



INTRODUCTION
The remarkable diversity of parasitic taxa and lifestyles makes parasitism an attractive model in which to investigate evolutionary and ecological forces (Poulin and Morand, Reference Poulin and Morand2004; Criscione et al. Reference Criscione, Poulin and Blouin2005). However, up until the past two or three decades the predominantly descriptive and systematic field of parasitology had little overlap with the more theoretical disciplines of evolutionary biology and ecology (Poulin, Reference Poulin2007). The decreasing cost of molecular genetic tools within biological research has led to an increase in both micro- and macroevolutionary studies of parasites. This has expanded our understanding of parasite speciation, co-evolution and biogeography, among others (Morand et al. Reference Morand, Krasnov and Littlewood2015). However, the representation of parasitic taxa in the evolutionary literature has not been equal, with research on certain groups lagging far behind those of medical or economic importance.
Members of the phylum Nematomorpha, or hairworms, are an example of such a group, as they are considered to be one of the most understudied taxa of parasites (Hanelt et al. Reference Hanelt, Thomas and Schmidt-Rhaesa2005). Freshwater hairworms (Gordiida) are perhaps best known for their ability to induce water-seeking behaviour in their definitive hosts (Thomas et al. Reference Thomas, Schmidt-Rhaesa, Martin, Manu, Durand and Renaud2002). Gordiids also have a unique life cycle, which contains two hosts and extends across aquatic and terrestrial habitats (Hanelt and Janovy, Reference Hanelt and Janovy2004). Adult hairworms emerge from their definitive host, typically an orthopteran, coleopteran, or mantid, into a stream or puddle where they can form tangled masses of mating individuals that are referred to as Gordian knots, the etymological root of the taxon. Larvae emerge from eggs laid by female worms after mating, and encyst in a wide range of aquatic invertebrate hosts, many of which are thought to constitute ‘dead-ends’ (Hanelt and Janovy, Reference Hanelt and Janovy2003). The transfer of the nematomorph larva to land is accomplished via infection of an intermediate host with an aquatic pre-adult life stage (e.g. ephemeropteran, plecopteran or culicid insects), which then metamorphoses, exits the aquatic environment, and is eaten by a definitive host, thus achieving trophic transmission of the parasite (Hanelt and Janovy, Reference Hanelt and Janovy2004).
Molecular studies involving hairworms have only become more common in the past 15 years, though in the majority of cases molecular data has only been used to investigate systematic relationships between phyla or within phyla closely related to Nematomorpha, with hairworm sequence data often solely serving as an outgroup in phylogenetic analyses (Aguinaldo et al. Reference Aguinaldo, Turbeville, Linford, Rivera, Garey, Raff and Lake1997; Blaxter et al. Reference Blaxter, De Ley, Garey, Liu, Scheldeman, Vierstraete, Vanfleteren, Mackey, Dorris, Frisse and Vida1998; Sørensen et al. Reference Sørensen, Hebsgaard, Heiner, Glenner, Willerslev and Kristensen2008). However, the implementation of molecular genetics in nematomorph studies shows great potential for elucidating life cycles, discovering cryptic species and investigating biogeography (Hanelt et al. Reference Hanelt, Schmidt-Rhaesa and Bolek2015; Harkins et al. Reference Harkins, Shannon, Papeş, Schmidt-Rhaesa, Hanelt and Bolek2016).
The nematomorph fauna of New Zealand is comparatively well characterized (Poinar, Reference Poinar1991, Reference Poinar and Gordon2009; Schmidt-Rhaesa, Reference Schmidt-Rhaesa2008). One marine hairworm (Nectomatida) and four gordiid species have been reported: Nectonema zealandica (Poinar and Brockerhoff, Reference Poinar and Brockerhoff2001), Gordius dimorphus (Poinar, Reference Poinar1991), Gordius paranensis (Camerano, Reference Camerano1892), Parchordodes diblastus (Örley, 1881), and E. nigromaculatus (Poinar, Reference Poinar1991) (Poinar, Reference Poinar1991; Schmidt-Rhaesa et al. Reference Schmidt-Rhaesa, Thomas and Poulin2000; Poinar and Brockerhoff, Reference Poinar and Brockerhoff2001; see Schmidt-Rhaesa, Reference Schmidt-Rhaesa2008). An additional Gordius sp. has been found in New Zealand, but could not be formally described due to the lack of reliable diagnostic characters (Schmidt-Rhaesa, Reference Schmidt-Rhaesa2008). Because of deforestation and the conversion of lowlands to agriculture, nematomorphs are mostly found in montane and subalpine areas in New Zealand, and therefore occur in isolated areas separated by unsuitable habitats for their hosts. DNA sequence data has only been obtained from G. paranensis and E. nigromaculatus, which were used in a systematic study of the phylum (Bleidorn et al. Reference Bleidorn, Schmidt-Rhaesa and Garey2002). With the exception of a sole investigation by Hanelt et al. (Reference Hanelt, Schmidt-Rhaesa and Bolek2015) into cryptic species of Gordius robustus in North America, no studies have used genetic data to examine allopatric speciation in hairworms.
