INTRODUCTION
Representatives of the genus Leishmania cause a variety of clinical syndromes, ranging from self-healing cutaneous forms (CL) through to muco-cutaneous (MCL) and lethal visceral diseases (VL). The leishmaniases are transmitted via the bite of infected female sand flies of different Phlebotomus and Lutzomyia species and are widespread over the tropical and subtropical regions around the world. At least 21 species of Leishmania have been recorded as being infective to humans (WHO, 1990). There is no absolute correlation between a particular clinical form and causative species (Lainson and Shaw, Reference Lainson, Shaw, Cox, Kreier and Wakelin1998). Generally, parasites of the L. donovani complex are associated with VL in the Old and New Worlds but have also been found to cause CL. On the other hand, cases of VL have occasionally been attributed to species usually recognized as causative agents of CL, such as L. tropica (see Sacks et al. Reference Sacks, Kenney, Kreutzer, Jaffe, Gupta, Sharma, Sinha, Neva and Saran1995).
The current classification of Leishmania is still based on isoenzyme typing by using multilocus enzyme electrophoresis (MLEE), as reviewed by Schönian et al. (Reference Kuhls, Chicharro, Canavate, Cortes, Campino, Haralambous, Soteriadou, Pratlong, Dedet, Mauricio, Miles, Schaar, Ochsenreither, Radtke and Schönian2008). This approach has been the most widely used technique during the past 25 years for the identification of Leishmania to species and subspecific levels. The MLEE methods used in Europe and in South America are, however, based on different enzyme panels and cannot be compared directly (Rioux et al. Reference Rioux, Lanotte, Serres, Pratlong, Bastien and Perieres1990; Cupolillo et al. Reference Cupolillo, Grimaldi and Momen1994). In addition, MLEE has several limitations. Molecular studies have shown that differences in electrophoretic mobilities can be simply due to heterozygosity at a single nucleotide position (Jamjoom et al. Reference Jamjoom, Ashford, Bates, Chance, Kemp, Watts and Noyes2004) or are not a consequence of nucleotide diversity of the particular gene (Mauricio et al. Reference Mauricio, Yeo, Baghaei, Doto, Pratlong, Zemanova, Dedet, Lukes and Miles2006; Zemanova et al. Reference Zemanova, Jirku, Mauricio, Horak, Miles and Lukes2007). On the other hand, indistinguishable zymodeme phenotypes have been shown to be produced by distinct genotypes (Mauricio et al. Reference Mauricio, Yeo, Baghaei, Doto, Pratlong, Zemanova, Dedet, Lukes and Miles2006; Alam et al. Reference Alam, Haralambous, Kuhls, Gouzelou, Sgouras, Soteriadou, Schnur, Pratlong and Schönian2009a). Consequently, other molecular studies do not always agree with the classification of Leishmania parasites by MLEE. For instance, the existence of 3 visceralizing species in East Africa, namely L. donovani, L. infantum and L. archibaldi, was not supported by analysis with many different molecular markers (Lukes et al. Reference Lukes, Mauricio, Schönian, Dujardin, Soteriadou, Dedet, Kuhls, Tintaya, Jirku, Chocholova, Haralambous, Pratlong, Obornik, Horak, Ayala and Miles2007), a separate species status was not confirmed for L. killicki (Schönian et al. Reference Schönian, Schnur, El Fari, Oskam, Kolesnikov, Sokolowska-Kohler and Presber2001; Schwenkenbecher et al. Reference Schwenkenbecher, Wirth, Schnur, Jaffe, Schallig, Al-Jawabreh, Hamarsheh, Azmi, Pratlong and Schönian2006), and the L. donovani zymodeme MON-37 was assigned to strains of different genetic background (Alam et al. Reference Alam, Haralambous, Kuhls, Gouzelou, Sgouras, Soteriadou, Schnur, Pratlong and Schönian2009a). The discriminatory power of MLEE for classifications below species level is limited because most of the L. infantum parasites causing visceral leishmaniasis in the Mediterranean and South America belong to the same zymodeme MON-1. Other important drawbacks of MLEE are that it requires bulk cultures of parasites, it is labour intensive and time-consuming, and it can only be performed in specialized laboratories.
Leishmaniasis is characterized not only by considerable clinical pleomorphism but also by varying epidemiology due to the remarkable diversity of Leishmania species and their vectors and, where applicable, their reservoir hosts. Recently, changes in the epidemiology of the leishmaniases have been reported from different endemic areas in the Old and New World. This includes amongst others, the emergence of new endemic foci (Karunaweera et al. Reference Karunaweera, Pratlong, Siriwardane, Ihalamulla and Dedet2003; Rhajaoui et al. Reference Rhajaoui, Nasereddin, Fellah, Azmi, Amarir, Al-Jawabreh, Ereqat, Planer and Abdeen2007; Sharma et al. Reference Sharma, Mahajan, Ranjan, Verma, Negi and Mehta2009): the spread of parasites to new areas (Jacobson et al. Reference Jacobson, Eisenberger, Svobodova, Baneth, Sztern, Carvalho, Nasereddin, El Fari, Shalom, Volf, Votypka, Dedet, Pratlong, Schönian, Schnur, Jaffe and Warburg2003; Capelli et al. Reference Capelli, Baldelli, Ferroglio, Genchi, Gradoni, Gramiccia, Maroli, Mortarino, Pietrobelli, Rossi and Ruggiero2004; Nasereddin et al. Reference Nasereddin, Baneth, Schönian, Kanaan and Jaffe2005); the sympatric presence of multiple species of Leishmania with overlapping clinical pictures (Al-Jawabreh et al. Reference Al-Jawabreh, Schnur, Nasereddin, Schwenkenbecher, Abdeen, Barghuthy, Khanfar, Presber and Schönian2004; Sharma et al. Reference Sharma, Mahajan, Kanga, Sood, Katoch, Mauricio, Singh, Parwan, Sharma and Sharma2005); the identification of new parasite-vector associations (Svobodova et al. Reference Svobodova, Votypka, Peckova, Dvorak, Nasereddin, Baneth, Sztern, Kravchenko, Orr, Meir, Schnur, Volf and Warburg2006a), indications of anthroponotic transmission of L. infantum (Svobodova et al. Reference Svobodova, Alten, Zidkova, Dvorak, Hlavackova, Myskova, Seblova, Kasap, Belen, Votypka and Volf2009) and of zoonotic transmission for L. tropica (Svobodova et al. Reference Svobodova, Volf and Votypka2006b); and the isolation of new as yet unclassified members of the genus Leishmania from CL cases (Noyes et al. Reference Noyes, Pratlong, Chance, Ellis, Lanotte and Dedet2002; Villinski et al. Reference Villinski, Klena, Abbassy, Hoel, Puplampu, Mechta, Boakye and Raczniak2008). The (re-)emergence and spread of these diseases observed worldwide have been attributed to 3 main risk factors: (i) environmental changes of human origin, (ii) immunosuppression, and (iii) treatment failure and drug resistance (Dujardin, Reference Dujardin2006). A sound and consensual taxonomical background based on the knowledge of the population structure and phylogenetic diversity is needed for a better understanding of these epidemiological changes (Banuls et al. Reference Banuls, Hide and Tibayrenc1999). For this, reliable, reproducible and user-friendly tools are required that allow differentiation of Leishmania parasites at species and strain levels. Table 1 summarizes key epidemiological questions concerning the leishmaniases that can be addressed by species- and strain-specific markers.
Table 1. Epidemiological questions to be addressed by tools differentiating Leishmania species and strains

Abbreviations:
a MLEE, multilocus enzyme electrophoresis; b RFLP, restriction fragment length polymorphism; c MLST, multilocus sequence typing; d MLMT, multilocus microsatellite typing. (X) limited application.
MOLECULAR EPIDEMIOLOGY AND MOLECULAR MARKERS
Molecular epidemiology is the application of molecular tools to answer epidemiological questions, particularly for pathogens. Molecular data are mainly analysed in 2 different ways, by population genetics or phylogenetics. Population genetic analyses provide a snapshot of the current structure of genetic variation within and between populations whereas phylogenetic analyses present information on the history of populations or organisms in the form of trees or networks. Most published studies are based on empirical analyses and, unfortunately, population genetic or phylogenetic approaches to data analysis have as yet been rarely used. This is perhaps due to the fact that the analytical methods and most available software are not very user-friendly and/or not well-suited for thorough analyses of the large datasets produced by molecular epidemiological studies (Constantine, Reference Constantine2003; Tibayrenc, Reference Tibayrenc2005).
