b). Data for each locus and inbred line were stored as two consecutive rows for population structure analysis (online Supplementary Table S2).
Population structure
Lines were subdivided into genetic clusters using the Bayesian model-based approach implemented in the software package STRUCTURE 2.3.3 (Pritchard et al., Reference Pritchard, Stephens and Donnelly2000). For a given number of subpopulations (clusters), this method assigns lines from the entire sample to clusters in such a way that Hardy–Weinberg disequilibrium and LD are minimized. No prior information regarding the pedigree origin of the inbred lines was used to infer subpopulations. As recommended by Pritchard et al. (Reference Pritchard, Wen and Falush2010), the admixture model was used as a starting point for data analysis. Under this model, each individual draws some fraction of its genome from each of the k subpopulations and, conditional on the ancestry vector, q (i), the origin of each allele is independent. That is, this model assumes that all markers are unlinked and provides independent information on an individual's ancestry. Two independent runs of STRUCTURE were performed by setting the number of k from 1 to 5. For each run, the burn-in time and the replication number were both set to 1,000,000. The program CLUMPP (Jakobsson and Rosenberg, Reference Jakobsson and Rosenberg2007) was used to line up the cluster labels across the two different runs. The program STRUCTURE HARVESTER was used to process STRUCTURE outputs and to implement the Evanno's method to select the optimal number of clusters k (Evanno et al., Reference Evanno, Regnaut and Goudet2005, Earl and VonHoldt, Reference Earl and vonHoldt2012). To assign inbred lines into clusters, lines with membership probabilities ≥0.80 were considered to belong to discrete clusters, whereas inbred lines with membership probabilities <0.80 were assigned to the ‘mixed’ subpopulation. Known pedigree records, graphical results, maximum likelihood and the rate of change in the log probability of data between successive k values (Δk) were taken into account to infer the true value of k.
Mini-core set design
The SA algorithm implemented in PowerMarker (Liu and Muse, Reference Liu and Muse2005) was used to design mini-core sets of 30, 40 and 50 inbred lines. The total allele number was used as the objective function to maximize. The number of evaluations for each annealing schedule, R, was set to 500, and the cooling coefficient ρ was set to 0.9. The SA algorithm was then used under the constraint that at least five representative lines from each of the discrete subpopulations inferred by STRUCTURE were included in the final core sets. The Mixed group was not constrained. The analysis was replicated ten times for each core set. The effective size of mini-cores, i.e. the number of inbreds, which were always included in all ten replicates, was indicated in Tables 1 and 2 between parentheses.
Table 1. Variation of the proportion of the linked loci pairs in significant LD across chromosomes within the Argentine public maize inbred line collection and subsets
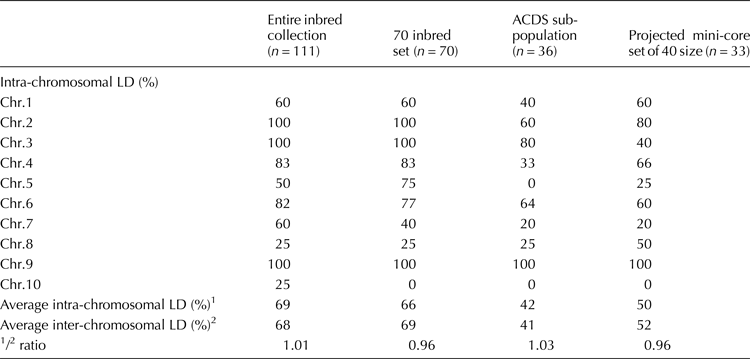
Table 2. Mini-core sets designs using the SA algorithm to maximize total allele number

Genetic diversity
The PowerMarker software was used to calculate major allele frequencies, residual heterozygosity (observed heterozygosity), and average gene diversity. The program ADZE: Allelic Diversity Analyser Version 1.0 (Szpiech et al., Reference Szpiech, Jakobsson and Rosenberg2008) with default parameters was used to calculate allelic richness. The rarefaction method implemented in ADZE trims unequal samples to the same standardized sample size. To compare subpopulations defined by STRUCTURE, a maximum standardized sample size of 20 without missing-filtered loci was used.