Molecular tools can also shed light on intraspecific patterns of genetic variation. Indeed, the comparative population structure between host and parasite populations has become an increasingly popular topic within evolutionary parasitology in recent years (Criscione and Blouin, Reference Criscione and Blouin2004; Criscione, Reference Criscione2008; Bruyndonckx et al. Reference Bruyndonckx, Henry, Christe and Kerth2009; Keeney et al. Reference Keeney, King, Rowe and Poulin2009). The close association between host and parasite and the dependence upon host movement for parasite dispersal has led to various predicted outcomes regarding local adaptation, population structure and co-speciation (Nadler, Reference Nadler1995; Jarne and Theron, Reference Jarne and Theron2001; Gandon and Michalakis, Reference Gandon and Michalakis2002). These predictions have started to be tested, and while the results of these various studies are not always congruent, there are some general patterns beginning to emerge (Blasco-Costa and Poulin, Reference Blasco-Costa and Poulin2013; Mazé-Guilmo et al. Reference Mazé-Guilmo, Blanchet, McCoy and Loot2016). As genetic information from hairworms is largely lacking, the addition of host–parasite co-structure data from nematomorph systems should lead to a more precise understanding of evolutionary trends within parasites in general, as opposed to the current focus on a smaller subset of parasitic taxa. Given their reliance on insect hosts with limited mobility for dispersal and the fragmented nature of their habitat in New Zealand, one might expect nematomorphs to show significant genetic structure even on relatively small spatial scales.
We collected free-living adult nematomorphs from a variety of montane and subalpine locations in central Otago, South Island, New Zealand, with the aims of: (i) characterizing genetic diversity of the New Zealand nematomorph fauna, and (ii) investigating parasite population structure in the most geographically widespread species recovered. This study provides new insights into the phylogenetic relationships among hairworm taxa and the existence of potential cryptic species, as well as a discussion of comparative population structure between hairworm parasites and their orthopteran hosts.
MATERIALS AND METHODS
Nematomorph collection
Free-living, post-parasitic adult nematomorphs were collected by hand from narrow mountain streams and pools in the alpine cushionfields and subalpine tussock grasslands of five mountain ranges in Central Otago: the Rock and Pillar Range (RP), the Crown Range (CR), the Remarkables (RK), the Ida Range (ID) and the Old Man Range (OM) between February and May, 2016 (Supplementary Data S1; Fig. 1). Specimens were transported to the laboratory in containers with stream water or 70% ethanol. All specimens were placed in 70% ethanol and left overnight at 4 °C. The following day specimens were sexed and measured. For species identification by scanning electron microscopy (SEM), a ~5 mm segment of the mid-section and the posterior end were removed from all individuals and placed in a 1·5 mL Eppendorf tube containing 70% ethanol. The remaining tissue was preserved in 95% ethanol for genetic analyses. Genetic analyses and species identification through SEM were done independently from each other, i.e. their results were only compared after they were completed.

Fig. 1. Map of study area. RK, Remarkables; CR, Crown Range; ID, Ida Range; OM, Old Man Range; RP, Rock and Pillar Range.
Scanning electron microscopy
Pieces for SEM were dehydrated in an increasing ethanol series, critical point dried and coated with gold in a sputter coater. Observations were made using a LEO SEM 1524, from which digital images were taken.
DNA extraction, PCR amplification and sequencing
Genomic DNA from all nematomorphs was extracted using a modified Chelex method (Walsh et al. Reference Walsh, Metzger and Higuchi1991). For each hairworm, a ~5 mm section was removed and the cuticle was sliced longitudinally with a scalpel. The internal tissue was then scraped out and placed in 300 µL of an aqueous 5% Chelex 100 (BioRad) solution containing 0·2 mg mL−1 of proteinase K (Roche). Extractions were incubated at 55 °C overnight and then at 94 °C for 10 m to deactivate the proteinase K. Tubes were then centrifuged at 14 000 rpm for 10 m to pellet Chelex resin and undegraded cuticle. The supernatant was then transferred to a fresh 1·5 mL Eppendorf tube or 96-well plate for storage at 4 °C.
Unless otherwise stated, all polymerase chain reactions (PCRs) were performed in a final volume of 15 µL, comprising 3 µL of MyTaq™ Red 5× reaction buffer (Bioline), 0·75 µL of 10 µ m stock (75 pm) of each primer, 0·2 µL (1 U) of MyTaq™ Red DNA Polymerase, and 3 µL of extracted nematomorph genomic DNA. PCR thermal cycling profiles are provided in Supplementary Data S2. Products were purified of remaining dNTPs and oligonucleotides using the ExoSap method. Sanger sequencing by capillary electrophoresis was performed by Genetic Analysis Services, Department of Anatomy, University of Otago (Dunedin, New Zealand) or Macrogen Inc. (Seoul, Republic of Korea).
Amplification of a 709 bp partial fragment of the mitochondrial gene cytochrome c oxidase subunit 1 (CO1) was attempted for all hairworm samples. Initially, either the ‘universal’ primers LCO1498 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) (Folmer et al. Reference Folmer, Black, Hoeh, Lutz and Vrijenhoek1994), or the degenerate primers HHW-cox1-F2 (5′-TTTCWACYAATCAYAARGAYATTGG-3′) and HHW-cox1-R2 (5′-TWACTTCAGGRTGACCAAAAAAYC-3′) (Hanelt et al. Reference Hanelt, Schmidt-Rhaesa and Bolek2015) were used. Initial attempts with these primer sets resulted in poor amplification success and/or sequencing of contaminants for all but 18 specimens. The partial CO1 sequences from these 18 individuals were aligned in Geneious v8.1.8 (http://www.geneious.com, Kearse et al. Reference Kearse, Moir, Wilson, Stones-Havas, Cheung, Sturrock, Buxton, Cooper, Markowitz, Duran, Thierer, Ashton, Meintjes and Drummond2012) and the consensus sequence was used to design the degenerate primers NZHW_CO1_F (5′-DTTYTGRTCTGCHTTTTTRGG-3′) and NZHW_CO1_R (5′-RAAAAAWGAWGTDTTAAYATTTCG-3′), which amplify a 592 bp fragment of CO1. PCRs were performed as previously stated, but with 1·5 µL of 10 µ m stock (150 pm) of each primer.