Since the advent of PCR numerous molecular tools have been published that distinguish species and strains of Leishmania parasites. The tools range from the amplification and subsequent restriction fragment length polymorphism (RFLP) or DNA sequence analysis of multicopy targets or multigene families, including coding and non-coding regions and PCR-fingerprinting techniques, to the recently developed multilocus sequence typing (MLST) and multilocus microsatellite typing (MLMT) – for review see Schönian et al. (Reference Kuhls, Chicharro, Canavate, Cortes, Campino, Haralambous, Soteriadou, Pratlong, Dedet, Mauricio, Miles, Schaar, Ochsenreither, Radtke and Schönian2008). Each of these molecular markers has its specific discriminatory power, advantages and drawbacks. The kind of marker that is most suitable depends on the research question to be asked (Table 1). This is especially important for the choice of the appropriate level of resolution. If the marker detects too much variation it may not faithfully represent more distant relationships and will fail to identify useful traits. If it is only moderately variable it will not explore the differences between closely related samples. Population genetic and phylogenetic approaches depend on neutral markers that are not affected by natural selection, although, in some situations non-neutral markers may identify a useful trait, e.g. virulence or drug resistance. Thus, markers should always be tested for neutrality. Co-dominant markers allowing for the detection of all 3 possible allele combinations in a diploid are preferred for population genetic studies. The markers should remain stable during in vitro or in vivo passage of the pathogens. The typing results should be reproducible, comparable between different laboratories and storable in databases. It should be possible to test intrinsic assumptions by comparing results from different methods of analyses and/or by examining the robustness of results using re-sampling techniques. The use of molecular markers directly in host tissues may be preferred because culture of Leishmania parasites often fails and may select for particular genotypes. Furthermore, direct methods facilitate sampling, although culture allows isolation of biological clones. Last but not least, since molecular epidemiological studies normally involve large sample sets, methods that are cost-effective and allow for high-throughput analyses are desirable.
How many markers are needed for molecular epidemiological and population genetic studies depends largely on the mode of reproduction. In clonal organisms, there is a correlation between the number of markers needed and the sample size to be investigated. Increasing the number of samples and loci will improve the estimates of genetic distance. However, if enough appropriate loci are available, reliable estimates of genetic distances can be obtained from few individuals (Kalinowski, Reference Kalinowski2005). In contrast, in recombining organisms larger sets of independent markers are needed to detect intraspecific variation since each locus might have a separate evolution. Frequent genetic recombination would result in unstable multilocus genotypes, in which case only individual genes can be analysed individually for epidemiological tracking (Tibayrenc, Reference Tibayrenc2005) or tests applied should incorporate analysis of recombination. There may be, however, a limit to the number of useful markers. In clonal populations the number of different genotypes seems to increase linearly with an increase in the number of markers, whereas a plateau will be reached for recombining organisms, after which no more genotypes are uncovered even if more loci are added (Halkett et al. Reference Halkett, Simon and Balloux2005). Genetic exchange has recently been demonstrated in insect stages of Leishmania, but it seems to be sporadic in natural populations (Akopyants et al. Reference Akopyants, Kimblin, Secundino, Patrick, Peters, Lawyer, Dobson, Beverley and Sacks2009), although rates may vary between populations.
MOLECULAR MARKERS RESOLVING LEISHMANIA PARASITES AT SPECIES LEVEL
The ability to distinguish between Leishmania species is crucial for the correct diagnosis and prognosis of the disease as well as for making decisions regarding treatment and control measures. This is especially useful for studies in areas with various co-existing Leishmania species such as the southern Mediterranean Basin where CL cases may be due to L. major, L. tropica or L. infantum or in South America where CL may be caused by L. mexicana and L. amazonensis as well as by the numerous often overlapping species of the subgenus L.(Viannia). In addition, due to increasing international travel and population migration Leishmania parasites are imported into other regions of the world, including areas non-endemic for the disease (Harms et al. Reference Harms, Schönian and Feldmeier2003; Schönian et al. Reference Schönian, Nasereddin, Dinse, Schweynoch, Schallig, Presber and Jaffe2003; Johnston et al. Reference Johnston, Stockley, Dockrell, Warrell, Bailey, Pasvol, Klein, Ustianowski, Jones, Beeching, Brown, Chapman, Sanderson and Whitty2009). Furthermore, the fact that lower trypanosomatids related to the monoxenous parasites of insects of the genera Leptomonas or Herpetomonas, have been identified as causative agents of VL in southern Europe (Jimenez et al. Reference Jimenez, Lopez-Velez, Molina, Canavate and Alvar1996), South America (Pacheco et al. Reference Pacheco, Marzochi, Pires, Brito, Madeira Mde and Barbosa-Santos1998) and in the Indian subcontinent (Bhattarai et al. Reference Bhattarai, Das, Rijal, Van der Auwera, Picado, Khanal, Roy, Speybroeck, Berkvens, Davies, Coosemans, Boelaert and Dujardin2009) points to the need for species identification even in areas where, so far, only one species had been thought to cause the disease.
The advantage of molecular approaches based on PCR or other amplification techniques is that they combine high sensitivity for direct detection of the infecting parasites in various human, animal and sand fly tissues, with species specificity. Numerous PCR approaches have been published based on different coding and non-coding regions in the Leishmania genome, not all of them are, however, useful for identification at the species level. PCR assays amplifying the conserved region of kinetoplast minicircle DNA (∼10 000 copies per cell) or SSU rDNA (40–200 copies per cell) have been shown to be most sensitive, but can identify leishmanial parasites only to the generic and/or subgeneric level (Van Eys et al. Reference Van Eys, Schoone, Kroon and Ebeling1992; Yurchenko et al. Reference Yurchenko, Kolesnikov and Lukes2000; Lachaud et al. Reference Lachaud, Marchergui-Hammami, Chabbert, Dereure, Dedet and Bastien2002; Schönian et al. Reference Schönian, Nasereddin, Dinse, Schweynoch, Schallig, Presber and Jaffe2003; Disch et al. Reference Disch, Pedras, Orsini, Pirmez, De Oliveira, Castro and Rabello2005; Bensoussan et al. Reference Bensoussan, Nasereddin, Jonas, Schnur and Jaffe2006). On the other hand, amplification of species-specific DNA sequences (Salotra et al. Reference Salotra, Sreenivas, Pogue, Lee, Nakhasi, Ramesh and Negi2001; Jirku et al. Reference Jirku, Zemanova, Al-Jawabreh, Schönian and Lukes2006; Laurent et al. Reference Laurent, Van der Auwera, Hide, Mertens, Quispe-Tintaya, Deborggraeve, De Doncker, Leclipteux, Banuls, Buscher and Dujardin2009) has serious limitations if not combined with a genus-specific assay to rule out false negatives and co-infections with other Leishmania species or interspecific hybrid infections. In areas where many species are sympatric several different approaches might be necessary, which would increase the cost of diagnostics. Approaches based on initial amplification of genus-specific sequences followed by subsequent differentiation of Leishmania species by RFLP, hybridization with specific probes or sequencing of the amplified sequences have proven most useful. Different targets have been used for this, such as the ribosomal internal transcribed spacer (ITS) (Cupolillo et al. Reference Cupolillo, Grimaldi, Momen and Beverley1995; Schönian et al. Reference Schönian, Nasereddin, Dinse, Schweynoch, Schallig, Presber and Jaffe2003; Nasereddin et al. Reference Nasereddin, Bensoussan-Hermano, Schönian, Baneth and Jaffe2008); the mini-exon gene (Harris et al. Reference Harris, Kropp, Belli, Rodriguez and Agabian1998); repetitive nuclear DNA sequences (Piarroux et al. Reference Piarroux, Azaiez, Lossi, Reynier, Muscatelli, Gambarelli, Fontes, Dumon and Quilici1993); the glucose-6-phosphate dehydrogenase gene (Castilho et al. Reference Castilho, Shaw and Floeter-Winter2003); gp63 genes (Victoir et al. Reference Victoir, Banuls, Arevalo, Llanos-Cuentas, Hamers, Noel, De Doncker, Le Ray, Tibayrenc and Dujardin1998); hsp70 genes (Garcia et al. Reference Garcia, Kindt, Bermudez, Llanos-Cuentas, De Doncker, Arevalo, Wilber Quispe Tintaya and Dujardin2004; Fraga et al. Reference Fraga, Montalvo, De Doncker, Dujardin and Van der Auwera2010); cytochrome b gene (Kato et al. Reference Kato, Uezato, Katakura, Calvopina, Marco, Barroso, Gomez, Mimori, Korenaga, Iwata, Nonaka and Hashiguch2005); and 7SL RNA gene sequences (Zelazny et al. Reference Zelazny, Fedorko, Li, Neva and Fischer2005). Most laboratories use, however, an in-house PCR. Few research groups have tried to standardize and validate their PCR assay by testing for sensitivity and specificity not only with DNA extracted from cultured promastigotes but also with DNA extracted from different types of clinical material and from tissue samples spiked with known numbers of parasites, or by controlling for inhibition and the quality of DNA extraction, or by comparing their assays to other PCR assays (Schönian et al. Reference Schönian, Nasereddin, Dinse, Schweynoch, Schallig, Presber and Jaffe2003; Garcia et al. Reference Garcia, Kindt, Bermudez, Llanos-Cuentas, De Doncker, Arevalo, Wilber Quispe Tintaya and Dujardin2004; Bensoussan et al. Reference Bensoussan, Nasereddin, Jonas, Schnur and Jaffe2006; Nasereddin et al. Reference Nasereddin, Bensoussan-Hermano, Schönian, Baneth and Jaffe2008).