Relatedness
The relative kinship (K) matrix was calculated on the basis of the 74 loci, using the method of Loiselle et al. (Reference Loiselle, Sork, Nason and Graham1995) implemented in SPAGeDI 1. 4 (Hardy and Vekemans, Reference Hardy and Vekemans2002). This method is adapted to heterozygous diploid individuals in the case of multiallele and multilocus data sets. Input data files contained the two alleles from each locus separated by any number of non-numerical characters other than a tab. Negative kinship values between inbreds in the resulting matrix were set to 0, as a negative value would indicate that they are less related than random individuals. Essentially, the degree of genetic covariance caused by polygenic effects was defined as 0 for a pair of individuals that are not related and as positive for a pair of individuals that are related. This threshold is similar to the pedigree-based coancestry matrix in which individuals with unknown relationship are set to 0 (Yu et al., Reference Yu, Pressoir, Briggs, Bi, Yamasaki, Doebley, McMullen, Gaut, Nielsen, Holland, Kresovich and Buckler2006).
LD analysis
LD was calculated separately for the 74 SSR loci on the same or different chromosomes (hereafter, linked and unlinked loci, respectively) with the correlation coefficient (r 2). Intra- and inter-chromosomal LD was studied using the Pairwise analysis of the PowerMarker software. LD was calculated in (i) the entire collection of 111 inbred lines, (ii) the 70 inbred lines discretely clustered by the STRUCTURE analysis, (iii) 36 inbreds discretely clustered in the largest subpopulation inferred by STRUCTURE and (iv) a set of lines selected as a result of the design of the mini-core sets.
To visualize LD patterns, SSR loci were ordered according to their known genomic position to represent ten linkage groups. The nominal level of LD was defined at r 2 = 0.20 according to Gaut and Long (Reference Gaut and Long2003). The significance of LD in the different data sets was assessed with an exact P-value test using PowerMarker default settings; the significance level was set up at P ≤ 0.01.
Results
Characterization of the Argentine public maize inbred line collection
Population structure
Bayesian analysis of population structure using the model-based approach of Pritchard et al. (Reference Pritchard, Stephens and Donnelly2000) provided support for the existence of genetic structure in our inbred panel. However, the inference of the number of gene pools, with k values ranging from 1 to 5, was not straightforward, given that the log-likelihood values for the data conditional on k, ln(X/k), increased progressively with larger values of k. STRUCTURE repetitions showed consistent results between replicate runs. The rate of change of the likelihood distribution and the second-order rate of change of the likelihood function with respect to k (Delta k) was highest at k = 2 (online Supplementary Table S3).
At k = 2, the Argentine public maize inbred line collection was separated into two main subpopulations with ≥0.80 membership (online Supplementary Table S3). The main group had 48 lines with flint origin and the other 37 lines with either BS13-BSSS background or flint combining ability; the remaining lines were mixed. The eight newly incorporated inbreds (coded from 104 to 111) were split into two discrete subpopulations; lines 104, 105, 106, 107 and 108 were assigned to the flint group whereas 109, 110 and 111 were clustered with the other group.
At k = 3, 73 lines were assigned into three subpopulations referred to as flints, BS13-BSSS and LP299-2. The main group comprised 46 inbreds with flint origin; some of them derived from the P465 inbred line and others from the Argentine x Caribbean Derived Stocks (ACDS). The second group, BS13-BSSS, comprised 16 inbreds with clear dent origin. The third group, LP299-2, clustered 11 members that mainly show flint combining ability. Coded Inbred lines 104, 105, 106, 107 and 108 were assigned into the flint group whereas, 109, 110 and 111 were clustered into the LP299-2 group.