A variable length fragment of the nuclear rDNA region containing partial sequence of the 3′ end of the 18S rRNA gene, internal transcribed spacer 1, 5·8S rRNA gene, internal transcribed spacer 2, and partial sequence of the 5′ end of the 28S rRNA were amplified using the primers HHW-ITS-F2 (5′-CACGCGCGCTACACTGAA-3′) and HHW-ITS-R2 (5′-GGCGCTGGACTCTTCCTATT-3′) (Hanelt et al. Reference Hanelt, Schmidt-Rhaesa and Bolek2015), or the novel primers HW_Grp5_ITS_F (5′-CTGGGTATCCGCTGAACTCC-3′) and HW_Grp5_ITS_R (5′-TAGTTTCTTTTCCTCCGCzTTATTAATATGC-3′).
Sequence processing and phylogenetics
All sequence data editing and aligning was performed using Geneious. All sequences were trimmed with the default error probability limit of 0·05, inspected by eye for incorrect base calls and ambiguities, and then edited manually. Alignments were created using the MAFFT algorithms (Katoh and Standley, Reference Katoh and Standley2013). For ribosomal regions, the specific MAFFT algorithm was selected using the ‘Auto’ function. Determination of boundaries of the various ribosomal fragments was performed by comparing regions of homology, and the sequences were annotated as such. For CO1, the ‘Translation Align’ feature was used, implementing the BLOSUM62 scoring matrix (Henikoff and Henikoff, Reference Henikoff and Henikoff1992). Sequences were inspected further to check for obvious incorrect base calls. Pairwise distance matrices of CO1 sequence data were created in MEGA v7 (Kumar et al. Reference Kumar, Stecher and Tamura2016) using the Kimura 2-parameter (K2P) measure of genetic divergence (Kimura, Reference Kimura1980), calculating the distance between each specimen as well as mean intra- and interspecific distances. For phylogenetic analyses, model selection and optimal partitioning schemes were determined using PartitionFinder v2.0.0 (pre-release 14) (Lanfear et al. Reference Lanfear, Calcott, Ho and Guindon2012), which utilizes the software PhyML v3.0 (Guindon et al. Reference Guindon, Dufayard, Lefort, Anisimova, Hordijk and Gascuel2010) and allows the user to restrict model testing to those implemented in MrBayes. The corrected Aikake information criterion (AICc) was used for evaluating the likelihood of the proposed models and partitioning schemes. For CO1, the optimal partitioning scheme consisted of one partition containing all codon positions. Phylogenetic tree building was performed with MrBayes v3.2.6 (Ronquist and Huelsenbeck, Reference Ronquist and Huelsenbeck2003) and RAxML v8 (Stamatakis, Reference Stamatakis2014), and was performed remotely using the CIPRES Science Gateway (Miller et al. Reference Miller, Pfeiffer and Schwartz2010). For Bayesian analyses of 18S and CO1, PartitionFinder selected the models SYM + I + Γ and GTR + I + Γ, respectively. Two runs, each consisting of four Markov chains, were allowed to run for 10 000 000 generations and were sampled every 1000. A chain heating parameter of 0·04 was used. The first 2500 trees (25%) were discarded as burn-in and posterior probabilities were obtained from a majority-rule consensus. Tracer v1.6 (Drummond and Rambaut, Reference Drummond and Rambaut2007) was used to assess convergence of the two runs and ensure high effective sample size. For maximum likelihood, the RAxML default model of GTR + Γ was used for all markers. Bootstrap support values were obtained using the rapid bootstrap method with 1000 replicates. Outgroups (two nematode species) and nematomorph reference sequences used in phylogenetic analyses were obtained from GenBank, and can be found in Supplementary Data S3. All sequences from this study are available on GenBank; accession numbers are provided in Supplementary Data S4.
Population genetics
Arlequin v3.5.2.2 (Excoffier and Lischer, Reference Excoffier and Lischer2010) was used to calculate the number of haplotypes (N h), number of polymorphic sites (N p), the haplotype diversity (h) and nucleotide diversity (π) for both markers for each hairworm species. For the most spatially widespread species, E. nigromaculatus, statistical parsimony networks of the CO1 and ITS data were created with the software PopArt (Leigh and Bryant, Reference Leigh and Bryant2015) using the TCS method (Clement et al. Reference Clement, Posada and Crandall2000). Clustering of individual haplotypes into groups was performed with BAPS v6.0 (Corander et al. Reference Corander, Waldmann and Sillanpää2003) with the number of estimated populations set to 2 ⩽ k ⩾ 20. For each k value, five independent runs were performed. To test for population differentiation of E. nigromaculatus, a single-level AMOVA (i.e. specimens from the same range treated as a distinct population with no higher-level grouping) (Excoffier et al. Reference Excoffier, Smouse and Quattro1992) was performed in Arlequin, implementing the TrN + Γ model of nucleotide substitution with a shape parameter α = 0·021 for each marker, as determined by jModelTest2 (Darriba et al. Reference Darriba, Taboada, Doallo and Posada2012). Significance of AMOVA results was determined by 10 000 random permutations.