To our knowledge, the PCR-RFLP of the internal transcribed spacer 1 (ITS1) is the most widely used assay for direct detection and identification of Leishmania species in the Old World. It has been applied to the distinction of sympatric species, especially in the Mediterranean region, as well as for the identification of imported cases (Harms et al. Reference Harms, Schönian and Feldmeier2003; Al-Jawabreh et al. Reference Al-Jawabreh, Schnur, Nasereddin, Schwenkenbecher, Abdeen, Barghuthy, Khanfar, Presber and Schönian2004; Rhajaoui et al. Reference Rhajaoui, Nasereddin, Fellah, Azmi, Amarir, Al-Jawabreh, Ereqat, Planer and Abdeen2007). By digesting the ITS1 PCR product with only 1 restriction enzyme, HaeIII, all medically relevant Leishmania species can be distinguished. Representatives of the L. donovani complex (L. donovani and L. infantum) or L. braziliensis complex (L. braziliensis, L. guyanensis, L. panamensis, L. peruviana etc.) have almost identical RFLP patterns with a great variety of restriction enzymes and cannot be resolved further by this approach (Schönian et al. Reference Schönian, Nasereddin, Dinse, Schweynoch, Schallig, Presber and Jaffe2003). This problem can, however be solved by sequencing the 350 bp ITS1 PCR product (Schönian and Kuhls, unpublished observations) which, in the case of the L. donovani complex, not only allows for clear separation of L. infantum from L. donovani, but also assigns strains of these species to different phylogenetic groups supported by differences in biology and clinical behaviour (Kuhls et al. Reference Kuhls, Mauricio, Pratlong, Presber and Schönian2005). A recently developed simple reverse line blot hybridization (RLB) assay based on ITS1 sequences seems to be very promising for leishmaniasis diagnostics because it distinguishes all Old World Leishmania species, even L. donovani from L. infantum, with a 10- to 100-fold enhanced sensitivity, comparable to that of kDNA PCR (Nasereddin et al. Reference Nasereddin, Bensoussan-Hermano, Schönian, Baneth and Jaffe2008). The primers used in these PCR assays amplify the ITS1 not only from all species of the genus Leishmania but also from other trypanosomatids, such as the genera Crithidia, Trypanosoma and Leptomonas (Schönian, unpublished observations). An ITS1 sequence that cannot be aligned to any of the Leishmania sequences available from GenBank might, therefore, be indicative for the presence of other trypanosomatids in the sample.
Recently, high-resolution melt (HRM) analysis of a real-time PCR product from the ITS1 region was used to identify and quantify Old World Leishmania species (Talmi-Frank et al. Reference Talmi-Frank, Nasereddin, Schnur, Schonian, Toz, Jaffe and Baneth2010). It is a closed tube assay that does not employ additional fluorescent probes but simply utilizes a DNA melting assay and computerized analysis of the results to produce a graphic output. When tested on 300 samples from human cases, reservoir hosts and sand flies, this approach distinguished all Old World Leishmania species causing human disease, except L. donovani from L. infantum.
The PCR-RFLP approach targeting hsp70 sequences has proven to be most useful for the differentiation between South American Leishmania (Viannia) species. The restriction endonuclease HaeIII produces diagnostic RFLP patterns for the L. guyanensis species complex as well as for L. lainsoni and L. shawi (Garcia et al. Reference Garcia, Kindt, Bermudez, Llanos-Cuentas, De Doncker, Arevalo, Wilber Quispe Tintaya and Dujardin2004; Da Silva et al. 2010). However, L. braziliensis and L. peruviana, both belonging to the L. braziliensis complex, as well as L. naiffi share an identical HaeIII RFLP pattern and can only be distingished by using a second restriction enzyme (Da Silva et al. 2010). The same is true for L. guyanensis and L. panamensis both belonging to the L. guyanensis complex (Fraga et al. Reference Fraga, Montalvo, De Doncker, Dujardin and Van der Auwera2010). This discrimination is not only of epidemiological importance but has consequences for the prognosis of the disease. So far, MCL has been principally associated with L. braziliensis (Lessa et al. Reference Lessa, Lessa, Castro, Oliveira, Scherifer, Machado and Carvalho2007), although other L. (Viannia) species have also been repeatedly suspected of causing MCL (Thomaz-Soccol et al. Reference Thomaz-Soccol, Velez, Pratlong, Agudelos, Lanotte and Rioux2000). The hsp70 PCR-RFLP has been successfully applied to direct identification of the infecting species in skin scrapings from Bolivian CL patients (Garcia et al. Reference Garcia, Parrado, De Doncker, Bermudez and Dujardin2007a) and in sand fly pools during field studies in the Amazonian lowlands of Bolivia (Garcia et al. Reference Garcia, Tellez, Parrado, Rojas, Bermudez and Dujardin2007b).
PCR assays combining detection of Leishmania parasites with species identification directly with clinical samples have proven useful in numerous field studies and should replace the current gold standard, multilocus enzyme electrophoresis (MLEE). The costs for PCR diagnosis are higher than for microscopy but comparable to those of culturing Leishmania. Species identification by PCR followed by RFLP, hybridization or sequencing is clearly less laborious and less costly than that by MLEE. However, the results of PCR diagnosis should always be evaluated in conjunction with clinical diagnosis, as PCR has been shown to be sensitive enough to detect parasite DNA in apparently parasitologically negative people living in areas endemic for leishmaniasis. A positive PCR result is thus a marker for infection or recent exposure rather than for disease (Deborggraeve et al. Reference Deborggraeve, Boelaert, Rijal, De Doncker, Dujardin, Herdewijn and Buscher2008a). However, the potential problem of PCR detecting naked DNA can probably be virtually disregarded, because Leishmania DNA has been shown to degrade quite quickly after the death of the parasites (Prina et al. Reference Prina, Roux, Mattei and Milon2007).
Assays using alternative amplification technologies such as quantitative nucleic acid sequence-based amplification (QT-NASBA) based on amplification of 18S RNA and loop-mediated isothermal amplification (LAMP) of kinetoplast minicircle DNA do not allow discrimination of different Leishmania species (Van der Meide et al. 2005; Takagi et al. Reference Takagi, Itoh, Islam, Razzaque, Ekram, Hashighuchi, Noiri and Kimura2009). Commercialized assays that have been developed for direct detection of Leishmania are not able to identify the infecting species. These commercial tests still rely on PCR for the amplification of the target and detection is achieved by hybridization to a genus-specific probe covalently linked to a dipstick (Deborggraeve et al. Reference Deborggraeve, Laurent, Espinosa, Van der Auwera, Mbuchi, Wasunna, El-Safi, Al-Basheer, Arevalo, Miranda-Verastegui, Leclipteux, Mertens, Dujardin, Herdewijn and Buscher2008b). Thus, for Leishmania species identification we still depend on validated in-house PCR assays. The use of these assays requires careful selection, comparison and evaluation of reagents, proper training of the operators, a laboratory with some standard equipment, monitoring of quality control, and tests may not be readily applicable under field conditions.
An alternative technique, which might be promising for use in Leishmania, has been developed by Hamilton et al. (Reference Hamilton, Adams, Malele and Gibson2008) for species identification in Trypanosoma. This method called fluorescent fragment length barcoding (FFLB) is based on length variation in regions of the 18S and 28Sα ribosomal DNA. Fluorescently tagged primers annealing to conserved regions were used to amplify fragments in the 18S and 28Sα ribosomal DNA with inter-species size variations. Sizes were accurately determined using an automated sequencer. By using 4 sets of primers amplifying 2 regions in each of the rRNA genes all trypanosome species could be distinguished and putative new species were recognized. FFLB has proven more sensitive than other PCR techniques and allows a high throughput of samples with up to 96 samples tested within 24 h.