At k = 4, 70 lines were assigned into subpopulations with ≥0.80 membership. The distribution of the 70 inbred lines into the four groups was as follows: 7 in the P465 group, 36 in the ACDS group, 11 in the LP299-2 group and 16 in the BS13-BSSS group (online Supplementary Table S1). The first two subpopulations (P465 and ACDS) shared mostly inbreds with known flint origins (online Supplementary Table S1). P465 has well-known combining ability with US dent lines, whereas ACDS comprised members with variable heterotic patterns, 22% of which have known dent combining ability. LP299-2 comprised mostly local inbreds with flint combining ability, and BS13-BSSS comprised inbreds with exotic, US dent origin, although the heterotic pattern of most of them has not been studied yet. The eight newly incorporated inbreds (104, 105, 106, 107 and 108) were reassigned to the ACDS, whereas the remainder three (109, 110 and 111) clustered with the LP299-2-derived lines.
At k = 5, 76 lines were assigned into subpopulations with ≥0.80 membership into the P465, ACDS, LP299-2 and BS13-BSSS groups. In addition, a new group was created by the split of ten inbred lines which have been previously clustered in the ACDS group at k = 4. Also, the Cuarentín synthetic lines coded 109, 110 and 111 shifted from the LP299-2 into the BS13-BSSS group as occurred at k = 2.
Because a better differentiation among the newly incorporated flints and the remaining inbreds was obtained at k = 4, this value was chosen as the best to capture the main structure in the entire collection (Fig. 1).

Fig. 1. Population structure in the Argentine public maize inbred line at k = 4 subpopulations (ACDS, BS13-BSSS, LP299-2 related lines and P465).
Genetic diversity
All 74 microsatellite loci were found to be polymorphic across the complete inbred panel. A total of 483 alleles were detected (online Supplementary Table S4). The number of alleles per locus was variable, ranging from 2 (umc1240, umc1583, umc1938) to 17 (bnlg1325 and bnlg1270), with an average of 6.5 alleles per locus.
In general, allele frequencies were low. We found that 378 out of 483 alleles (78%) occurred at a frequency of 0.25 or less, predicting high gene diversity, and that 11 were private alleles found only in a single inbred line. Average gene diversity and residual heterozygosity were 0.68 (online Supplementary Table S5) and 0.06, respectively (online Supplementary Table S4). Average residual heterozygosity was low, as expected for inbred materials. However, almost all SSRs, except umc2319 and phi057, detected residual heterozygosity. Among these loci, umc2059 detected the minimum (0.01) and umc2190 the maximum residual heterozygosity (0.20).
Relatedness
Relatedness estimation provided additional information about allelic covariance among loci. Kinship coefficients ranged from 0 to 0.47 (Fig. 2). In the Argentine public maize inbred line collection 93% of the pairwise kinship coefficients were ≤0.05. The distribution of classes of pairwise kinship coefficients among the 70 inbred lines clustered at k = 4 was variable among subpopulations. The P465 subpopulation yielded the smallest variation and highest coancestry, with coefficients ranging from 0.16 to 0.29, whereas the LP299-2, BS13-BSSS and ACDS subpopulations had kinship coefficients withi – the 0–0.1875, 0–0.1375 and 0–0.0750 classes, respectively.

Fig. 2. Distribution of kinship coefficients classes within the Argentine public maize inbred line and subpopulations discretely clustered at k = 4.
LD analysis
Most of the r 2 values between linked loci in the entire inbred collection were low (all below the 0.2 nominal level), but statistically significant (online Supplementary Fig. S1a, Supplementary Table S6). Forty-eight out of 64 (75%) pairwise intra-chromosomal comparisons showed significant LD (P ≤ 0.01). All pairwise r 2 values within chromosomes 2 (r 2 ≤ 0.05), 3 (r 2 ≤ 0.05) and 9 (r 2 ≤ 0.20) were significant (Table 1). In chromosome 6, where a denser mapping was conducted, 82% of pairwise comparisons were significant (r 2 ≤ 0.20). The chromosome 6 segment between umc2208 and umc1857 showed the highest proportion of pairwise loci in highly significant LD (P ≤ 0.01). However, no trend was found between p-values and physical distance (bp) among markers in chromosome 6 (online Supplementary Table S6).