RESULTS
Collections and morphology
In total, 87 nematomorphs were collected (Table 1). With the exception of G. dimorphus, morphological identification by SEM revealed all previously reported New Zealand hairworms. Euchordodes nigromaculatus was the most commonly encountered species, with a total of 34 individuals from three of the five ranges, RK, RP and OM. This species has polygonal cuticular areoles and is the only New Zealand species in which males have an undivided posterior end (Fig. 2A and B). Parachordodes diblastus was found at three locations, the CR, RP and OM, but was most heavily represented at CR, where it comprised 12 of the 13 collected individuals. Parachordodes diblastus can be identified by its elevated dark superareoles (Fig. 2C and D), which are characteristic of this genus. Gordius paranensis was collected from the ID and RP in low numbers. This species is identified by its smooth cuticle and the presence of a semicircular pre-cloacal row of bristles, in addition to the post-cloacal crescent that is characteristic of the genus (Fig. 2E and F). Individuals of a previously reported but undescribed Gordius sp. were also collected (Schmidt-Rhaesa, Reference Schmidt-Rhaesa2008). This species displays a post-cloacal crescent but lacks the pre-cloacal bristles characteristic of G. paranensis (Fig. 2G and H). In addition to the previously reported New Zealand species, seven individuals of an undescribed species were also found (Fig. 2I and J). This species exhibits areoles surrounded by fine bristles and possesses a row of large bristles on the ventral side of the posterior end in males. These characters are typical of the genus Gordionus. A formal description of this species will be provided elsewhere. One specimen (OM 10) could not be identified by SEM or molecular methods.
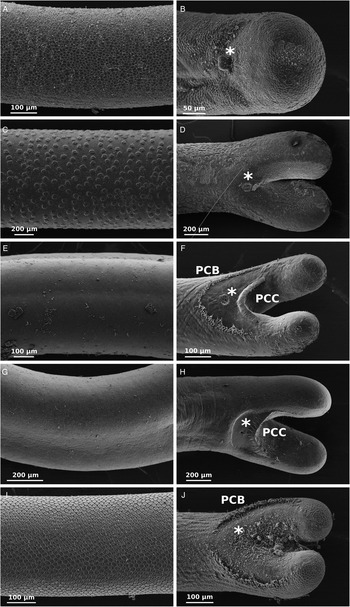
Fig. 2. SEM of nematomorph mid-body cuticular features (A, C, E, G, I) and male posterior ends (B, D, F, H, J). (A, B), Euchordodes nigromaculatus. (C, D), Parachordodes diblastus. (E, F), Gordius paranensis. (G, H), undescribed Gordius sp. (I, J), Gordionus n. sp. *=cloaca (in B, sperm is coming out of the cloaca), PCB, precloacal bristles; PCC, postcloacal crescent.
Table 1. Summary of nematomorph collections by species, sex and location

Mean lengths of each hairworm species are provided in Supplementary Data S5, in which data only from intact specimens of known sex are included. The longest specimen was a female Gordius sp. at 565 mm.
DNA sequences and phylogenetics
Partial CO1 sequences were successfully obtained from 71 of the 87 hairworms. The partial 18S/ITS1/5·8S/ITS2/partial 28S rDNA region was amplified and sequenced from 83 hairworms, although not all sequences were complete due to insufficient read length. Some sequences were not entirely of high quality due to long stretches of ambiguities in the ITS regions, likely driven by the bi-allelic nature of nuclear rDNA. Thus, not all of the rDNA sequences could be used for subsequent population genetic analyses. Of the 83 rDNA sequences, all but two contained high-quality sequence data for the 3′ end of the 18S rRNA gene. These 81 sequences were aligned with available nematomorph 18S reference sequences and nematode outgroups, creating a 200 bp alignment for phylogenetic analysis. The final alignment for CO1 consisted of 519 bp, with Paragordius spp. selected as outgroups based on the results of the 18S tree.
Bayesian and maximum-likelihood (ML) methods yielded near identical topologies for both the 18S (Fig. 3) and CO1 (Fig. 4) datasets. Therefore, the Bayesian trees are shown with posterior probabilities and bootstrap support values at branch nodes. Separate Bayesian and ML trees can be found in Supplementary Data S6–S9. Exclusion of the third codon position of CO1 did not significantly affect the topology (data not shown). The results of the phylogenetic trees are in complete agreement with the morphological identifications by SEM. With the exception of the displacement of the genus Paragordius, both trees support the traditional division of Gordiida into Gordiidae and Chordodidae. This is most clearly observed in the CO1 tree, which shows a well-supported bifurcation, with one branch containing only members of the genus Gordius and the other containing representatives from all of the remaining included genera. In the 18S tree both Gordionus alpestris and the putative Gordionus n. sp. from the present study do not form a monophyletic clade with the other Gordionus species. This can also be observed in the CO1 tree, although there is no available CO1 data for G. alpestris. Both trees support the division of Gordiidae into two groups, one containing G. paranensis and the other containing the unidentified Gordius sp. from this study. However, the G. paranensis 18S reference sequence is found on the branch opposite from those collected during this study. Furthermore, both trees indicate the presence of two distinct clades within the G. paranensis individuals from this study. Strangely, in the CO1 tree these two clades are separated by an unidentified Gordius sp. from New Mexico.
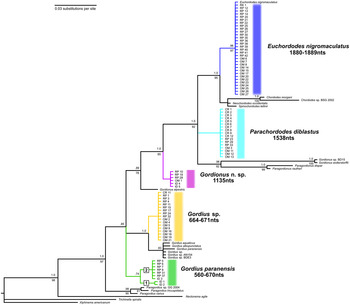
Fig. 3. Composite ML/Bayesian inference phylogenetic tree for 18S rRNA gene. Length of ITS 1, 5·8S, ITS2 region listed below species names from this study. Boxed numbers indicate two sub-clades of Gordius paranensis from this study.