MOLECULAR MARKERS TO IDENTIFY LEISHMANIA PARASITES AT STRAIN LEVEL
Different DNA-based techniques have been used to differentiate Leishmania parasites at the strain level. These include DNA and PCR fingerprinting approaches, e.g. Randomly Amplified Polymorphic DNA (RAPD) techniques, which require cultured parasites because probes and primers used in this context are not specific for Leishmania. DNA fingerprinting with the human multilocus minisatellite probe 33.15 was applied to follow an outbreak of VL in central Israel (Nasereddin et al. Reference Nasereddin, Baneth, Schönian, Kanaan and Jaffe2005). PCR fingerprinting and RAPD techniques have been successfully applied to characterize L. tropica strains in a new Israeli focus (Jacobson et al. Reference Jacobson, Eisenberger, Svobodova, Baneth, Sztern, Carvalho, Nasereddin, El Fari, Shalom, Volf, Votypka, Dedet, Pratlong, Schönian, Schnur, Jaffe and Warburg2003) as well as to detect intraspecies variation in the L. donovani complex (Hide et al. Reference Hide, Banuls and Tibayrenc2001; Zemanova et al. Reference Zemanova, Jirku, Mauricio, Miles and Lukes2004). These techniques are relatively simple, rapid and do not need prior sequence information, but suffer from poor reproducibility. Importantly, the results are not comparable between laboratories.
Numerous epidemiological studies in leishmaniasis, only few of which can be cited here, have used PCR-RFLP approaches based on sequence polymorphisms in coding and non-coding regions of multigene families, including cysteine protease B (cpB) (Garcia et al. Reference Garcia, Kindt, Quispe-Tintaya, Bermudez, Llanos, Arevalo, Banuls, De Doncker, Le Ray and Dujardin2005; Quispe-Tintaya et al. 2005); major surface glycoprotein (gp63) (Mauricio et al. Reference Mauricio, Gaunt, Stothard and Miles2001; Garcia et al. Reference Garcia, Kindt, Quispe-Tintaya, Bermudez, Llanos, Arevalo, Banuls, De Doncker, Le Ray and Dujardin2005); intergenic spacers of the ribosomal operon (Cupolillo et al. Reference Cupolillo, Grimaldi, Momen and Beverley1995, Reference Cupolillo, Brahim, Toaldo, De Oliveira-Neto, De Brito, Falqueto, De Farias Naiff and Grimaldi2003; Schönian et al. Reference Schönian, Schnur, El Fari, Oskam, Kolesnikov, Sokolowska-Kohler and Presber2001; Rotureau et al. Reference Rotureau, Ravel, Nacher, Couppie, Curtet, Dedet and Carme2006); miniexon sequences (Mauricio et al. Reference Mauricio, Stothard and Miles2004; Quispe Tintaya et al. Reference Garcia, Kindt, Bermudez, Llanos-Cuentas, De Doncker, Arevalo, Wilber Quispe Tintaya and Dujardin2004), and kinetoplast minicircles (Noyes et al. Reference Noyes, Reyburn, Bailey and Smith1998; Morales et al. Reference Morales, Chicharro, Ares, Canavate, Barker and Alvar2001; Chicharro et al. Reference Chicharro, Morales, Serra, Ares, Salas and Alvar2002; Cortes et al. Reference Cortes, Mauricio, Almeida, Cristovao, Pratlong, Dedet and Campino2006; Laurent et al. Reference Laurent, Rijal, Yardley, Croft, De Doncker, Decuypere, Khanal, Singh, Schönian, Kuhls, Chappuis and Dujardin2007). These approaches are relatively simple and rapid, and can be used directly with clinical isolates. The PCR-RFLP of whole minicircle DNA is a highly polymorphic assay that can differentiate between closely related organisms, such as L. infantum MON-1, and has been used to distinguish between recrudescence and re-infection (Morales et al. Reference Morales, Cruz, Rubio, Chicharro, Canavate, Laguna and Alvar2002). The fragment patterns obtained are, however, difficult to analyse. They are sometimes very complex and difficult to compare between laboratories and between different observers even when the same systems have been used. The patterns are not fully reproducible and artefacts due to partial restriction cannot be excluded. Finally, changes in the RFLP profiles have been observed during in vivo and in vitro passages of Leishmania parasites (T. Laurent, personal communication). These techniques depend therefore, on careful standardization and are recommended for comparative studies involving few strains rather than for large-scale epidemiological studies. It should also be mentioned that in vitro selection for drug-resistant L. amazonensis lead to changes in kDNA minicircle RFLP due to a switch of minicircle class dominance (Lee et al. Reference Lee, Tarn and Chang1993). The minicircle RFLP pattern of Nepalese isolates of L. donovani did not, however, show any correlation with treatment outcome in patients (Laurent et al. Reference Laurent, Rijal, Yardley, Croft, De Doncker, Decuypere, Khanal, Singh, Schönian, Kuhls, Chappuis and Dujardin2007).
All the strain-specific DNA markers mentioned so far have one disadvantage in common, they are not co-dominant. The only co-dominant markers that currently differentiate within the Leishmania species and can detect all 3 diploid allele combinations possible in a diploid are MLEE, multilocus sequence typing (MLST) and multilocus microsatellite typing (MLMT). A detailed discussion of the latter 2 approaches is given in the next sections.
MULTILOCUS SEQUENCE TYPING (MLST)
Multilocus sequence typing (MLST) refers to analysis based on the DNA sequence of multiple gene targets. The term was adopted for a specific system developed initially for bacteria (Maiden et al. Reference Maiden, Bygraves, Feil, Morelli, Russell, Urwin, Zhang, Zhou, Zurth, Caugant, Feavers, Achtman and Spratt1998) and which is applied like multilocus enzyme electrophoresis (MLEE). In the strict bacterial MLST context, short DNA sequences of 300–500 bp for 7–12 targets are generated by direct sequencing in both directions. MLST is able to detect co-dominant single nucleotide polymorphisms (SNP), as mentioned above, and although indels can complicate the analysis and require cloning or other allele-specific analysis, in our experience they are extremely rare in protein-coding genes. Each sequence is scored as a haplotype, bacteria being haploid, and the combination of the haplotypes for all gene targets is the sequence type (ST). Targets must be selectively neutral, given that a single gene subject to high positive or negative selection may disrupt phylogenies. Sequence data are highly reproducible and easily comparable between laboratories and thus have been amenable to sharing in databases via the internet. A public website (www.mlst.net) is dedicated to host the web interface for most of such existing databases, hosted at Imperial College, UK. Via this and similar websites, users can perform several tasks such as downloading alleles and STs, comparing profiles to reference datasets and concatenating sequences. Users are encouraged to submit new alleles or STs to the curated database. A new development includes maps through which the global distribution of isolates and STs can be visualized by country in combination with Google Maps or Google Earth. Online access is also provided to data analysis software such as BURST, NRDB (Non-redundant databases), SplitsTree and linkage disequilibrium.
Recently MLST has been applied to diploid organisms, notably to fungal pathogens of the genus Candida, in which case it is the diploid sequence type that is coded, using the codes for ambiguous nucleotides (Bougnoux et al. Reference Bougnoux, Morand and D'Enfert2002). Diploids pose particular problems for analysis in that they possess 2 alleles per locus and there is no physical linkage between loci located in different chromosomes. Diploid MLST data may be analysed with programmes such as SplitsTree that accommodate ambiguous nucleotides or by concatenating datasets with duplicated nucleotides so that heterozygous sites become 2 sites with a single base at each. This latter method makes assumptions about the origin of each single nucleotide polymorphism (SNP), as identical nucleotides may be identical by descent (homologous) or by state (homoplasy) and should thus be used with caution.
In Leishmania, tentative steps have been taken to develop an MLST system, although an internet accessible database has not yet been created. The L. donovani complex has been studied by 2 sets of 5 loci for genes coding for enzymes used in MLEE (Mauricio et al. Reference Mauricio, Yeo, Baghaei, Doto, Pratlong, Zemanova, Dedet, Lukes and Miles2006; Zemanova et al. Reference Zemanova, Jirku, Mauricio, Horak, Miles and Lukes2007): one set with asat, gpi, nh1, nh2 and pgd and the other with icd, me, mpi, g6pdh, and fh. Together those 10 targets should form a complete MLST system applicable to the L. donovani complex. Although results from MLST of Leishmania are largely in agreement with the results from MLEE, some key discrepancies were found and increased resolution was obtained. Thus silent SNPs were found that provide further resolution, such a single SNP in gpi that distinguishes between strains of L. infantum (Mauricio et al. Reference Mauricio, Yeo, Baghaei, Doto, Pratlong, Zemanova, Dedet, Lukes and Miles2006). However, SNPs responsible for amino acid changes were also found in genes coding for enzymes giving indistinguishable electrophoretic profiles, notably in nh2, which has the same protein band size for all L. donovani complex strains.
Four gene targets (gpi, mdh, mpi and 6pgd) have been described for Leishmania of the New World (Tsukayama et al. Reference Tsukayama, Lucas and Bacon2009), not for analysis by MLST data, but for diagnostic tools. Unfortunately, the analysis did not include sufficient diversity of strains for each species, and unpublished analyses (Mauricio, unpublished observations; Cupolillo, personal communication) have shown that some potentially species-diagnostic SNPs are actually present in other species. In addition, 6 gene targets that are not associated with MLEE have been used to characterize L. major/L. infantum hybrids (Ravel et al. Reference Ravel, Cortes, Pratlong, Morio, Dedet and Campino2006), although their diversity within each species has not yet been published.
