


Introduction
Identifying both environmental and genetic determinants of fetal growth is important, not only in terms of the clinical management of pregnancies, but also because of the apparent significance of early development in establishing health patterns later in life. Associations found between low birth weight and increased risks of cardiovascular disease, stroke, hypertension, diabetes and other illnesses in adulthood are now well established and have generated a vast area of research looking at the fetal and developmental origins of adult disease.Reference Godfrey1 Some of the most robustly reproduced associations are those of low birth weight with insulin resistance and type 2 diabetes (T2D) in adulthood. The mechanism driving these associations in humans, however, is still uncertain.
The majority of research to date has focused on the theory of intrauterine programming, whereby exposure in utero to an insult, such as malnutrition, during critical periods of development, ‘programs’ the fetus to adapt to this adverse environment, with long-term health consequences.Reference Barker2–Reference Poulsen, Vaag, Kyvik, Moller Jensen and Beck-Nielsen5 An alternative, but complementary, hypothesis is the fetal insulin hypothesis, which proposes that low birth weight and T2D could both be phenotypes of the same genetic predisposition to insulin resistance and diabetesReference Hattersley and Tooke6 (see Fig. 1).

Fig. 1 Genes or environment? The two explanations for the association between low birth size and type 2 diabetes and insulin resistance (adapted from Frayling and HattersleyReference Frayling and Hattersley67).
Research to date has found evidence in support of both fetal programming and genetics as explanations of the association between low birth weight and T2D. It is essential to determine the extent to which genes account for this association as it will impact on the success of any strategies for prevention and intervention. In this review, we aim to discuss the recent evidence for an important genetic contribution to fetal growth and its influence on susceptibility to T2D in later life.
Genetic influence on fetal growth
Two clear examples of genetic influences on fetal growth are seen when looking at gender or comparing different racial groups. Girls are consistently lighter than boys,Reference Kramer7, Reference Cogswell and Yip8 and there are clear differences in their body composition at birth, with girls being lighter, but having more adipose tissue.Reference Guihard-Costa, Grange, Larroche and Papiernik9–Reference Shields, Knight, Powell, Hattersley and Wright13 Asian and black newborns tend to be lighter on average than white and Hispanic newborns,Reference Kramer7, Reference Cogswell and Yip8, Reference Shiono, Klebanoff, Graubard, Berendes and Rhoads14 and again, it is not only the weight that differs, but also the body composition.Reference Yajnik, Fall and Coyaji12, Reference Yajnik, Lubree and Rege15, Reference Denham, Schell, Gallo and Stark16
Familial trends provide further evidence for the role of genetics in normal fetal growth. There is a strong correlation between siblings;Reference Tanner, Lejarraga and Turner17 although when looking at half-siblings, the correlation is higher in those who share a mother, compared to those who share a father (intraclass correlation coefficients of 0.581 and 0.102, respectively),Reference Morton18 reflecting the stronger influence of the mother on her offspring.
Further support for a genetic influence is seen from associations between parental and offspring birth weights.Reference Langhoff-Roos, Lindmark, Gustavson, Gebre-Medhin and Meirik19, Reference Hennessy and Alberman20 Many other familial associations have been reported, but those between paternal factors and birth size provide the strongest evidence for genetic regulation of growth, as the father has very little effect on the intrauterine environment. Relationships between offspring birth size and paternal height, weight and birth weight indicate that genes are important in fetal growth.Reference Kramer7, Reference Shields, Knight, Powell, Hattersley and Wright13, Reference Catalano, Drago and Amini21–Reference Klebanoff, Mednick, Schulsinger, Secher and Shiono24 Paternal height appears to have the strongest effect on birth length, and it has been suggested that genetic factors are more likely to alter skeletal size rather than fat mass.Reference Shields, Knight, Powell, Hattersley and Wright13, Reference Catalano, Drago and Amini21, Reference Knight, Shields and Turner23
The complex interaction between the intrauterine environment and fetal and maternal genetic effects has meant quantifying the genetic contribution to birth weight has proved difficult, with estimates of heritability varying considerably. Twin and intergenerational studies have been inconsistent, with the proportion of variance in birth weight attributed to genes estimated to lie between 25 and 80%.Reference Magnus25–Reference Johnston, Clark and Savage33 The majority of recent studies suggest that it is around 40%.Reference Gielen, Lindsey and Derom29–Reference Vlietinck, Derom and Neale32
Certain regions of the genome that are imprinted (i.e. expressed from only one parental chromosome of origin) are known to be important in fetal growth. This may be illustrated by rare abnormalities of chromosome 11p15.5. For example, Beckwith–Wiedemann syndrome is a fetal overgrowth disorder associated with biallelic expression of IGF2, which may arise when an individual inherits two copies of the chromosome from the father and none from the mother.Reference Li, Squire and Weksberg34 Conversely, Silver Russell syndrome is characterized by low birth weight and has been associated with maternal duplication of the region that represses IGF2 expression.Reference Fisher, Thomas and Cockwell35 Despite their known importance, however, the extent to which variants at imprinted loci explain common variation in birth weight is not known.