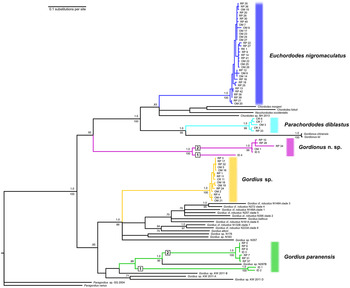
Fig. 4. Composite ML/Bayesian inference phylogenetic tree for cytochrome oxidase subunit 1 (CO1) gene. Boxed numbers indicate two sub-clades of Gordius paranensis and of Gordionus n. sp. from this study.
The rDNA region displayed substantial variation in length between the five species (Fig. 3). The longest ITS1/5·8S/ITS2 region was that of E. nigromaculatus, at 1880–1889 nts, whereas the shortest was that of G. paranensis clade 2, at 560 nts. Interestingly, there was a trend towards longer rDNA region length in species that have diverged more recently, based on the 18S tree.
CO1 sequence data for specimens OM 12 and RP 15 were excluded from phylogenetic analyses and calculations of pairwise genetic distance, as they were presumed to be nuclear pseudogene copies. The CO1 data for OM 12 contained many ambiguities, one of which led to a potential early stop codon. The CO1 data for RP 15 also contained many ambiguities and was very distant from other members of the Gordius sp. This coupled with the observation that the ribosomal data for this individual was highly similar to those of the other individuals of this species supports the conclusion that it is a pseudogene (data not shown). Calculations of pairwise K2P genetic distances were performed treating G. paranensis clades 1 and 2 as separate taxa (Table 2). Additionally, specimen ID 4 was treated as belonging to a separate clade of Gordionus n. sp., clade 1, due to its high degree of sequence divergence, as evidenced by its long branch length in the CO1 tree. The CO1 sequence of this specimen is unlikely to be a pseudogene due to the high quality of sequence data and additional observed differences in ribosomal sequence data and morphology. The ribosomal sequence of this specimen had numerous insertions/deletions in the ITS regions, as well as many polymorphisms in the 5·8S rRNA gene (data not shown). Mean intraspecific distance ranged from 0·01 in G. paranensis clade 1 to 0·044 in G. paranensis clade 2. Mean interspecific distance among all clades was 0·328 ± 0·040. The range of intraspecific (0·002–0·059) and interspecific (0·206–0·373) distances did not overlap, yielding a so-called ‘barcoding gap’ of 0·147.
Table 2. Mean CO1 K2P genetic distances of nematomorph species/clades

Mean intraspecific distance on diagonal, mean interspecific below.
Population genetics
The final dataset for analyses of population genetics was composed of 68 CO1 sequences and 66 full-length ITS1/5·8S/ITS2 sequences. Standard indices of genetic diversity for each species and sampling sites can be found in Table 3. Due to the broad range of species collected, the number of individuals of a particular species at a particular site was often low, limiting the resolution of the diversity estimates. For most species there was substantial genetic variation in both CO1 and the ITS1/5·8S/ITS2 region. In all cases nucleotide diversity (π) was higher for CO1 than for the ribosomal region. The overall number of unique haplotypes of CO1 and ITS1/5·8S/ITS2 was 54 and 45, respectively. No reliable full-length ITS1/5·8S/ITS2 sequence data were obtained from Gordionus n. sp. clade 2.
Table 3. Standard population genetic indices of nematomorph species/clades of both markers

bp, length of fragment in base pairs; n, number of individual sequences; N h, number of haplotypes; N p, number of polymorphic sites; h, haplotype diversity; π, nucleotide diversity.
Due to the greater numbers in which E. nigromaculatus was collected, this species was selected for further analysis. Two statistical parsimony networks were created to show the relationships among CO1 and ITS haplotypes (Fig. 5A and B). Twenty-six of the 34 CO1 haplotypes and 20 of the 23 ribosomal haplotypes were unique. Optimal BAPS clustering yielded three haplogroups for both the CO1 and ribosomal datasets. BAPS clustering solutions were generally inconsistent between the two markers, as the corresponding haplogroups did not tend to contain the same individuals. Proportions of haplogroups represented at each site (Fig. 5C and D) illustrate the general lack of population structure. AMOVA tests of both datasets confirmed the lack of population structure. Fixation indices ΦST for CO1 and the ribosomal region were quite low (0·036 and 0·030, respectively) and were not significant (P = 0·2745 and 0·2604, respectively).

Fig. 5. Haplotypic graphics for Euchordodes nigromaculatus. (A, B), statistical parsimony networks of CO1 (A) and ITS1/5·8S/ITS2 (B) haplotypes. Hatch marks indicate number of mutational steps. Coloured boxes indicate haplogroups identified by BAPS. (C, D), proportion of haplogroup representation at each sampling location for CO1 (C) and ITS1/5·8S/ITS2 (D).
DISCUSSION
With the exception the 18S rRNA genes of G. paranensis and E. nigromaculatus (Bleidorn et al. Reference Bleidorn, Schmidt-Rhaesa and Garey2002), no sequence data had been published for New Zealand hairworms. In this study, we present the first mitochondrial and nuclear ribosomal sequence data for P. diblastus and a previously reported but undescribed Gordius sp. (Schmidt-Rhaesa, Reference Schmidt-Rhaesa2008), as well as contribute mitochondrial sequences for those species already sequenced for 18S. Individuals assigned to the genus Gordionus were also found and molecularly characterized. This brings the total number of New Zealand gordiid species to six. We developed two novel primer sets, which may prove useful for future molecular studies of hairworms. More importantly, results from this study have implications for the systematics of the phylum Nematomorpha and the potential for cryptic species, which will be addressed below. We also compare the population structures of E. nigromaculatus and its potential hosts using previously published data, and provide a discussion of possible mechanisms for reduced parasite population differentiation.