The main advantage of MLST over MLMT is the possibility of generating genus-wide phylogenies and, given high quality sequencing, unequivocal comparability between laboratories. Similar and further targets are being analysed for the subgenus Leishmania (Viannia), aiming to produce a share panel of MLST targets, although possibly with different primers, that can be applied to the entire genus. MLST is likely to become the gold standard basis for taxonomy and thus identification of Leishmania. For example, a preliminary analysis (Mauricio, unpublished observations) of the gene gpi for a number of samples of different species has shown that comparisons of genetic diversity within and across species may help decide on validity of controversial species. In comparison with MLEE, MLST does not necessarily require sterile culture of parasites, does not require simultaneous typing of reference strains, can be done commercially without in-house specialized equipment and, as mentioned above, the data are portable.
New generation sequencing (NGS) enables fast sequencing of large numbers of genes. MLST may be extended to 100 or more targets, including neutral targets and those known to have roles in key biological features such as virulence, drug resistance, shock resistance, etc. NGS on long read platforms also has the advantage of cloning samples so that reads are haplotypes and true heterozygotes can be distinguished from mixed samples. Such studies are likely to enhance in-depth knowledge of the natural history, evolution and population genetics of Leishmania.
MULTILOCUS MICROSATELLITE TYPING (MLMT)
Microsatellite sequences, also called simple sequence repeats (SSRs) or short tandem repeats (STRs), are repeated motifs of 1–6 nucleotides found in all eukaryotic and prokaryotic genomes (Toth et al. Reference Toth, Gaspari and Jurka2000; Mrazek et al. Reference Mrazek, Guo and Shah2007). They are present in coding, albeit rarely, and in non-coding regions constitute a large fraction of sequences. Analysis of length polymorphisms of microsatellite-containing sequences has, recently, become an important tool for population and genetic studies of many species including humans. Microsatellites mutate at rates 5–6 orders of magnitude higher than the bulk of DNA, which makes them particularly useful for studying variation between closely related organisms. Microsatellite sequence variation results from the gain and loss of single repeat units, which can easily be detected after amplification with primers annealing specifically to their flanking regions. The results of these analyses are theoretically reproducible and exchangeable between laboratories. Selection does not seem to act on polymorphisms in microsatellite length, and allelic variants are detectable because of the co-dominant nature of these markers.
The mutation rate at a given microsatellite locus is influenced by various factors: the repeated motif itself, allele size, chromosome position, GC content in flanking DNA, and the efficacy of the mismatch repair system (MMR), which is critical for the stability of microsatellites. Recombination may change the repeat number by unequal crossing-over or by gene conversion (Li et al. Reference Li, Korol, Fahima, Beiles and Nevo2002; Ellegren, Reference Ellegren2004). However, the most plausible explanation for variation in repeat numbers is slippage of polymerase during DNA replication (Schlotterer and Tautz, Reference Schlotterer and Tautz1992). The transient dissociation of the replicating DNA strands followed by misaligned re-association leads to gain or loss of repeat units. Most of these primary mutations in vivo are corrected by the MMR system, and only the small fraction that was not repaired or not correctly repaired ends up as variable microsatellites. Although there is a general tendency for gain of new repeats, natural selection acts against very long repeats (Li et al. Reference Li, Korol, Fahima, Beiles and Nevo2002; Ellegren, Reference Ellegren2004). This means that the evolutionary history of a particular repeat sequence may be uncertain and that these markers are prone to homoplasy. To overcome this main obstacle in the use of microsatellite analysis it is recommended that all studies should use a panel of 10–20 unlinked microsatellite markers.
The genome of Leishmania was found to be relatively rich in microsatellites with about 600 (CA)n loci per haploid genome (Rossi et al. Reference Rossi, Wincker, Ravel, Blaineau, Pages and Bastien1994). During recent years the development of microsatellite-based approaches has been attempted for strain typing within the genus Leishmania to overcome the lack of discriminatory power of MLEE and of many other molecular typing methods. It very soon became obvious that microsatellite markers highly discriminatory within one species of Leishmania could not be amplified, were even absent or not informative in others (Jamjoom et al. Reference Jamjoom, Ashford, Bates, Kemp and Noyes2002a; Schwenkenbecher et al. Reference Schwenkenbecher, Frohlich, Gehre, Schnur and Schönian2004). Regions flanking the repeats were not strongly conserved between closely related species of Leishmania and repeats polymorphic in one species were often interrupted by single nucleotide substitutions and insertions in other species. Thus, species-specific and/or species-complex-specific marker sets are needed in the genus Leishmania.
So far, microsatellite loci with high discriminatory power and suitable for characterizing closely related strains have been published for the L. donovani complex (Bulle et al. Reference Bulle, Millon, Bart, Gallego, Gambarelli, Portus, Schnur, Jaffe, Fernandez-Barredo, Alunda and Piarroux2002; Jamjoom et al. Reference Jamjoom, Ashford, Bates, Kemp and Noyes2002b; Ochsenreither et al. Reference Ochsenreither, Kuhls, Schaar, Presber and Schönian2006), L. major (Jamjoom et al. Reference Jamjoom, Ashford, Bates, Kemp and Noyes2002a; Al-Jawabreh et al. Reference Al-Jawabreh, Diezmann, Mueller, Wirth, Schnur, Strelkova, Kovalenko, Razakov, Schwenkenbecher, Kuhls and Schoenian2008), L. tropica (Schwenkenbecher et al. Reference Schwenkenbecher, Wirth, Schnur, Jaffe, Schallig, Al-Jawabreh, Hamarsheh, Azmi, Pratlong and Schönian2006) and for species of the subgenus L. (Viannia) (Russell et al. Reference Russell, Iribar, Lambson, Brewster, Blackwell, Dye and Ajioka1999; Rougeron et al. Reference Rougeron, Waleckx, Hide, De Meeûs, Arevalo, Llanos-Cuentas and Banuls2008; Oddone et al. Reference Oddone, Schweynoch, Schönian, De Sousa Cdos, Cupolillo, Espinosa, Arevalo, Noyes, Mauricio and Kuhls2009). Recently, a searchable database of microsatellite loci within the genome has been established at http://www.genomics.liv.ac.uk/tryps/Microsatellites.V1.html, which allows the development of more microsatellite markers for the L. donovani complex, L. major and L. braziliensis (Fakhar et al. Reference Fakhar, Motazedian, Daly, Lowe, Kemp and Noyes2008).
The multilocus microsatellite typing (MLMT) approaches developed so far for Leishmania make use of sets of 14–20 unlinked microsatellite loci. For application in large-scale epidemiological and population genetic studies, size variations of fluorescence-labelled PCR products should be detectable by using the fragment analysis tool of automated sequencers which have the potential for multiplexing and high-throughput analyses. Markers requiring sequence analysis of repeat number variation are not appropriate (Bulle et al. Reference Bulle, Millon, Bart, Gallego, Gambarelli, Portus, Schnur, Jaffe, Fernandez-Barredo, Alunda and Piarroux2002) and, for that reason, most marker sets use primers closely flanking the repeat. Repeat numbers estimated for the different loci are assembled into a multilocus microsatellite profile for every strain under study.
Distance-based methods and models based on Bayesian statistics have been applied in the analysis of these profiles. Microsatellite based genetic distances may be calculated using the software MSA (Dieringer and Schlötterer, Reference Dieringer and Schlötterer2003) and POPULATIONS (http://bioinformatics.org/~tryphon/populations) by applying different distance measures appropriate for microsatellites. Based on the resulting distance matrix Neighbour-joining (NJ) trees are constructed with programmes such as POPULATIONS and MEGA (Kumar et al. Reference Kumar, Tamura and Nei2004). The model-based method to uncover population structure implemented in STRUCTURE (Pritchard et al. Reference Pritchard, Stephens and Donnelly2000) has advantages over distance-based data evaluation because its algorithm uses patterns of allele frequencies to identify distinct subpopulations and it determines fractions of each genotype within each subpopulation. However, panmixia is one of the essential assumptions in the STRUCTURE algorithm. Nevertheless, even if used with organisms not in Hardy-Weinberg equilibrium, STRUCTURE results have so far always corroborated those obtained by genetic distance, have accurately inferred individual ancestries, have been appropriate for characterization of population structure and have provided information on population relationships and history (e.g. see Schwenkenbecher et al. Reference Schwenkenbecher, Wirth, Schnur, Jaffe, Schallig, Al-Jawabreh, Hamarsheh, Azmi, Pratlong and Schönian2006; Al-Jawabreh et al. Reference Al-Jawabreh, Diezmann, Mueller, Wirth, Schnur, Strelkova, Kovalenko, Razakov, Schwenkenbecher, Kuhls and Schoenian2008; Wirth et al. Reference Wirth, Hildebrand, Allix-Beguec, Wolbeling, Kubica, Kremer, Van Soolingen, Rusch-Gerdes, Locht, Brisse, Meyer, Supply and Niemann2008).