Genetic influences on the predisposition to T2D
T2D is a complex, multifactorial disease, and is the consequence of both genetic susceptibility and environmental influence. There is increasing evidence for a strong genetic component, and understanding this genetic background will be vital in terms of assessing an individual’s risk of developing T2D.
Variability in the prevalence of diabetes in different racial groups provides clear evidence for a genetic influence on the predisposition to T2D. T2D is relatively rare in the Mapuche Indian or Chinese populations, but in the Naruan and Pima Indian populations, the prevalence is as high as 50%.Reference King and Rewers36 Although environmental factors may, in part, explain this disparity in risk, migration studies and studies of genetic admixture provide strong support for an important genetic contribution.Reference Barroso37, Reference Gloyn and McCarthy38
Twin and family studies have also indicated that there is a substantial genetic component to T2D. Concordance for T2D is high in monozygotic and dizygotic twins, although estimates of concordance rates have varied considerably.Reference Barroso37, Reference Gloyn and McCarthy38 The relative risk for the sibling of an affected individual, compared with the population prevalence, is estimated at 3.5.Reference Rich39 Lifetime risk for T2D is increased in individuals with a single parent affected with T2D, and is even higher in those in whom both mother and father have the disease.Reference Stumvoll, Goldstein and van Haeften40
The most compelling evidence, however, is from recent research identifying specific genetic loci associated with T2D. It is now well established that T2D is a polygenic disease arising from the interaction of multiple susceptibility genes and environmental exposure. Candidate gene and linkage approaches have revealed some associations between common genetic variants and T2D risk.Reference Altshuler, Hirschhorn and Klannemark41–Reference Gudmundsson, Sulem and Steinthorsdottir46 More recently, genome-wide association studies have enabled even more T2D loci to be identifiedReference Prokopenko, Langenberg and Florez47–Reference Rung, Cauchi and Albrechtsen59 (Table 1). Associations found between these genetic variants and T2D are robust (P < 5 × 10−8) and have been replicated widely. Individually, these polymorphisms contribute very little to disease risk, with <10% of the overall variance in T2D susceptibility being accounted for by the variants identified to date.Reference Prokopenko, McCarthy and Lindgren60 However, studies looking at their combined effects show promising potential for identifying subgroups of individuals at the highest risk for developing T2D.Reference Weedon, McCarthy and Hitman61–Reference Lin, Song and Lim66 This research is still in its infancy, and it is clear that further variants will need to be identified to improve the predictive value of common polymorphisms beyond clinical characteristics.
Table 1 Genetic loci identified to date with a role in type 2 diabetes susceptibility

OR, odds ratios; SNP, single-nucleotide polymorphism.
Adapted from McCarthy and HattersleyReference McCarthy and Hattersley76, with additional data from more recent publicationsReference Prokopenko, Langenberg and Florez47, Reference Yasuda, Miyake and Horikawa55–Reference Rung, Cauchi and Albrechtsen59. Risk allele frequencies and ORs are estimated from studies of European populations.