Nematomorph systematics and cryptic species
The results of the phylogenetic analyses are largely in agreement with previously published phylogenies within Nematomorpha. One noticeable difference, however, is the placement of the genus Paragordius. Our results indicate that Paragordius is basal to all remaining freshwater nematomorphs, whereas previous studies placed the genus within the family Chordodidae (Bleidorn et al. Reference Bleidorn, Schmidt-Rhaesa and Garey2002; Efeykin et al. Reference Efeykin, Schmatko and Spiridonov2016), although this was not always well supported (Bleidorn et al. Reference Bleidorn, Schmidt-Rhaesa and Garey2002). This discrepancy could be due in part to the relatively small size of the 18S alignment. However, this is unlikely since initial phylogenetic reconstructions using nematode outgroups for the CO1 dataset yielded the same result (data not shown). Additionally, a phylogenetic tree by Sato et al. (Reference Sato, Watanabe, Tamotsu, Ichikawa and Schmidt-Rhaesa2012) placed Paragordius outside of Gordiidae and Chordodidae, although the tree is unrooted and therefore whether or not Paragordius is basal cannot be determined. This disagreement should be resolved as sequence data becomes available from additional nematomorph genera.
The presence of two well-supported clades within the G. paranensis specimens from this study suggests the existence of cryptic species. Hanelt et al. (Reference Hanelt, Schmidt-Rhaesa and Bolek2015) similarly found evidence of nematomorph cryptic species during an investigation of genetic lineages of Gordius cf. robustus across the contiguous USA, in which they argued the case for eight distinct species. They found a minimum among-clade CO1 K2P distance of 0·06; the distance between G. paranensis clades 1 and 2 of 0·306 is well above that threshold. Furthermore, the lengths of the ITS1/5·8S/ITS2 region in these two clades differ by 110 bp, much greater than the intraspecific differences observed in other species from the current study and among the eight G. cf. robustus lineages. Gordius paranensis has also been reported from Chile, Paraguay and Brazil, where it was originally described (Camerano, Reference Camerano1892, Reference Camerano1894; Montgomery, Reference Montgomery1898; De Villalobos et al. Reference De Villalobos, Zanca and Ibarra-Vidal2005). Given the findings of the current study, it is possible that South American individuals represent cryptic species within G. paranensis as well, though there is no available genetic data for specimens from that region. In both the CO1 and 18S phylogenies, the genus Gordius was found to comprise two main clades, one containing the G. paranensis specimens from this study along with undescribed Gordius spp. from Japan (Sato et al. Reference Sato, Watanabe, Tamotsu, Ichikawa and Schmidt-Rhaesa2012), and the other containing all remaining Gordius spp. One obvious inconsistency in the 18S tree regarding the position of G. paranensis is the displacement of individuals from the current study from the sequence of a New Zealand G. paranensis specimen included in Bleidorn et al. (Reference Bleidorn, Schmidt-Rhaesa and Garey2002). If G. paranensis is a monophyletic group, it is difficult to explain how a true G. paranensis sequence could be included in a clade of Gordius spp. that lack pre-cloacal bristles. The most parsimonious explanation for this contradiction is that the specimen for that study was not properly identified, although it cannot be ruled out that pre-cloacal bristles may have evolved independently in separate lineages. This alternative scenario is supported by the separation of the two G. paranensis clades in the CO1 tree by an unidentified Gordius sp. from New Mexico (Hanelt et al. Reference Hanelt, Schmidt-Rhaesa and Bolek2015). Although images of this specimen could not be obtained, it can be assumed that it lacked pre-cloacal bristles since the presence of this trait would have identified it as G. paranensis or G. difficilis, which also possesses pre-cloacal bristles but has cuticular areoles, instead of an undescribed species (Montgomery, Reference Montgomery1898; Bolek and Coggins, Reference Bolek and Coggins2002). This suggests that in addition to the potential of this trait having evolved independently more than once, it can also be subsequently lost in certain lineages. The addition of genetic data for G. difficilis and the newly described G. jorriti, which also possesses a pre-cloacal row of bristles, would shed light upon the potential multiple evolution and loss of this character (Schmidt-Rhaesa and Schwarz, Reference Schmidt-Rhaesa and Schwarz2016). The plasticity of cuticular features in nematomorphs has been illustrated previously by Hanelt et al. (Reference Hanelt, Schmidt-Rhaesa and Bolek2015). They found that two Gordius spp. with distinct areolar traits clustered within the Gordius cf. robustus species complex, which was classically identified by its lack of areoles. Together these findings could have implications for the interpretation of cuticular features in species delimitation for nematomorphs.
Seven specimens were identified by SEM as belonging to Gordionus, a genus new to New Zealand. However, monophyly of these specimens with other Gordionus sequences was not observed in either the CO1 or 18S trees. Schmidt-Rhaesa (Reference Schmidt-Rhaesa and Schmidt-Rhaesa2013) previously suggested that Gordionus may not comprise a monophyletic group due to inconsistencies within the genus and similarities to other genera regarding its supposedly characteristic areolar pattern. A revision of this genus may be necessary, but is outside the scope of this study. This issue will be addressed in a forthcoming formal species description of the specimens reported herein. From a morphological perspective, it is conceivable that slight differences in the areoles between Gordionus n. sp. clades 1 and 2 (data not shown) are an example of intraspecific variation, as noted elsewhere for this genus (Schmidt-Rhaesa, Reference Schmidt-Rhaesa2001). However, the genetic distance (0·206) is much greater than that used for species delimitation in the genus Gordius (see above), despite being the smallest observed inter-clade distance in the present study. Therefore, we conclude that these two clades represent distinct species. Unfortunately, only one specimen was found for Gordionus n. sp. clade 1, and it was not intact, preventing a comparison of the male posterior end.