Once the population structure is defined, F-statistics are of high value for population studies of diploid organisms including Leishmania. F is, a measure of the inbreeding of individuals resulting from the deviation from panmixia, and F st, a measure of the relatedness between individuals due to the structure of the population, provide information about the mode of reproduction and population differentiation (De Meeus et al. Reference De Meeus, Lehmann and Balloux2006). Clonal diploids are expected to accumulate heterozygosity over time at every locus and should therefore exhibit negative F is values. Heterozygote deficiency (positive F is values) seen in many MLMT studies of Leishmania can be due to the presence of different factors, such as population subdivision (Wahlund effect) or a high rate of gene conversion. The impact of population substructure can be studied with the Bayesian analysis of genetic population structure (BAPS) software (Corander et al. Reference Corander, Gyllenberg and Koski2007). Using this method Rougeron et al. (Reference Rougeron, De Meeus, Hide, Waleckx, Bermudez, Arevalo, Llanos-Cuentas, Dujardin, De Doncker, Le Ray, Ayala and Banuls2009) have demonstrated that the high F is values found in their MLMT analysis of Bolivian and Peruvian L. braziliensis are only partly explained by population subdivision.
It is advantageous that MLMT can be used directly on biological material without culture of the parasite. DNA extracted from specimens spotted on filter paper or glass slides or from old Giemsa-stained microscope slides was successfully applied in MLMT approaches (Alam et al. Reference Alam, Kovalenko, Kuhls, Nasyrova, Ponomareva, Fatullaeva, Razakov, Schnur and Schönian2009b,Reference Alam, Kuhls, Schweynoch, Sundar, Rijal, Shamsuzzaman, Raju, Salotra, Dujardin and Schönianc). Moreover, accurate, quality controlled microsatellite profiles can be stored in databases and compared between different laboratories.
POPULATION STRUCTURE OF KEY LEISHMANIA SPECIES AS REVEALED BY MLST AND MLMT
MLMT brought about the first true population genetics studies in Leishmania. The first study was done on L. tropica (Schwenkenbecher et al. Reference Schwenkenbecher, Wirth, Schnur, Jaffe, Schallig, Al-Jawabreh, Hamarsheh, Azmi, Pratlong and Schönian2006). It showed the existence of genetically different populations with geographical associations, as well as genetically isolated sympatric populations in rather small territories, e.g. in Israel and the Palestinian Authority and in Morocco. The population structure of L. tropica was found to be complex, with new variants spreading to distant foci, a zoonotic focus and with a heterozygous clade (putative hybrid lineage) achieving a widespread distribution. Unpublished MLST results by one of the authors (Mauricio) have shown consistency within groups identified by MLMT of L. tropica (Schwenkenbecher et al. Reference Schwenkenbecher, Wirth, Schnur, Jaffe, Schallig, Al-Jawabreh, Hamarsheh, Azmi, Pratlong and Schönian2006) and L. major (Al-Jawabreh et al. Reference Al-Jawabreh, Diezmann, Mueller, Wirth, Schnur, Strelkova, Kovalenko, Razakov, Schwenkenbecher, Kuhls and Schoenian2008).
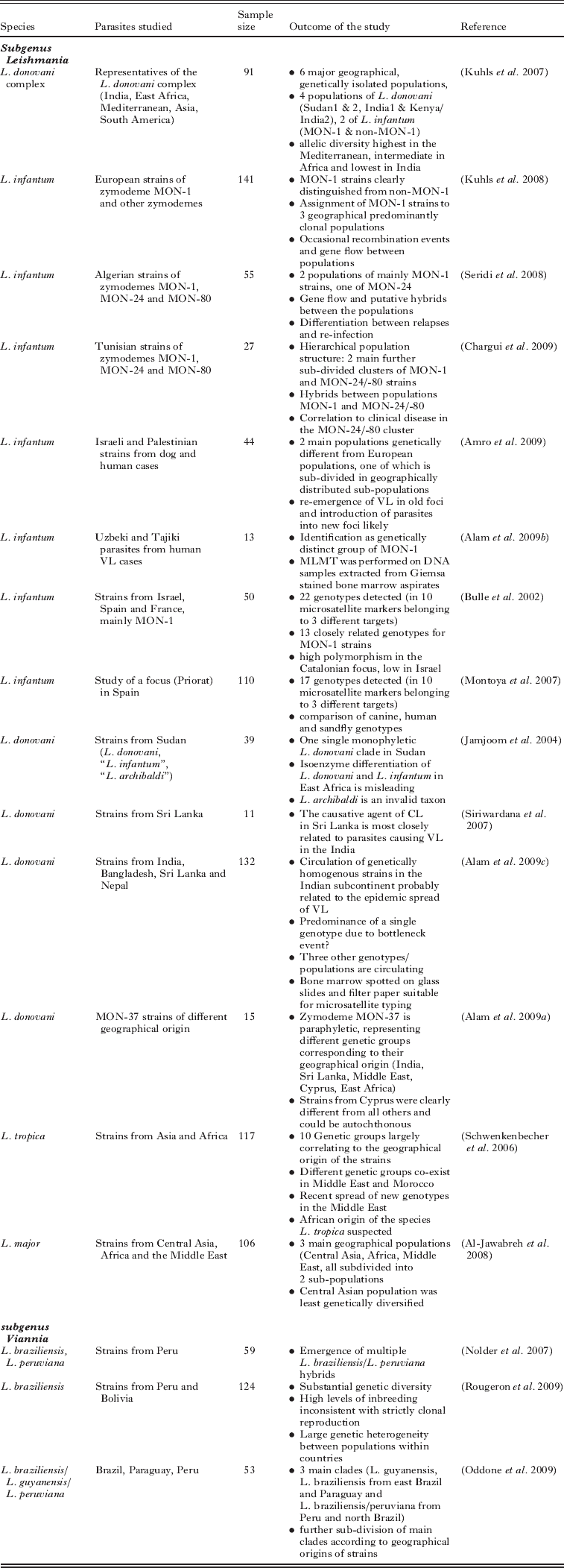
Most of the MLMT studies published so far (Table 2) have addressed epidemiological and population genetic questions related to the L. donovani complex. A set of 15 microsatellite markers has been applied to type strains of L. donovani and L. infantum isolated from the main regions endemic for VL (Kuhls et al. Reference Kuhls, Keilonat, Ochsenreither, Schaar, Schweynoch, Presber and Schönian2007). Six principal genetically distinct populations were identified: 2 populations of L. infantum from the Mediterranean area and South America comprising the MON-1 and non-MON-1 strains, respectively; 2 populations of L. donovani from Sudan and Ethiopia; 1 of L. donovani MON-2 from India; and 1 consisting of strains of L. donovani (MON-36, 37, 38) from Kenya and India. This was perhaps not very surprising but corroborated the fragmentary data published in numerous studies using other genetic markers (Mauricio et al. Reference Mauricio, Stothard and Miles2004; Kuhls et al. Reference Kuhls, Mauricio, Pratlong, Presber and Schönian2005; Quispe-Tintaya et al. 2005; Mauricio et al. Reference Mauricio, Gaunt, Stothard and Miles2007). Although 2 populations of L. donovani were found in Sudan and Ethiopia, like other molecular methods MLMT did not support the presence of other species, L. infantum and L. archibaldi, reported from MLEE in East Africa. Based on a combination of multiple genetic data, including MLMT data, a revised taxonomy was proposed for the L. donovani complex (Lukes et al. Reference Lukes, Mauricio, Schönian, Dujardin, Soteriadou, Dedet, Kuhls, Tintaya, Jirku, Chocholova, Haralambous, Pratlong, Obornik, Horak, Ayala and Miles2007).
Table 2. Overview of epidemiological and population genetic studies in leishmaniasis using MLMT
MLST produced similar results and confirmed the division of the L. donovani complex into a number of genetic groups (Mauricio et al. Reference Mauricio, Yeo, Baghaei, Doto, Pratlong, Zemanova, Dedet, Lukes and Miles2006), equivalent to those found for MLMT (Kuhls et al. Reference Kuhls, Keilonat, Ochsenreither, Schaar, Schweynoch, Presber and Schönian2007). An exception was the subdivision of the Sudanese group, although one of these groups included strains placed in a separate MLST group. Notably, the MLST analysis was able to identify introgression of the ASAT allele typical of L. infantum into some Sudanese L. donovani strains, able to detect a strain with L. infantum/L.donovani gene mosaics, in addition to a clear hybrid between 2 L. donovani subgroups, heterozygous at all genes with different alleles.