Can low birth weight and T2D be two phenotypes of the same genotype?
The fetal insulin hypothesis proposes that low birth weight and T2D are two phenotypes of the same genotype.Reference Hattersley and Tooke6 Insulin is a key growth factor in utero, and T2D occurs as a result of insulin resistance and/or beta-cell dysfunction. Genes impacting on insulin secretion and insulin action are, therefore, likely to alter both fetal growth and susceptibility to T2D (Fig. 1).
Two tests of this hypothesis were proposed.Reference Hattersley and Tooke6 First, an inverse correlation between paternal insulin resistance and offspring birth weight would support a role for fetal genes altering fetal insulin-mediated growth. Paternal insulin resistance reflects the paternal genotype, and as paternal factors are independent of the maternal intrauterine environment, any association observed is likely to reflect genes inherited by the fetus from the father, which result in insulin resistance and reduced insulin-mediated growth (Fig. 2). Second, polymorphisms and mutations in genes associated with insulin resistance and T2D should also be associated with reduced birth weight.

Fig. 2 Factors affecting fetal insulin secretion and fetal growth.
Tests of these hypotheses have now been carried out, and the latest studies suggest that it is genes involved in beta-cell mass and beta-cell function that are important, rather than genes related to insulin resistance. We will summarize the early work in support of the fetal insulin hypothesis (reviewed previously by Frayling and HattersleyReference Frayling and Hattersley67) below, and then present the findings of more recent epidemiological study and the latest supporting evidence that has arisen as a result of genome-wide association studies.
Evidence from monogenic disorders
Table 2 details a number of monogenic disorders affecting fetal insulin secretion or insulin resistance and their impact on birth weight. The first evidence for the fetal insulin hypothesis came from observations in patients with heterozygous mutations in the glucokinase (GCK) gene, which causes life-long mild fasting hyperglycemia.Reference Froguel, Zouali and Vionnet68 These subjects have reduced insulin secretion due to impaired glucose sensing and have been found to be approximately 500 g lighter than unaffected siblings.Reference Hattersley, Beards and Ballantyne69 Importantly, it was found that the gene could alter birth weight either directly through changes to fetal insulin secretion, or indirectly, through changes to maternal glucose levels. Babies without the mutation, born to mothers with a GCK mutation, tended to be heavier (by a mean of 600 g), in line with what is seen in diabetic pregnancies where hyperglycemia in utero causes increased fetal insulin secretion and a corresponding increase in insulin-mediated growth. In contrast, if the baby was born to a non-diabetic mother, but had inherited the mutation from the father, it was born 533 g lighter on average, due to reduced glucose sensing and, hence, reduced insulin secretion associated with the mutation.Reference Hattersley, Beards and Ballantyne69
Table 2 Monogenic disorders affecting fetal insulin secretion and insulin resistance and their effect on birth weight

Mutations in other genes associated with renal cysts and diabetes syndrome (HNF1B) and transient and permanent neonatal diabetes (KCNJ11, ABCC8) also demonstrate how mutations resulting in decreased insulin secretion lead to reduced birth weight and a diabetic phenotypeReference Edghill, Bingham and Slingerland70–Reference Babenko, Polak and Cave73 (Table 2). The opposite is seen in patients with HNF4A diabetes and activating mutations in KCNJ11 and ABCC8, which have been found to be associated with hyperinsulinism in utero and early life resulting in macrosomia and hypoglycemia at birth and in the neonatal period.Reference Pearson, Boj and Steele74, Reference Aparicio, Carpenter, Schwartz and Gruppuso75
Most monogenic diabetes is caused by mutations altering beta-cell function. This suggests that the healthy beta-cell is usually able to sufficiently compensate for insulin resistance.Reference McCarthy and Hattersley76 One exception is insulin receptor mutations that result in severe insulin resistance, which leads to reduced birth weight (Table 2).
These disorders have established the principle that genes affecting fetal insulin secretion can alter fetal growth, but these conditions are rare, and therefore do not explain the association between low birth weight and T2D seen in the general population.