Inter-chromosomal pairwise r 2 values were also low (≤0.20) (online Supplementary Fig.S1a). Despite the low magnitude of r 2 between unlinked loci, overall, 68% had highly significant LD. In particular, significant LD between chromosome 9 and the remaining chromosomes was observed (online Supplementary Table S7, A, upper triangle). In contrast, chromosome 10 had the lowest proportion of loci in significant LD in inter-chromosomal comparisons (online Supplementary Table S7).
Characterization of subpopulations and mini-core sets
Subpopulations
Genetic diversity analyses were conducted across the 70 inbreds with >0.80 memberships to any of the subpopulations defined at k = 4. As a result, the mean gene diversity was 0.67, whereas gene diversity values for each subpopulation were 0.34, 0.65, 0.49 and 0.51, for the P465, ACDS, LP299-2 and BS13-BSSS subpopulations, respectively.
To analyse allelic richness within subpopulations, we used the minimum sample size of P465 (Fig. 3). Thus, at equal sample size (i.e. n = 14), the allelic richness for ACDS was almost twice that for P465. The mean number of distinct alleles per locus varied as follows: P465: 2.4; ACDS: 4.2; LP299-2: 2.9; and BS13-BSSS: 3.0.

Fig. 3. Variation of the mean number of distinct alleles per locus as a function of standardized sample size for four subpopulations discretely clustered at k = 4. The maximum standardized sample size across subpopulations was equal to 20.
The LD among the 70 inbreds and within the ACDS subpopulation was also analysed. In the 70 inbred set, r 2 values were below the nominal level, 69% of inter-chromosomal comparisons resulted in highly significant LD (online Supplementary Fig.S1b), and the proportion of linked loci in significant LD was similar to that in the entire collection (Table 1).
LD analysis within subpopulations was only possible in ACDS because of the existence of several monomorphic loci in the remaining subpopulations. In this subpopulation, linked and unlinked locus pairs had r 2 values below the nominal level (online Supplementary Fig. S1c). The exception was the nc009 and umc1014 linked pair, which mapped in chromosome 6 and reached an r 2 = 0.22. Most pairs of loci had r 2 values in the 0.01–0.05 range. In comparison with the 70-inbred set, the ACDS subpopulation showed an increase in r 2 values in the 0.051–0.200 range, which represents 12% of the total. However, the significance of LD was reduced (as seen with the increase in colour-filled squares in the lower triangle section). Only 41% of the inter-chromosomal SSR pairs were found to be in significant LD.
Mini-core sets
To design mini-core sets of 30, 40 and 50 members, the Design Line application of PowerMaker was used to include at least five lines discretely assigned by STRUCTURE to each subpopulation at k = 4 (Table 2). The constrained inbreds were as follows: 1, 2, 3, 4 and 5 (P465); 6, 10, 11, 12 and 13 (ACDS); 14, 15, 16, 17 and 26 (LP299-2-related lines); and 80, 81, 82, 90 and 103 (BS13-BSSS).
The core set selection algorithm was replicated independently ten times for each of the designed mini-core sets. An output set, which always included the constrained set of representative lines, was obtained in all ten replicates (Table 2). The effective size of mini-cores, was 26, 33 and 42 for the 30, 40 and 50 nominal sets, respectively. Inbreds which were not included in all ten replicates were excluded from further analysis. During this simulation, there were six inbreds (LP916, LPB2, LP153, L4637, (LP562 × 3584)-1-39 and P21) that were always pooled in all ten replicates and designed sets (Table 2). These inbreds clustered mostly into the ACDS group or had mixed but higher membership to this subpopulations (LP916 and LPB2). The designed mini-core set with 50 members mostly pooled inbred lines from ACDS and incorporated two additional inbred lines from the P465 (LP661) and BS13-BSSS (L5605) subpopulations.
Analysis of the genetic diversity in the 33-line mini-core set showed that this reduced set solely captured 385 out of 483 alleles (80% of total). Allele frequencies were low; 269 out of 385 alleles (70%) occurred at a frequency of 0.25 or less, resulting in relatively high gene diversity (0.65). This gene diversity value was similar to that for the Argentine maize inbred line collection (0.68), the 70 inbreds set (0.67) and the ACDS subpopulation (0.65).