Interestingly, the highest microsatellite diversity was observed for L. infantum from the Mediterranean basin and the lowest for L. donovani from India. To test whether the latter finding might be attributed to a sampling bias or to the fact that a strain of L. infantum had been used for the design of the microsatellite markers used (Ochsenreither et al. Reference Ochsenreither, Kuhls, Schaar, Presber and Schönian2006), we conducted a more comprehensive study by including more strains from India, Nepal and Bangladesh and by designing new microsatellite markers with a library made from an Indian strain of L. donovani (Alam et al. Reference Alam, Kuhls, Schweynoch, Sundar, Rijal, Shamsuzzaman, Raju, Salotra, Dujardin and Schönian2009c). The outcome was, however, the same, even when 34 additional microsatellite sequences had been analysed. The homogeneity of L. donovani from the Indian subcontinent was remarkable, 125 of the 132 strains tested belonged to the same population regardless of geographical origin, clinical manifestation, and whether they presented in vitro or in vivo susceptibility to antimonial drugs. Identical multilocus microsatellite profiles were found for 108 strains. The most plausible explanation is that this population emerged only recently with a very short time for subsequent evolution. However, MLST-based networks suggest sustained and ancient reproductive isolation from other L. donovani lineages. A bottleneck event related to the insecticide spraying under the Malaria Control programme in the 1960s might have exterminated the original L. donovani population(s) leaving only a small pocket of survivors that started to spread after the campaign was finished.
Because substructures were detected in the main populations of the L. donovani complex according to place and time of strain isolation (Kuhls et al. Reference Kuhls, Keilonat, Ochsenreither, Schaar, Schweynoch, Presber and Schönian2007) we attempted to use the MLMT for discriminating further the strains within the L. infantum MON-1 population. A big disadvantage of all previously used genetic approaches, except for kDNA-RFLP, was that they could not differentiate further the strains of this predominating zymodeme of L. infantum (up to 70% are MON-1). When we applied MLMT to 107 L. infantum MON-1 strains mainly from Portugal, Spain and Greece they were always at first separated from non-MON-1 strains included for comparison, but secondly split into 3 different populations comprising strains from Greece, the Balearic Islands and the Iberian peninsula, respectively (Kuhls et al. Reference Kuhls, Chicharro, Canavate, Cortes, Campino, Haralambous, Soteriadou, Pratlong, Dedet, Mauricio, Miles, Schaar, Ochsenreither, Radtke and Schönian2008).
Different genetic groups of strains of L. infantum were also observed when strains from Israel and the Palestinian Authority, Tunisia and Algeria were subjected to MLMT. In the 2 North African countries, different zymodemes of L. infantum can cause both visceral and cutaneous disease. Microsatellite typing of strains belonging to zymodemes MON-1, MON-24 and MON-80 identified 3 different populations in both countries (Seridi et al. Reference Seridi, Amro, Kuhls, Belkaid, Zidane, Al-Jawabreh and Schönian2008; Chargui et al. Reference Chargui, Amro, Haouas, Schönian, Babba, Schmidt, Ravel, Lefebvre, Bastien, Chaker, Aoun, Zribi and Kuhls2009). The MON-1 strains were assigned to 2 different populations one of which contained only local strains and the other local and European strains of MON-1. The non-MON-1 strains were always separated from the MON-1. Gene flow was detected between the 2 MON-1 populations and the local MON-1 and the non-MON-1 populations, respectively. The existence of hybrid strains between different populations representing different zymodemes has been suspected in Algeria and was verified in Tunisia by analysis of clones of one of these strains.
The Israeli and Palestinian strains of L. infantum, all belonging to zymodeme MON-1, were assigned to 2 different populations (Amro et al. Reference Amro, Schönian, Al-Sharabati, Azmi, Nasereddin, Abdeen, Schnur, Baneth, Jaffe and Kuhls2009). One population comprised all but one strain isolated from human and canine cases in the Palestinian West Bank and central Israel. The second population, consisting of canine isolates from central and northern Israel, was further divided into 3 subpopulations according to the place of isolation. Canine leishmaniasis (CanL) and human VL are re-emerging in central and northern Israel and in Palestinian foci, respectively. The re-emergence of CanL is most likely due to increased dog and human contact with sylvatic cycles of parasitic infection, whereas the spread of the second population towards the centre of Israel seems to result from recent introduction of parasites from older foci of northern Israel. Interestingly, none of the Israeli and Palestinian strains were found to group with European MON-1 or non-MON-1 strains.
A preliminary analysis of the MLMT profiles obtained for 398 strains of L. infantum uncovered a hierarchical population structure (Kuhls and Schonian, unpublished observations), with 3 different populations. One population included all non-MON-1 strains, which were from North Africa, with some from southwest Europe, but none from eastern Mediterranean foci. MON-1 strains were divided into 2 populations. One population had isolates from southwest Europe and South America and the other was of eastern and southern Mediterranean origin (Fig. 1). Both MON-1 populations showed further geographical clustering.

Fig. 1. MLMT analysis of 398 strains of Leishmania infantum revealed the existence of three main populations in the Mediterranean basin. According to data analysis based on Bayesian statistics (Structure software) the strains belonging to the predominating zymodeme MON-1 were assigned to two different populations, shown in green and blue, whereas all other strains (non-MON-1) grouped together in the red population. The charts show the percentages of strains belonging to respective populations. The MON-1 populations correlate with the geographical origin of the isolates.
CONTRIBUTION OF MLST AND MLMT TO THE RESOLUTION OF MLEE ZYMODEMES
MLMT, and also MLST to a lesser extent, are further able to differentiate below the zymodeme level. As discussed above, MLMT revealed a remarkable genetic heterogeneity of L. infantum strains belonging to the zymodeme which is predominant in the Mediterranean area and in South America, MON-1. On the other hand, MLMT always assigned strains of zymodemes MON-77, 108 and 98 to the MON-1 cluster (Kuhls et al. Reference Kuhls, Chicharro, Canavate, Cortes, Campino, Haralambous, Soteriadou, Pratlong, Dedet, Mauricio, Miles, Schaar, Ochsenreither, Radtke and Schönian2008). This was confirmed by MLST for the zymodeme MON-98, for which the MLEE phenotype could not be explained by respective changes in the amino acid sequence (Zemanova et al. Reference Zemanova, Jirku, Mauricio, Horak, Miles and Lukes2007).
MLMT, as well as MLST, has also resolved dramatic cases of MLEE convergence. Recently, strains of L. donovani MON-37 were identified as the causative agent of VL and CL cases in Cyprus (Antoniou et al. Reference Antoniou, Haralambous, Mazeris, Pratlong, Dedet and Soteriadou2008). This zymodeme had previously been identified in Sri Lanka (Karunaweera et al. Reference Karunaweera, Pratlong, Siriwardane, Ihalamulla and Dedet2003) and earlier in India, Middle East and Kenya (Moreno et al. Reference Moreno, Rioux, Lanotte, Pratlong, Serres and Rioux1986; Moreno, Reference Moreno1989; Schnur et al. Reference Schnur, Eisenberger, Nasereddin, Dedet, Pratlong, Jaffe and Benami2001). It was suggested that human migration had been responsible for the introduction of this zymodeme in both countries, previously unknown to be foci of L. donovani. However, the MON-37 strains from each location were found to be paraphyletic (Alam et al. Reference Alam, Haralambous, Kuhls, Gouzelou, Sgouras, Soteriadou, Schnur, Pratlong and Schönian2009a). The ultimate origin of the different clades of this zymodeme, particularly in Cyprus, remains to be clarified.
CONTRIBUTIONS OF MLST AND MLMT TO THE ‘CLONALITY VS RECOMBINATION’ DEBATE
Leishmania parasites are still thought to reproduce predominantly clonally, although there is growing evidence of gene flow and recombination (Mauricio et al. Reference Mauricio, Yeo, Baghaei, Doto, Pratlong, Zemanova, Dedet, Lukes and Miles2006; Kuhls et al. Reference Kuhls, Chicharro, Canavate, Cortes, Campino, Haralambous, Soteriadou, Pratlong, Dedet, Mauricio, Miles, Schaar, Ochsenreither, Radtke and Schönian2008; Akopyants et al. Reference Akopyants, Kimblin, Secundino, Patrick, Peters, Lawyer, Dobson, Beverley and Sacks2009; Chargui et al. Reference Chargui, Amro, Haouas, Schönian, Babba, Schmidt, Ravel, Lefebvre, Bastien, Chaker, Aoun, Zribi and Kuhls2009).