Evidence from epidemiological studies
Evidence for the fetal insulin hypothesis in the general population comes from epidemiological studies looking at the effect of family history of diabetes and insulin resistance on birth size.
A central idea behind tests of the fetal insulin hypothesis is that associations found between paternal factors and offspring birth size must be largely independent of the intrauterine environment, and therefore reflect the effects of genes inherited by the fetus from the father (Fig. 2). This is particularly important as maternal diabetes is associated with macrosomia in her offspring, and thus complicates efforts to examine associations with low birth weight.
Data from a study of the Pima Indians showed that babies born to only a father who developed diabetes had lower birth weights than those with no parents with diabetes or only a mother who developed diabetes.Reference Lindsay, Dabelea and Roumain77 Furthermore, those in the lowest birth weight tertile born to fathers with diabetes were themselves at a higher risk of developing diabetes than those in the middle or highest tertile (Fig. 3). This association between risk of paternal diabetes and lower offspring birth weight has also been confirmed in other populations.Reference Davey Smith, Sterne, Tynelius and Rasmussen78, Reference Hypponen, Smith and Power79

Fig. 3 Mean birth weight and parental diagnosis of diabetes (adapted from Lindsay et al.Reference Lindsay, Dabelea and Roumain77).
The relationship between offspring birth weight and paternal insulin resistance is less clear. Two studies in the United Kingdom have shown contrasting findings, with one reporting an inverse association between offspring birth weight and paternal insulin resistance in late adulthood,Reference Wannamethee, Lawlor and Whincup80 whereas the other, found no association between offspring birth weight and father’s insulin resistance when assessed at the time of pregnancyReference Knight, Shields and Hill81 (Fig. 4). It is possible that this difference could be explained by the ages of the two cohorts; the younger fathers may not manifest clear signs of insulin resistance until they are older, reflecting the association between insulin resistance and increasing age. In addition this, however, a positive relationship between paternal insulin resistance and birth weight was observed in an Indian population, although, this was not independent of paternal body mass index (BMI).Reference Yajnik, Coyaji, Joglekar, Kellingray and Fall82

Fig. 4 Paternal log insulin resistance, adjusted for body mass index, is not associated with offspring birth weight (Knight et al.Reference Knight, Shields and Hill81).
The lack of a clear association between birth weight and paternal insulin resistance may be explained by fetal compensation for inherited insulin resistance. A positive association between paternal insulin resistance and umbilical cord insulin in the offspring has been observed, which suggests that an insulin-resistant fetus may increase its insulin secretion in order to maintain normal growth.Reference Shields, Knight and Turner83 This pattern is also consistent with the finding that girls, who are smaller and more insulin resistant than boys, also have higher cord insulin levels.Reference Shields, Knight and Hopper84 Similarly, comparisons between Indian and White European babies have shown that Indian babies, despite being smaller and more insulin resistant, have increased umbilical cord insulin concentrations.Reference Yajnik, Lubree and Rege15
Evidence from genome-wide association studies
Genome-wide association studies (Table 3) have identified new common genetic variants associated with T2D and fasting glucose and some of these polymorphisms have been found to influence fetal growth, either directly or indirectly. Recent research has begun to focus on associations between these DNA variants and birth weight, looking at both the genotype of the mother and of the fetus. These studies have provided some of the clearest evidence for the fetal insulin hypothesis in the general population.
Table 3 Common risk alleles for T2D or Fasting Plasma Glucose (FPG) and their effect on birth weight

T2D, type 2 diabetes; SNP, single-nucleotide polymorphism; CI, confidence interval.
One of the first studies with consistent results examined how single-nucleotide polymorphisms (SNPs) altering fasting glucose could impact on birth weight. A common haplotype of the GCK gene, rs1799884, was found to be associated with glucose concentrations with an increase of 0.07 mmol/l for each copy of the A-allele.Reference Weedon, Frayling and Shields85 In line with this, maternal GCK genotype was associated with a mean increase of 64 g(95% confidence interval (CI) = 25–102; P = 0.001) in birth weight for the A-allele carriers over GG homozygotes.Reference Weedon, Frayling and Shields85 These findings were replicated in a further meta-analysis where a 27 g increase in birth weight for the presence of a maternal A-allele was observed.Reference Weedon, Clark and Qian86 This genetic regulation of birth weight appeared to be entirely mediated through maternal glucose. In contrast with the monogenic mutation in GCK, no direct effect of fetal genotype was observed.