Despite the reduced number of members in the 33-line mini-core set, only few values of r 2 were above the 0.2 nominal level (online Supplementary Fig. S1d). Most of the locus pairs had r 2 values in the 0.01–0.20 range.
Intra-chromosomal r 2 values in the 33 line mini-core set were low, with locus pairs at bins 3.07–3.09 (umc2050–phi047) and 6.04 (nc009–umc1014) being the only ones reaching a value of r 2 = 0.10. Thirty-four of the 64 pairwise linked SSRs (53%) were in significant LD at the P ≤ 0.01 level. In chromosome 6, 64% of pairwise locus comparisons showed significant LD. Intra-chromosomal LD within this mini-core set was not increased relative to the complete Argentine public maize inbred line collection or the 70-inbred set (Table 1).
Inter-chromosomal pairwise r 2 values had larger values instead. Eleven unlinked loci from chromosomes 5, 6, 7, 8, 9 and 10 had statistically significant r 2 values in the 0.201–0.500 range, whereas 52% of inter-chromosomal SSR pairs were in highly significant LD. The number of locus pairs in significant LD in the mini-core set was lower than that of the ACDS subpopulation (n = 36) (online Supplementary Fig. S1d). Moreover, as seen in online Supplementary Table S7B, significant LD occurrences were reduced when compared with the Argentine public maize inbred line collection and remaining sets.
Discussion
In the present study, we extended the characterization of the Argentine public maize inbred line collection previously reported (Olmos et al., Reference Olmos, Delucchi, Ravera, Negri, Mandolino and Eyhérabide2014a). Eight historic inbred lines with flint origin were incorporated. In particular, five new inbreds had a Cuarentín-type background related to Cateto flint races from South America (Paterniani and Goodman, Reference Paterniani and Goodman1977). In addition, SSR density was increased, particularly at chromosome 6, bin 6.04. Experimental records of known combining ability of 62 of the 111 inbred lines were also provided to test for the correspondence of genetic structure with the heterotic response of inbred lines, thus providing additional tools for optimum exploitation of heterosis in breeding (Reif et al., Reference Reif, Hallauer and Melchinger2005).
Population structure and genetic diversity
The Bayesian clustering approach was used to infer the optimal number of subpopulations in the Argentine public maize inbred line collection. As a result, k = 4 was selected as the optimal choice to assign the eight historic flint inbred lines into the entire collection. Two subpopulations (i.e. P465 and ACDS) with mainly dent combining ability and two subpopulations (i.e. LP299-2 and BS13-BSSS) with mainly flint combing ability were identified. The eight historic flints were split into two separated clusters, the ACDS (inbred lines coded 104–108) and the LP299-2-related lines (inbred lines 109–111).
Modern maize production is based on heterosis, which results from crossing inbred lines from contrasting heterotic group pairs, being specific pairs called heterotic patterns (Melchinger and Gumber, Reference Melchinger, Gumber, Lamkey and Staub1998). In the present study, genetic clustering was in agreement with the combining ability of inbred lines within groups, suggesting the existence of four heterotic groups and two heterotic patterns (flint and dent) within the Argentine public maize inbred line collection.
