


Tetralogy of Fallot occurs in about four per 10,000 live births and accounts for approximately one-tenth of important congenital heart defects. The cardiac morphology is variable. There is usually pulmonary outflow tract obstruction with confluent central pulmonary arteries, but more complex forms may be associated with pulmonary atresia, major aortopulmonary collateral arteries, absent pulmonary valve syndrome, or atrioventricular septal defect. Trisomy 21 and chromosome 22q11.2 deletion are the most frequent associated genetic anomalies.Reference Rauch, Hofbeck and Zweier 1
Associations between Trisomy 21 and aberrant right subclavian artery, and between right aortic arch and chromosome 22q11.2 deletion, have been described. However, there are few systematic studies in clearly defined populations that have examined the relationship between variation in aortic arch anatomy and chromosomal or genetic abnormalities. Prenatal diagnosis of tetralogy of Fallot is common.Reference Poon, Huggon, Zidere and Allan 2 , Reference Bull 3 Surgical repair of tetralogy of Fallot with pulmonary stenosis affords very good long-term prognosis from the cardiac perspective.Reference Bailliard and Anderson 4 We investigated the relationship between variation in aortic arch anatomy and chromosomal or genetic abnormalities in patients with tetralogy of Fallot with pulmonary stenosis, principally to assess whether this could inform counselling with regard to decisions about invasive foetal chromosomal investigation after prenatal diagnosis of tetralogy of Fallot with pulmonary stenosis.
Materials and methods
Design
A retrospective analysis was undertaken of 257 consecutive patients with tetralogy of Fallot with pulmonary stenosis who underwent intracardiac repair from January, 2003 to March, 2011 at Great Ormond Street Hospital. There were 149 male and 108 female patients with median age at surgery of 8 months (range 1 month to 10 years).
Exclusion criteria
Tetralogy of Fallot associated with pulmonary atresia; major aortopulmonary collateral arteries requiring intervention, that is, ligation, occlusion, or unifocalisation; absent pulmonary valve syndrome; or atrioventricular septal defect, that is, common atrioventricular junction. These variant forms of tetralogy of Fallot are known to have a higher incidence of chromosomal anomalies and genetic syndromes than tetralogy of Fallot with pulmonary stenosis.
Data
Data were collected from medical records, department database, and archived echocardiography images in all patients. Computerised tomography, cardiac magnetic resonance, or angiographic images were reviewed in those in whom additional imaging had been performed. Echocardiography was performed according to a standard protocol, which included documentation of aortic arch laterality and branching pattern. The echocardiographic images were reviewed and interpretation confirmed in unit meetings at least twice before surgical repair.
Chromosomal study was requested in all patients with aortic arch abnormality, absent or hypoplastic thymus, dysmorphic features, or other extracardiac abnormality. In the remaining patients, these studies were performed according to physician and family decision. Informed consent was obtained from parents before analysis. Standard G-banding karyotype with fluorescent in situ hybridisation using a probe for the critical region at 22q11.2 was performed for all referred until 1 January, 2011; subsequently, patients received genome-wide array comparative genome hybridisation analysis at a resolution of 0.2 MB with Nimblegen array. Additional genetic studies were targeted according to clinical features.
Aortic arch anatomy
We classified aortic arch anatomy into four groups:
-
1) left aortic arch with normal branching pattern (Fig 1);