The gene-encoding transcription factor 7-like-2 (TCF7L2) is one of the most important diabetes susceptibility genes discovered to date.Reference Grant, Thorleifsson and Reynisdottir43, Reference Groves, Zeggini and Minton87–Reference Chandak, Janipalli and Bhaskar94 Individuals who inherit one copy of the T-allele of the rs7903146 SNP (44% of Europeans) are 36% more likely to develop T2D than those inheriting none, and the 9% of Europeans who inherit two copies are nearly twice as likely to develop T2D.Reference Groves, Zeggini and Minton87 Studies in non-diabetic subjects have indicated that TCF7L2 is involved in insulin secretion,Reference Damcott, Pollin and Reinhart88, Reference Saxena, Gianniny and Burtt95, Reference Munoz, Lok and Gower96 making it an ideal candidate gene for influencing fetal growth. Results from a meta-analysis of 24,053 individuals across six studies showed an 18 g (95% CI = 7–29 g) increase in birth weight associated with each copy of the fetal risk allele (P = 0.001).Reference Freathy, Weedon and Bennett97 Examination of the effect of maternal genotype, available in three of the cohorts studied, showed a 30 g (95% CI = 15–45 g) increase in birth weight per risk allele (P = 2.8 × 10−5).Reference Freathy, Weedon and Bennett97 However, the fetal association was confounded by the 50% correlation between maternal and fetal genotypes; after adjusting for maternal genotype, there was no evidence for an independent effect of the fetal genotype on birth weight.
The first substantial evidence for an effect of common fetal variants on birth weight comes from a more recent study of polymorphisms at the CDKAL1 and HHEX-IDE loci.Reference Freathy, Bennett and Ring98 In this study of 19,200 offspring, the fetal risk alleles of the SNPs rs10946398 (CDKAL1) and rs1111875 (HHEX-IDE) were associated with a reduction in birth weight of 21 g (95% CI = 11–31; P = 2 × 10−5) and 14 g (95% CI = 4–23; P = 0.004) per risk allele, respectively. In an analysis of a subgroup of these individuals, where both maternal and fetal genotypes were available (n = 5342–5507), there was some evidence for an opposing effect of maternal genotype in CDKAL1 (P = 0.04), but this will require further confirmation in larger studies. Findings from more recent studies of US and Finnish individualsReference Zhao, Li and Bradfield99, Reference Pulizzi, Lysseako and Jonsson100 support the associations with CDKAL1 and HHEX-IDE, respectively.
Individually, the contribution of these genes to variation in birth weight is small, but in combination, we are starting to see effect sizes comparable to recognized environmental influences. Combined analysis of maternal GCK and TCF7L2 risk alleles has shown that the 4% of mothers with three or four risk alleles are an estimated 119 g heavier at birth than the 32% of mothers with no risk alleles.Reference Freathy, Weedon and Bennett97 A fetal risk allele score for both CDKAL1 and HHEX-IDE shows a decrease in birth weight of 80 g for the 4% of offspring carrying four risk alleles compared with the 8% carrying no risk alleles.Reference Freathy, Bennett and Ring98 These observations equate to an effect size comparable with smoking three or four cigarettes a day in the third trimester of pregnancy.Reference Bernstein, Mongeon and Badger101
Not all of the common T2D susceptibility genes that were identified appear to have an effect on fetal growth. In similar large-scale studies, common variants in PPARG, KCNJ11, SLC30A8, IGF2BP2 and CDKN2A/2B showed no association with birth weight.Reference Freathy, Bennett and Ring98–Reference Pulizzi, Lysseako and Jonsson100, Reference Bennett, Sovio and Ruokonen102
How do genes alter fetal growth