Previous field experiments with representative inbred lines from the Argentine public maize inbred line collection showed that two flint inbreds (LP612 and LP122-2) had contrasting heterotic patterns when crossed to the US dent B73 and MO17, respectively (Delucchi et al, Reference Delucchi, Eyhérabide, Lorea, Presello, Otegui and López2012). This result and the fact that, in the present study, LP612, LP122-2 and B73 clustered in the P465, ACDS and BS13-BSSS subpopulations, respectively, may explain the occurrence of the three of four heterotic groups mentioned above within the Argentine public maize inbred line collection. However, there is no evidence so far that the LP299-2 subpopulation might be directly related to the US dent MO17, since MO17 (a representative of the ‘Lancaster SureCrop’ heterotic group) has not yet been included in the Argentine public maize inbred line collection. The genetic background of the LP299-2 family is not totally clear because the LP299-2 inbred line was obtained by self-pollinating a Pioneer Hi-Bred commercial hybrid. However, one of the most relevant findings in this work was the clear distinction of the LP299-2 subpopulation from the B73-derived lines. LP299-2 members were previously set as mixed, mostly between P465 and BS13-BSSS subpopulations, whereas only LP509 inbred shared ancestry in ACDS (Olmos et al., Reference Olmos, Delucchi, Ravera, Negri, Mandolino and Eyhérabide2014a). Tracing back the history of the genetic improvement of the Pioneer Hi-Bred breeding programme might indicate a link between Pioneer Hi-Bred hybrids with Argentine germplasms. Thus, the Argentine ‘maíz amargo’ landrace, registered later as US B96 inbred line (Roberts et al., Reference Roberts, Grant, Ramirez, Hatheway and Smith1957; Guthrie et al., Reference Guthrie, Russell, Bing and Dicke1991; Goodman, Reference Goodman, Coors and Pandey1999), played with B73 an important role in the creation of a unique Stiff Stalk (SS) germplasm within Pioneer Hi-Bred (Mikel and Dudley, Reference Mikel and Dudley2006). Although the Argentine public maize inbred line collection did not include B96, previous studies have shown that a ‘maíz amargo’ landrace had good combining ability when crossed with the LP122-2, LP612 and B73 inbred lines but a bad performance when crossed with MO17 (Delucchi et al., Reference Delucchi, Eyhérabide, Lorea, Presello, Otegui and López2012), implying that the LP299-2 subpopulation would fit into a MO17-like heterotic group.
The degree of genetic diversity detected varied across subpopulations. The smallest and least diverse group P465 had a clear flint origin and good combining ability in crosses with US dent lines. The large ACDS group evidenced high allelic richness, which agreed with the heterogeneous origin and variable heterotic patterns of its members. The LP299-2 subpopulation, which included inbred lines with good combining ability when crossed with Argentine flint genetic background and comprised a mix of semi-flint and semi-dent texture grain types, had intermediate gene diversity values similar to those of the BS13-BSSS subpopulation. Finally, the composition of inbred lines of the BS13-BSSS subpopulation was conserved as compared to that observed previously (Olmos et al., Reference Olmos, Delucchi, Ravera, Negri, Mandolino and Eyhérabide2014a), with the exception of inbred lines coded 58, 79, 96, 97 and 98, which, in the present study, turned into the BS13-BSSS subpopulation.
The addition of the eight historic flint inbred lines did not result in the delimitation of new genetic clusters and did not increase the average gene diversity of the entire collection. However, it helped to differentiate the current LP299-2 subpopulation and to provide preliminary evidence about the genetic background of such subpopulation. Indeed, the LP509 inbred that shared membership in the LP299-2 subpopulation is known to carry a Cuarentín and BSSS background. This allows us to suggest that the Pioneer Hi-Bred hybrid from which the LP299-2 subpopulation derives might partially share a common genetic background with the Cuarentín and BS13-BSSS inbred lines, which in turn have been preserved through the development of LP299-2-derived lines. In contrast, the assignment of two historic inbred lines with Argentine Caribbean × Cuarentín Synthetics background (coded 107 and 108) within the ACDS may be related to the ancestral origin of the Cuarentín type (Paterniani and Goodman, Reference Paterniani and Goodman1977), a group that, according to Blumenschein (Reference Blumenschein1973), shows strong phenotypic similarity to Cuban flints. The close relationship of Cuban and Argentinean flints was recorded in early times when it was proposed that Cuban flints were introgressed with flint corn from Argentina in the early 1900's (Hatheway, Reference Hatheway1957). Indeed, Bayesian analysis based on microsatellite markers has highlighted the close affiliation of the Argentine race Orgullo Cuarentón with germplasm from the Caribbean (Lia et al., Reference Lia, Poggio and Confalonieri2009), supporting the relationship of these two historic flint inbred lines with the ACDS subpopulation.