Figure 1 Left aortic arch with normal branching. Diagram of left aortic arch with normal branching pattern. The first branch is the brachiocephalic trunk bifurcating into RSA and RCCA. The second aortic branch is the LCCA. The third aortic branch is the LSA. Asc Ao = ascending aorta; Des Ao = descending aorta; LCCA = left common carotid artery; LSA = left subclavian artery; RCCA = right common carotid artery; RSA = right subclavian artery.
-
2) right aortic arch with mirror image branching pattern (Fig 2a and b);
Figure 2 Right aortic arch with mirror image branching. (a) Suprasternal coronal echocardiographic image illustrates the superior part of the Asc Ao and the first aortic branch. This is the left brachiocephalic artery, dividing into LCCA and LSA. (b) Suprasternal right oblique echocardiographic view in the long axis of the right aortic arch illustrates the origin of the second aortic branch, which is RCCA, and the origin of the third aortic branch, which is RSA. Asc Ao = ascending aorta; LCCA = left common carotid artery; LSA = left subclavian artery; RCCA = right common carotid artery; RSA = right subclavian artery.
-
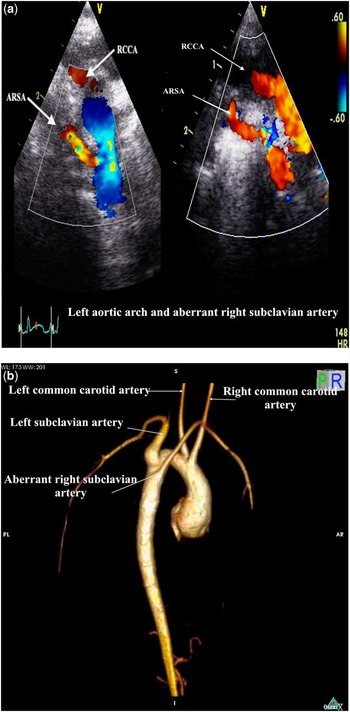
3) left aortic arch with aberrant right subclavian artery (Fig 3a and b);
Figure 3 Left aortic arch and ARSA. (a) Suprasternal coronal echocardiographic images. Left: posterior imaging plane demonstrating origin of an ARSA from Des Ao. Right: a more anteriorly angled imaging plane shows the parallel courses of the non-dividing first aortic branch (RCCA), with the aberrant RSA coursing inferior to this. (b) Three-dimensional reconstruction of cardiac magnetic resonance image. Right posterior oblique view to illustrate the posterior origin of the ARSA, which is the fourth aortic arch branch and arises from the superior part of the thoracic Des Ao. Note that the first branch, which is the right common carotid artery, does not have proximal bifurcation. ARSA aberrant right subclavian artery; Des Ao = descending aorta; RCCA = right common carotid artery; RSA = right subclavian artery.
-
4) right aortic arch with aberrant left subclavian artery (Fig 4).
Figure 4 Right aortic arch and ALSA. Suprasternal coronal echocardiographic image angled posteriorly to show the origin of the ALSA from the superior part of the thoracic descending aorta. The first branch of the arch courses to the left but does not divide (LCCA, origin not seen), and the ALSA courses inferior to this. ALSA = aberrant left subclavian artery; LCCA = left common carotid artery.



Statistical analysis
The association between the four groups of aortic arch anatomy and documented chromosomal or genetic abnormality was assessed using odds ratios; the most common aortic arch anatomy – left aortic arch with normal branching – was assigned an odds ratio of 1. The odds ratio for the other groups was considered significant if the 95% confidence intervals did not include zero. Proportions were compared using Fisher's exact test.
Results
A total of 257 consecutive patients with tetralogy of Fallot with pulmonary stenosis underwent intracardiac repair between January, 2003 and March, 2011. Left aortic arch with normal branching was identified in 203 (79%), right aortic arch with mirror image branching pattern in 36 (14%), left aortic arch with aberrant right subclavian artery in 8 (3%), and right aortic arch with aberrant left subclavian artery in 10 (4%) patients.
Chromosomal analysis was performed in 161 of the 257 (63%) patients. Chromosomal analysis was not performed in 96 of the 257 (37%) patients, all of whom had left aortic arch with normal branching, normal thymus appearance, normal phenotypic appearance, and no clinical suspicion of chromosomal or genetic abnormality on follow-up.
A chromosomal or genetic abnormality was identified in 49 of the 257 (19%) patients (Table 1). Of these 49 patients, chromosome 22 q11.2 deletion was present in 16 of 49 (32%) and trisomy 21 was detected in 14 of 49 (29%) patients. There were nine (18%) patients who had other chromosomal anomalies. These included deletion of chromosome 2, deletion of chromosome 17p, deletion of chromosome 7p, deletion of chromosome 18p, unbalanced translocation of chromosomes 4 and 11, 46XXXY, partial small deletion of chromosomes 2 and 11 – paternally inherited, balanced translocation between chromosomes 20 and 22, and balanced Robertsonian translocation of chromosomes 13 and 14 – paternally inherited. The last three of these were of doubtful phenotypic significance. The rest of the identifiable genetic abnormalities are shown in Table 1.
Table 1 Chromosomal abnormalities and syndromes in all patients.