The high F is values found in the MLMT analysis of Bolivian and Peruvian L. braziliensis (Rougeron et al. Reference Rougeron, De Meeus, Hide, Waleckx, Bermudez, Arevalo, Llanos-Cuentas, Dujardin, De Doncker, Le Ray, Ayala and Banuls2009) point to frequent sexual crosses of individuals from the same strain (inbreeding). The substantial heterozygote deficiency and extreme inbreeding found in this study is not consistent with a strictly clonal reproduction (Rougeron et al. Reference Rougeron, De Meeus, Hide, Waleckx, Bermudez, Arevalo, Llanos-Cuentas, Dujardin, De Doncker, Le Ray, Ayala and Banuls2009). Moreover, a strong population structure was found at a micro-geographical scale as the populations within the different countries were genetically heterogenous. This is in accord with the results of earlier population studies of L. infantum (Kuhls et al. Reference Kuhls, Chicharro, Canavate, Cortes, Campino, Haralambous, Soteriadou, Pratlong, Dedet, Mauricio, Miles, Schaar, Ochsenreither, Radtke and Schönian2008; Seridi et al. Reference Seridi, Amro, Kuhls, Belkaid, Zidane, Al-Jawabreh and Schönian2008; Amro et al. Reference Amro, Schönian, Al-Sharabati, Azmi, Nasereddin, Abdeen, Schnur, Baneth, Jaffe and Kuhls2009; Chargui et al. Reference Chargui, Amro, Haouas, Schönian, Babba, Schmidt, Ravel, Lefebvre, Bastien, Chaker, Aoun, Zribi and Kuhls2009), L. tropica (Schwenkenbecher et al. Reference Schwenkenbecher, Wirth, Schnur, Jaffe, Schallig, Al-Jawabreh, Hamarsheh, Azmi, Pratlong and Schönian2006), and L. major (Al-Jawabreh et al. Reference Al-Jawabreh, Diezmann, Mueller, Wirth, Schnur, Strelkova, Kovalenko, Razakov, Schwenkenbecher, Kuhls and Schoenian2008). Recent research by MLMT on Sudanese L. donovani showed populations consistent with inbreeding, although it also detected the first population without departure from Hardy-Weinberg equilibrium (Baleela, personal communication). Moreover, the detection of strains with potential mosaic and heterozygous genotypes, and of hybrids within L. infantum – even between MON-1 and non-MON-1 populations, and of high inbreeding within and gene flow between the populations, pointed to at least occasional recombination events. Rougeron et al. (Reference Rougeron, De Meeus, Hide, Waleckx, Bermudez, Arevalo, Llanos-Cuentas, Dujardin, De Doncker, Le Ray, Ayala and Banuls2009) came to the conclusion that Leishmania parasites may alternate between clonal and sexual modes of reproduction with the latter most probably occurring in the vector. Sexual fusion may frequently take place between genetically related parasites or even identical members of the same strain with occasional recombination events between individuals of different genotypes.
MLMT on parasites of the L. (V.) braziliensis complex showed that recombination events are much more frequent in Leishmania than previously thought. L. braziliensis/L. peruviana hybrids were found to be quite common in a Peruvian focus where both species can occur sympatrically (Nolder et al. Reference Nolder, Roncal, Davies, Llanos-Cuentas and Miles2007). In some MCL patients the hybrids were the only organisms isolated. Whether the hybrids may give rise to epidemiologically important emergent genotypes needs to be further elucidated. Volf et al. (Reference Volf, Benkova, Myskova, Sadlova, Campino and Ravel2007) demonstrated recently that L. infantum/L. major hybrids could be transmitted by Ph. papatasi which is normally only competent to transmit L. major. This suggests that the hybrids may circulate using this sand fly vector and spread into new foci throughout the broad range of Ph. papatasi distribution.
MLST has so far been able to identify hybrids and genome mosaics (Mauricio et al. Reference Mauricio, Yeo, Baghaei, Doto, Pratlong, Zemanova, Dedet, Lukes and Miles2006; Zemanova et al. Reference Zemanova, Jirku, Mauricio, Horak, Miles and Lukes2007) much more reliably than MLEE, and even MLMT, which is more prone to homoplasy. Such results have also suggested that recombination is likely to have been much more frequent historically and in extant populations, than previously recognized.
CONCLUSIONS
Global epidemiological, taxonomic and population genetic studies of Leishmania require good sampling strategies and appropriate molecular markers that allow discrimination at the desired genetic level. Markers that are stable during in vitro or in vivo passages and can be tested directly on clinical samples would be advantageous. The markers should accommodate test assumptions, comparisons with different analytic methods and assessment of robustness of results by using re-sampling techniques. The typing results should be reproducible, comparable between laboratories and storable in databases. Finally, high-throughput cost-effective methods are needed for large-scale studies. This is necessary since sampling should include isolates from many endemic foci, all possible hosts, and representing different zymodemes and clinical forms of the disease. Sample size is crucial as it has a direct effect on the ability to test hypotheses.
Answering key epidemiological questions requires new or improved tools that allow for differentiation of Leishmania parasites at species and strain levels. The current gold standard, MLEE, has some drawbacks, such as the need for cultured parasites and the lack of discriminatory power. At the species level, it should be replaced by PCR assays that enable direct detection and identification of different species of Leishmania in human and animal samples and in infected sand flies. Many of the PCR assays described in the literature have proven useful in numerous field studies. There is, however, an urgent need for standardization and validation of diagnostic PCR assays and for comparisons of the sensitivity and specificity of different approaches under routine conditions.
When different DNA-based methods of strain typing were compared, PCR-RFLP of kinetoplast minicircle DNA and MLMT were found to be most discriminatory at intra-species level, allowing further characterization of the parasite diversity and establishment of genetic links between remote populations of L. infantum and L. donovani (Botilde et al. Reference Botilde, Laurent, Quispe Tintaya, Chicharro, Canavate, Cruz, Kuhls, Schönian and Dujardin2006, Bhattarai et al. Reference Bhattarai, Dujardin, Rijal, De Doncker, Boelaert and Van der Auwera2010). Because of its better reproducibility and possibility of data storage and exchange, MLMT currently seems to be the best candidate for becoming the gold standard for strain level differentiation. Since microsatellite markers are largely species-specific in Leishmania and different marker sets have to be used with different species, MLMT is not suited for inferring phylogeny.
Although less discriminatory, MLST is potentially the most powerful phylogenetic approach and will, most probably, advantageously replace MLEE in the future. Preliminary results show that the same targets can be used across the Leishmania genus which will enable comparisons of distances between the species but also of the degree of genetic diversity within species (Miles et al. Reference Miles, Llewellyn, Lewis, Yeo, Baleela, Fitzpatrick, Gaunt and Mauricio2009).
New high-throughput and cheaper sequencing technologies have opened the door for genome-wide multilocus genotyping in malaria research. Almost 47 000 single nucleotide polymorphisms (SNPs) were identified across the Plasmodium genome (Volkman et al. Reference Volkman, Sabeti, DeCaprio, Neafsey, Schaffner, Milner, Daily, Sarr, Ndiaye, Ndir, Mboup, Duraisingh, Lukens, Derr, Stange-Thomann, Waggoner, Onofrio, Ziaugra, Mauceli, Gnerre, Jaffe, Zainoun, Wiegand, Birren, Hartl, Galagan, Lander and Wirth2007). This allowed development of microarray-based platforms for screening more than 3000 SNPs that were successfully applied for population genetic analyses and genome-wide association studies in P. falciparum (Neafsey et al. Reference Neafsey, Schaffner, Volkman, Park, Montgomery, Milner, Lukens, Rosen, Daniels, Houde, Cortese, Tyndall, Gates, Stange-Thomann, Sarr, Ndiaye, Ndir, Mboup, Ferreira, Moraes Sdo, Dash, Chitnis, Wiegand, Hartl, Birren, Lander, Sabeti and Wirth2008; Mu et al. Reference Mu, Myers, Jiang, Liu, Ricklefs, Waisberg, Chotivanich, Wilairatana, Krudsood, White, Udomsangpetch, Cui, Ho, Ou, Li, Song, Li, Wang, Seila, Sokunthea, Socheat, Sturdevant, Porcella, Fairhurst, Wellems, Awadalla and Su2010). Research on Leishmania has already taken steps in similar directions, which are likely to finally enable researchers to answer some questions on population genetic questions, and ultimately tease out key aspects of drug resistance, virulence and other important issues relevant for control.
ACKNOWLEDGEMENTS
We are extremely grateful to all our collaborators, cited in the accompanying bibliography, for their friendship and support. We thank Michael Miles for his critical reading of the manuscript which helped to considerably improve it.
FINANCIAL SUPPORT
The work on which this review is based was funded in part by EC (contracts 01810 Leishmania genotyping and 015407 LeishEpiNetSA), the Deutsche Forschungsgemeinschaft (contracts Scho 448/6-1-3 and Scho 448/8-1), the Deutsche Akademische Austauschdienst and other financial contributors.