LD extent
In maize, LD generally decays rapidly with short nucleotide distances within genes, contrary to the high level of genome-wide LD observed by the SSRs (Remington et al., Reference Remington, Thornsberry, Matsuoka, Wilson, Whitt, Doebley, Kresovich, Goodman and Buckler2001). In addition, genome-wide LD assays with a high-density single nucleotide polymorphisms (SNP) array have demonstrated that LD extent varies according to the composition of the maize panels, the chromosomes and the genomic positions along chromosomes (Rincent et al., Reference Rincent, Nicolas, Bouchet, Altmann, Brunel, Revilla, Malvar, Moreno-Gonzalez, Campo, Melchinger, Schipprack, Bauer, Schoen, Meyer, Ouzunova, Dubreuil, Giauffret, Madur, Combes, Dumas, Bauland, Jamin, Laborde, Flament, Moreau and Charcosset2014). SNPs and SSRs markers with genome-wide distribution have revealed that, in maize, SSRs have a higher power than SNPs to detect unlinked LD (Van Inghelandt et al., Reference Van Inghelandt, Reif, Dhillon, Flament and Melchinger2011). In our study, the ratio of linked and unlinked locus pairs in significant LD was almost one and the proportion of significant LD was affected by chromosomes and the population structure. Linked SSR pairs from chromosomes 5, 8 and 10 had significant LD proportions below the overall inter-chromosomal LD (68%). In agreement, variation in LD significance according to chromosomes has also been reported for a diverse collection of 290 maize inbred lines (Liu et al., Reference Liu, Guo, Zhang, Zhao, Zhu, Huang and Chen2014) and a cotton panel (Abdurakhmonov et al., Reference Abdurakhmonov, Saha, Jenkins, Buriev, Shermatov, Scheffler, Pepper, Yu, Kohel and Abdukarimov2009) surveyed with SSRs. A similar ratio of linked to unlinked pairs in significant LD was obtained by Stich et al. (Reference Stich, Melchinger, Frisch, Maurer, Heckenberger and Reif2005). This indicates that LD in diverse maize panels is reached by forces such as relatedness, population stratification, and genetic drift, which cause a high risk of detecting false positives in association mapping. Controlling the population structure in the Argentine maize inbred collection resulted in a reduction in the number of locus in significant LD, for instance, within the ACDS subpopulation. However, the ratios of linked to unlinked pairs in significant LD did not vary. Stratification has also been found to reduce the significance of LD between SSR loci in diverse collections of maize inbred lines (Remington et al., Reference Remington, Thornsberry, Matsuoka, Wilson, Whitt, Doebley, Kresovich, Goodman and Buckler2001, Liu et al, Reference Liu, Goodman, Muse, Smith, Buckler and Doebley2003; Wang et al., Reference Wang, Yu, Zhao, Shi, Song, Wang and Li2008). The effect of the model-based clustering method was also evident on the assignment of relatively distantly related inbred lines in the ACDS subpopulation, since only 5% of kinship coefficients between inbred lines with full memberships in ACDS were found to be above 0.1. It has been proposed that the STRUCTURE model-based clustering method and kinship estimates account for different magnitudes of relatedness among individuals, with kinship revealing relationships in a finer scale (Yu et al., Reference Yu, Pressoir, Briggs, Bi, Yamasaki, Doebley, McMullen, Gaut, Nielsen, Holland, Kresovich and Buckler2006). In our work, we noticed that a high number of kinship coefficients were close to zero as also obtained in the diverse maize association panel characterized by Yu et al., (Reference Yu, Pressoir, Briggs, Bi, Yamasaki, Doebley, McMullen, Gaut, Nielsen, Holland, Kresovich and Buckler2006). The slight variation in kinship coefficients within the entire collection and subpopulations indicates the need to study kinships at a deeper scale to reveal subtle genetic relationships within and among subpopulations. Thus, the gene diversity and the allelic richness, together with the low relatedness and genetic structure, may have contributed to reducing the LD extent in the ACDS subpopulation.