We have demonstrated clear associations between common T2D polymorphisms in both the mother and fetus and birth weight. The mechanisms underlying these associations are likely to reflect alterations to pancreatic beta-cell function affecting glucose sensitivity and/or insulin secretion. Maternal genes predominantly affect birth weight through their impact on the intrauterine environment (Fig. 5). Maternal glucose crosses the placenta where it enters the fetal circulation and is sensed by the fetal pancreas. The fetus secretes insulin in response to this glucose stimulus, and the insulin acts as a fetal growth factor. Therefore, alterations to maternal glucose can have a substantial effect on the birth size of her offspring, and a clear demonstration of this is the macrosomia seen in diabetic pregnancies. Common variation in both GCK and TCF7L2 alter maternal fasting glucose. GCK is involved in pancreatic glucose sensing, and so has a direct influence on circulating blood glucose concentrations.Reference Byrne, Sturis and Clement103 Studies of non-diabetic subjects have shown that the TCF7L2 risk alleles are associated with reduced insulin secretion, resulting in an indirect effect on fasting glucose.Reference Saxena, Gianniny and Burtt95–Reference Freathy, Weedon and Bennett97
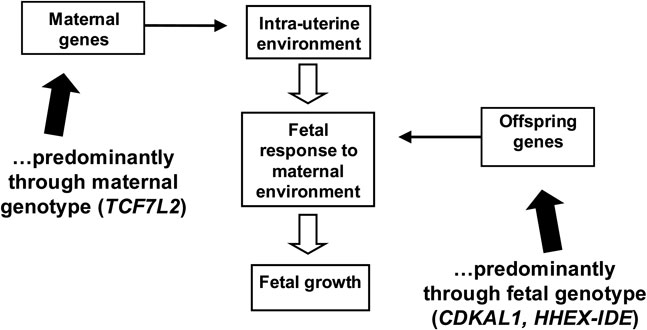
Fig. 5 Effects of maternal and offspring genes can alter fetal growth.
Fetal genes regulate birth weight by altering the response by the fetus to the intrauterine environment (Fig. 5). The T2D variants at CDKAL1 and HHEX-IDE, which show association with reduced birth weight through the fetal genotype, have each been shown to be associated with impaired insulin release in both healthy and T2D adults,Reference Pascoe, Tura and Patel104–Reference Palmer, Goodarzi and Langefeld106 so we hypothesize that they impact on birth weight by altering insulin secretion in the fetus as well.
The heterogeneity in the impact of common T2D susceptibility genes on fetal growth suggest that there may be different pathways to beta-cell dysfunction, some of which affect fetal insulin secretion and are associated with reduced birth weight (e.g. CDKAL1). Other common variants that are not associated with birth weight may either have only a very small impact on insulin secretion and insulin action during development, or their effects may not become apparent until later in life (e.g. TCF7L2). This may lead to some interesting insights into the nature of decline in beta-cell function and the extent to which particular gene variants impact on beta-cell development in utero.
Ongoing/future studies
To date, not all of the variants identified that have been found to be associated with T2D and fasting plasma glucose variants have been tested in large numbers and efforts are being made to examine the associations of these variants with birth weight. Future work is planned to carry out meta-analyses of genome-wide association studies, and it is hoped that these studies will yield even further T2D variants. A genome-wide analysis of loci associated with birth weight is also planned, and therefore, variants identified in common with the T2D genome-wide association studies will be of interest.
Conclusion
We have presented research from rare monogenic conditions, epidemiological studies and genome-wide analysis that has provided evidence for a genetic contribution to the association between low birth weight and susceptibility to T2D in later life. The evidence to date suggests that part of this association could be explained by genes that alter beta-cell function, rather than insulin resistance, and that there may be some compensation for insulin resistance by the fetus. This genetic background should be taken into consideration when investigating the impact of environmental determinants and when developing strategies for intervention or prevention.
Acknowledgements
BS and ATH are employed as core members of the Peninsula NIHR Clinical Research Facility. RMF is funded by Sir Henry Wellcome Postdoctoral Fellowship (Wellcome Trust Grant 085541/Z/08/Z).