Both CO1 and ribosomal molecular data support the expected placement of P. diblastus within the family Chordodidae but outside the subfamily Chordodinae, which includes only species without bilobed male posterior ends (Heinze, Reference Heinze1935; Bleidorn et al. Reference Bleidorn, Schmidt-Rhaesa and Garey2002; Schmidt-Rhaesa, Reference Schmidt-Rhaesa and Schmidt-Rhaesa2013). Euchordodes nigromaculatus was confirmed as belonging to Chordodinae using both mitochondrial and ribosomal datasets as shown by Bleidorn et al. (Reference Bleidorn, Schmidt-Rhaesa and Garey2002) and as classically defined due to its lack of tail lobes.
Lack of host–parasite co-structure
Tests of genetic differentiation of E. nigromaculatus using both mitochondrial and nuclear data failed to demonstrate even low levels of population structure. Because hairworms in this study were all collected in the post-parasitic adult stage, no host genetic data were obtained and therefore a direct comparison between host and parasite population structures could not be undertaken. There are, however, numerous previously published studies investigating the phylogeographical patterns of montane and subalpine insects in New Zealand (see Trewick et al. Reference Trewick, Wallis and Morgan-Richards2011). While there are only two confirmed definitive hosts for E. nigromaculatus, the wētā Deinacrida parva (Orthoptera: Anostostomatidae) and the grasshopper Sigaus australis (Poinar, Reference Poinar1991; Poulin, Reference Poulin1995), it is presumed that E. nigromaculatus infects a broader range of New Zealand orthopterans (Poinar, Reference Poinar1991; Thomas et al. Reference Thomas, Schmidt-Rhaesa and Poulin1999). The wētā species Deinacrida connectens and Hemideina maori are both well-studied taxa from a phylogeographical perspective (Trewick et al. Reference Trewick, Wallis and Morgan-Richards2000; Trewick and Wallis, Reference Trewick and Wallis2001; King et al. Reference King, Kennedy and Wallis2003; King, Reference King2015). Both species have been shown to display a substantial level of regional population structure, with distinct haplotype groups occurring in neighbouring ranges, even over relatively small spatial scales (~50–100 km). Genetically distinct lineages of D. connectens among nearby ranges in Central Otago and across the South Island were identified by both single-stranded conformation polymorphism and sequencing of CO1 (Trewick et al. Reference Trewick, Wallis and Morgan-Richards2000; Trewick and Wallis, Reference Trewick and Wallis2001). Using CO1 sequence data, King (Reference King2015) similarly found genetic differentiation of H. maori between populations in the RP and the Black Umbrella Range, which is within the same larger mountain range as the OM, two locations from which E. nigromaculatus specimens were collected for the current study. Trewick (Reference Trewick2008) also found differentiated lineages of S. australis populations between these two ranges. Allopatric speciation of members of the endemic cockroach genus Celatoblatta among South Island ranges has also been demonstrated (Trewick and Wallis, Reference Trewick and Wallis2001; Chinn and Gemmell, Reference Chinn and Gemmell2004), with this cockroach genus being a known host taxon of New Zealand hairworms (Zervos, Reference Zervos1989).
A frequent assumption in studies of parasite population structure is that it should mirror that of their hosts and that, in general, parasite populations should be more differentiated (Jarne and Theron, Reference Jarne and Theron2001). This is based on the concept that the dispersal potential of a parasite should be dictated by its most mobile host (Prugnolle et al. Reference Prugnolle, Liu, De Meeûs and Balloux2005a ), and that the low effective population sizes and short generation times typical of many parasites render them more susceptible to genetic drift (Gandon and Michalakis, Reference Gandon and Michalakis2002; Criscione and Blouin, Reference Criscione and Blouin2005; Prugnolle et al. Reference Prugnolle, Théron, Pointier, Jabbour-Zahab, Jarne, Durand and Meeûs2005b ). Varying levels of parasite population structure as a result of differential host vagility has been demonstrated in several systems (McCoy et al. Reference McCoy, Boulinier, Tirard and Michalakis2003; Criscione and Blouin, Reference Criscione and Blouin2004; Blasco-Costa et al. Reference Blasco-Costa, Waters and Poulin2012). For many parasite species with complex life cycles the most mobile host is the final host, especially in the case of parasites of vertebrates. New Zealand hairworm dispersal, however, may depend more on intermediate hosts, as flightless alpine insects such as acridid grasshoppers and wētā are known to have limited dispersal potential (White, Reference White1978; Leisnham and Jamieson, Reference Leisnham and Jamieson2002). Gordiid larvae can encyst in a wide variety of aquatic organisms, although many of these probably constitute ‘dead-end’ hosts (Hanelt and Janovy, Reference Hanelt and Janovy2003, Reference Hanelt and Janovy2004). Suitable intermediate hosts must be able to provide a bridge between aquatic and terrestrial habitats, and may include midges, mayflies and stoneflies (Hanelt and Janovy, Reference Hanelt and Janovy2003). While no intermediate hosts of New Zealand hairworms have been experimentally confirmed, Winterbourn (Reference Winterbourn2005) found that adult stoneflies of the species Spaniocerca zelandica and Cristaperla fimbria exhibit a high prevalence of infection (69–90%) by nematomorph larvae. Winterbourn and Pohe (Reference Winterbourn and Pohe2016) subsequently found nematomorph larvae in two species of another New Zealand stonefly genus, Stenoperla. Previous studies of stonefly dispersal have demonstrated that adult stoneflies can travel hundreds or even thousands of metres along and between stream corridors (Briers et al. Reference Briers, Gee, Cariss and Geoghegan2004; MacNeale et al. Reference MacNeale, Peckarsky and Likens2005). Correspondingly, phylogeographical analyses of two of the aforementioned New Zealand stonefly genera found little to no population differentiation/allopatric speciation over the geographical range of the current study (McCulloch et al. Reference McCulloch, Wallis and Waters2010). These findings together suggest that the dispersal capabilities of the potential intermediate hosts of E. nigromaculatus may be a factor in its observed lack of population structure.