Design of mini-core sets
Another strategy to reduce the complexity of a plant collection is the use of mini-core sets, which consist of a limited set of varieties or lines (about 10% of the full collection) that represent the genetic diversity of a species with a minimum of repetitiveness (Brown, Reference Brown1989). To date, mini-core sets have been exclusively developed in crops with autogamous reproduction including rice (Li et al., Reference Li, Yan, Agrama, Hu, Jia, Jia, Jackson, Moldenhauer, McClung and Wu2010; Zhang et al., Reference Zhang, Zhang, Wang, Sun, Qi, Li, Wei, Han, Qiu, Tang and Li2011), peanut (Wang et al., Reference Wang, Sukumaran, Barkley, Chen, Chen, Guo, Pittman, Stalker, Holbrook, Pederson and Yu2011, Jiang et al., Reference Jiang, Huang, Ren, Chen, Zhou, Xia, Huang, Lei, Yan, Wan and Liao2014), sorghum (Upadhyaya et al., Reference Upadhyaya, Wang, Gowda and Sharma2013), soybean (Kaga et al., Reference Kaga, Shimizu, Watanabe, Tsubokura, Katayose, Harada, Vaughan and Tomooka2012; Guo et al., Reference Guo, Li, Hong and Qiu2014), sesame (Zhang et al., Reference Zhang, Zhang, Che, Wang, Wei and Li2012) and mungbean (Schafleitner et al., Reference Schafleitner, Nair, Rathore, Wang, Lin, Chu, Lin, Chang and Ebert2015). Similar types of studies in allogamous plants are scarce; in the case of maize, core and mini-core definitions are limited to collections from Europe (Gouesnard et al., Reference Gouesnard, Dallard, Bertin, Boyat and Charcosset2005), China (Wang et al., Reference Wang, Yu, Zhao, Shi, Song, Wang and Li2008) and Mexico (Wen et al., Reference Wen, Franco, Chavez-Tovar, Yan and Taba2012).
To our knowledge, this is the first report using the SA algorithm to choose a mini-core set of lines from the Argentine collection of 111 temperate inbred lines, constrained with 20 representative inbred lines selected by the model-based approach following Liu and Muse (Reference Liu and Muse2005). The result was the selected set of lines that have maximized the amount of genetic diversity, while the population structure was controlled by the mini-core set design itself. As compared with the overall Argentine public maize inbred line collection, the resulting mini-core set design with 33 members reduced overall allelic richness only by 20%. At the same time, it reduced the amount of statistically significant LD between unlinked loci. Moreover, average pairwise r 2 = 0.04 for all markers pairs on a same chromosome was in the same magnitude of the LD level found within the diverse-26 maize nested association mapping founders (Yu et al., Reference Yu, Holland, McMullen and Buckler2008). Furthermore, because the size of the mini-core set with 33 inbreds is still small, the collection enrichment with representative donor inbred lines with desirable agronomic and nutritional traits, such as disease resistance and grain quality, or the parental RIL population mentioned above, will be desirable. The mini-core sets identified in the present study can provide a useful tool for affordable high-density SNP genotyping and sequencing-based allele mining (Ashkani et al., Reference Ashkani, Yusop, Shabanimofrad, Azadi, Ghasemzadeh, Azizi and Latif2015). These mini-core sets could also be useful to search for consistent phenotypic variation and to explore the genetic basis of underlying phenotypic responses with the integration of functional genomics (Ishitani et al., Reference Ishitani, Rao, Wenzl, Beebe and Tohme2004; Andrade et al., Reference Andrade, Sala, Pontaroli, León, Sadras and Calderini2015).
Conclusion
In conclusion, our findings indicate that the Argentine public maize inbred line collection is a diverse panel that has accumulated numerous historical recombinations leading to an expected low extent of LD, suggesting that it provides a valuable tool for association mapping purposes.
Supplementary material
The supplementary material for this article can be found at http://dx.doi.org/10.1017/S1479262116000228.
Acknowledgements
This research was conducted at the National Institute of Agricultural Technology (INTA) Pergamino station, Argentina, and financed by INTA projects and PICT 2009 No. 0036 (ANPCyT). We thank all staff members that participated during laboratory analysis.