CHARGE: Coloboma of the eye, heart defect, atresia of choanae, retarded growth and development, genital and urinary abnormalities, ear abnormalities and deafness
PHACE: Posterior fossa malformation, haemangioma, arterial lesions, cardiac defect, eye abnormalities
In patients with left arch and normal branching pattern, chromosomal or genetic abnormality was identified in 33 of 203 (16%) patients. These included chromosome 22q11.2 deletion (n = 10), trisomy 21 (n = 10), other chromosomal anomalies (n = 6), Pierre Robin (n = 2), CHARGE (n = 1), Alagille (n = 1), Kaufman McKusick (n = 1), Goldenhar (n = 1), and Kabuki (n = 1). In the group with right aortic arch and mirror image branching pattern, three of 36 (8%) patients had chromosomal anomalies, comprising chromosome 22q11.2 deletion (n = 1), balanced translocation between chromosomes 20 and 22 (n = 1), and Chromosome 7p deletion, that is, Smith–Magenis syndrome (n = 1).
An aberrant subclavian artery origin was identified in 18 of 257 (7%) patients, and 13 of those 18 (72%) patients were diagnosed with chromosomal or genetic abnormality (Table 2). These included seven of eight patients with a left aortic arch and aberrant right subclavian artery, comprising chromosome 22q11.2 deletion (n = 3), trisomy 21 (n = 2), Holt–Oram (n = 1), and CHARGE syndrome (n = 1). A right aortic arch and aberrant left subclavian artery was present in 10 patients, six of whom had chromosomal or genetic abnormality, comprising chromosome 22q11.2 deletion (n = 2), trisomy 21 (n = 2), PHACE, and Chromosome 18p deletion (n = 1 each).
Table 2 Chromosomal abnormalities and syndromes in patients with aberrant subclavian artery.

The odds ratio for a chromosomal or genetic abnormality when there was an aberrant subclavian artery origin, compared with a left aortic arch and normal branching, was 15 (95% confidence interval 4.5–50; p < 0.0001, Fisher exact test, two-tailed value). In contrast, the odds ratio for right aortic arch and mirror image branching was 0.4 (95% confidence interval 0.1–1.6) (Table 3).
Table 3 Odds ratios for chromosomal abnormalities and syndromes according to aortic arch anatomy.

OR = odds ratio; CI = confidence interval
If analysis is confined to those patients in whom chromosomal study was performed, the odds ratios for associated chromosomal or genetic syndrome in the separate groups of aortic arch morphology are shown in Table 4.
Table 4 Odds ratios for chromosomal abnormalities and syndromes according to aortic arch anatomy confined to patients with chromosomal analysis.