However, intermediate host dispersal likely is not the only reason for this lack of population structure, for despite the stonefly's impressive flying ability, it is not sufficient to transport gordiid larvae directly between mountain ranges. Instead, dispersal of nematomorph lineages on this scale is likely a multi-generational process. Passive dispersal of nematomorph eggs or pre-parasitic larvae coupled with a lack of definitive host specificity may be one such avenue. Several species of hairworms deposit egg strings on debris (Hanelt and Janovy, Reference Hanelt and Janovy2002; Bolek et al. Reference Bolek, Schmidt-Rhaesa, Hanelt and Richardson2010), and these could be carried far downstream, as larval emergence can occur months after laying (Müller, Reference Müller1926; Schmidt-Rhaesa, Reference Schmidt-Rhaesa and Schmidt-Rhaesa2013). Free-living larvae could be similarly transported, as they remain infective for weeks (Hanelt et al. Reference Hanelt, Thomas and Schmidt-Rhaesa2005). Once downstream, a larva could infect an aquatic invertebrate and subsequently be transmitted to a lowland orthopteran definitive host. While we only found E. nigromaculatus individuals at elevations above 1300 m, this is likely a result of bias in sampling effort, as specimens of this species occur at much lower elevations (Poinar, Reference Poinar1991). After emerging from this lowland definitive host, the parasite could then complete its life cycle and its offspring subsequently infect another intermediate host. As aquatic insects with flying adult stages disperse upstream as a means of combating the so-called ‘drift paradox’ (Müller, Reference Müller1954; Hershey et al. Reference Hershey, Pastor, Peterson and Kling1993; Pachepsky et al. Reference Pachepsky, Lutscher, Nisbet and Lewis2005), hairworm lineages could use this host trait to spread upstream. This process could progress in a stepping-stone manner (Kimura and Weiss, Reference Kimura and Weiss1964) through multiple generations as seen in other parasite systems (Dybdahl and Lively, Reference Dybdahl and Lively1996; McCoy et al. Reference McCoy, Boulinier and Tirard2005), maintaining sufficient levels of gene flow between alpine hairworm populations to prevent population structuring. Surely more studies of New Zealand nematomorphs characterizing the complete breadth of their intermediate hosts are necessary before any definitive conclusions are made regarding the influence of intermediate host movement on parasite population structure (or lack thereof).
The absence of congruence between the population structure of E. nigromaculatus and its potential definitive hosts stands in contrast to a once prevalent paradigm, which presumes that: (1) host mobility is the main determinant of parasite population structure; (2) population structure of parasites should mirror that of their hosts; and (3) overall levels of differentiation should be higher for parasite than for host (Price, Reference Price1980; Jarne and Theron, Reference Jarne and Theron2001). Recent empirical studies and meta-analyses have highlighted the importance of considering other factors in the determination of host–parasite co-structure (Bruyndonckx et al. Reference Bruyndonckx, Henry, Christe and Kerth2009; Keeney et al. Reference Keeney, King, Rowe and Poulin2009; Blasco-Costa and Poulin, Reference Blasco-Costa and Poulin2013; Couchoux et al. Reference Couchoux, Seppä and van Nouhuys2016; Mazé-Guilmo et al. Reference Mazé-Guilmo, Blanchet, McCoy and Loot2016). Bruyndonckx et al. (Reference Bruyndonckx, Henry, Christe and Kerth2009) showed that while the mobility of the bat host is responsible for gene flow between populations of its tick parasite Spinturnix bechsteini, their population structures did not reflect one another because parasite transmission between populations did not occur during bat mating, and therefore did not result in gene flow between host populations. Their results also emphasize the influence of parasite demographic factors, such as population bottlenecks, in generating population structure. Keeney et al. (Reference Keeney, King, Rowe and Poulin2009) found contrasting levels of population structure between two trematode species that both use the same marine snail as an intermediate host and shorebirds as definitive hosts. This difference was attributed in part to differences in infection intensity as well as the use of different tissues within the definitive host, which may affect transmission rates. Meta-analyses have emphasized the impact of other factors such as the per cent of free-living stages in a parasite's life cycle, parasite sexual mode and geographical range on the outcome of studies of host–parasite co-structure (Blasco-Costa and Poulin, Reference Blasco-Costa and Poulin2013; Mazé-Guilmo et al. Reference Mazé-Guilmo, Blanchet, McCoy and Loot2016). These studies and others have remodelled our understanding of the relative influence of certain aspects of parasite life-history, ecology and demography on the genetic composition of parasite populations, and provide an interesting lens through which to view the results of the current study.
SUPPLEMENTARY MATERIAL
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182017000233.
ACKNOWLEDGEMENTS
We thank Tony Stumbo and Brenah Hearne for assistance with sampling, Tania King for support with molecular work, Fátima Jorge for advice on phylogenetic analyses, Renate Walter for assistance with SEM work, and members of the University of Otago Evolutionary and Ecological Parasitology Research Group for helpful comments during manuscript preparation.
FINANCIAL SUPPORT
This research was funded by internal grants to ZJCT and RP from the Department of Zoology, University of Otago. AKY participated as an Overseas Research Associate of the Department of Biotechnology, Govt. of India at University of Hamburg.