OR = odds ratio; CI = confidence interval
Discussion
Chromosomal abnormalities
In a consecutive series of patients with tetralogy of Fallot with pulmonary stenosis undergoing cardiac surgical repair, we demonstrated a 19% incidence of associated chromosomal anomalies or genetic syndrome. The most common chromosomal anomalies were trisomy 21 (5.5%) and 22q11.2 deletion (7%). This is a lower incidence of chromosomal abnormality than reported in most studies of tetralogy of Fallot.Reference Poon, Huggon, Zidere and Allan 2 , Reference Marino, Digilio and Toscano 5 , Reference Samanek, Slavik, Zborilova, Hrobonova, Voriskova and Skovranek 6 This is explained by case selection. The incidence of chromosomal anomalies is higher in patients with tetralogy of Fallot in the presence of associated anomalies such as pulmonary atresia, major aortopulmonary collateral arteries, atrioventricular septal defect, or absent pulmonary valve syndrome.Reference Rauch, Hofbeck and Zweier 1 , Reference Chessa, Butera and Bonhoeffer 7 , Reference Lammer, Chak and Iovannisci 8 Termination of pregnancy may also have contributed to a lower incidence of chromosomal abnormalities in our patients. Any effect of this is difficult to quantify, as geographic foetal cardiology referral patterns in the south of England do not match exactly postnatal cardiac surgical services. Termination of pregnancy is weighted heavily towards complex forms of tetralogy, associated extracardiac defects, or chromosomal abnormality.Reference Poon, Huggon, Zidere and Allan 2 Natural selection excluded lethal chromosomal abnormalities such as Trisomy 18 or 13 from our cohort. It is most unlikely that decisions regarding termination of pregnancy when Trisomy 21 or chromosome 22q11.2 deletion was detected were influenced by aortic arch anatomy, and there is no reason to believe that the distribution of aortic arch anatomy in those with tetralogy of Fallot with pulmonary stenosis would have differed from this postnatal series.
Aberrant subclavian artery origin
The incidence of aberrant right subclavian artery in the general population is in the range 0.1–2.3%.Reference Zapata, Edwards and Titus 9 – Reference Borenstein, Minekawa, Zidere, Nicolaides and Allan 11 It is higher in trisomy 21, with or without associated cardiac defects. In patients with a range of cardiac defects, the incidence of aberrant right subclavian artery is about 3% in those with normal karyotype, compared with about 17% in those with Trisomy 21.Reference Borenstein, Minekawa, Zidere, Nicolaides and Allan 11 The higher incidence of aberrant right subclavian artery in patients with trisomy 21 led to studies investigating whether prenatal detection of aberrant right subclavian artery would be a useful predictor of Trisomy 21 in population screening. A higher incidence of aberrant right subclavian artery in foetuses with Trisomy 21 was confirmed, with an incidence of 0.6–1.4% in large groups of “normal” foetuses, compared with 8–37.5% in the much smaller number of foetuses with Trisomy 21.Reference Borenstein, Minekawa, Zidere, Nicolaides and Allan 11 – Reference Zalel, Achiron, Yagel and Kivilevitch 13 Interestingly, the prevalence of aberrant right subclavian artery was higher in trisomy 21 foetuses without cardiac defects than in those with cardiac defects.Reference Chaoui, Heling, Sarioglu, Schwabe, Dankof and Bollmann 14 , Reference Borenstein, Cavoretto, Allan, Huggon and Nicolaides 15 However, the relatively high incidence of aberrant right subclavian artery in foetuses with normal karyotype, and the technical difficulty of diagnosis by anyone other than highly skilled ultrasonographers, hinders a potential role in unselected population screening for prenatal detection of Trisomy 21.
Right aortic arch
Right aortic arch has been reported to be an indicator of an increased risk of chromosomal abnormality. Right aortic arch with normal intracardiac anatomy was associated with chromosome 22q11.2 deletion in 11 of 37 (30%) patients in one report, with a similar incidence regardless of whether there was associated aberrant origin of the left subclavian artery.Reference McElhinney, Clark and Weinberg 16 The incidence of chromosomal abnormalities was 46%, that is, 23 of 50 cases, mainly 22q11.2 deletion, in a series of foetuses with right aortic arch in association with tetralogy of Fallot or common arterial trunk.Reference Zidere, Tsapakis, Huggon and Allan 17 The incidence of chromosomal abnormality was 59%, that is, 16 of 27 cases, when a right aortic arch was associated with tetralogy of Fallot in an overlapping foetal series, which included all variant forms of tetralogy of Fallot.Reference Poon, Huggon, Zidere and Allan 2 The branching pattern of the aortic arch was not described in either of these reports. Assessment confined to patients with tetralogy of Fallot with pulmonary stenosis reported chromosome 22q11.2 deletion in 41% with right aortic arch compared with 18% of those without.Reference Momma 18
Implications of this study
In our study, the incidence of aberrant origin of a subclavian artery (aberrant right subclavian artery or aberrant left subclavian artery) was 5% in the cohort with tetralogy of Fallot with pulmonary stenosis, which is similar to the incidence previously described.Reference Zapata, Edwards and Titus 9 We detected chromosomal or genetic abnormalities in 72% (13/18) of patients with either aberrant right subclavian artery or aberrant left subclavian artery. None of these had balanced or inherited chromosomal abnormalities of doubtful phenotypic significance. The incidence of chromosomal or genetic anomalies in the presence of right aortic arch with mirror image branching was not increased (three of 36 cases, odds ratio 0.4, 95% confidence interval 0.1–1.6, compared with left aortic arch with normal branching). This suggests that the previously reported association between right aortic arch and chromosomal abnormality in tetralogy of Fallot was not accounted for by the presence of a right aortic arch per se, but rather by the combination of right aortic arch with aberrant left subclavian artery, or an association with variant forms of tetralogy of Fallot other than tetralogy of Fallot with pulmonary stenosis.
We cannot exclude the possibility that subtle abnormalities may have been present in some patients who did not have formal chromosomal analysis. None of the patients in whom chromosomal analysis was not performed had dysmorphic or other features identified during follow-up to suspect a chromosomal or genetic syndrome. All of these patients had left aortic arch with normal branching pattern, with an apparently normal thymus identified on echocardiography and confirmed at surgery. It is reasonable to conclude that none of these patients would have had abnormality sufficient to influence a decision for termination of pregnancy if invasive chromosomal analysis had been performed after prenatal diagnosis.
We confined our study to consecutive patients with tetralogy of Fallot with pulmonary stenosis undergoing complete surgical repair. This represents a clearly defined and relatively homogeneous group at the less severe end of the morphological spectrum of tetralogy of Fallot, in whom a good long-term outcome would be anticipated from the cardiac viewpoint. There were only two (0.8%) deaths within 30 days of surgical repair in this cohort. The incidence of comorbid chromosomal or genetic abnormalities is lower in tetralogy of Fallot with pulmonary stenosis than in more “severe” variant forms of tetralogy of Fallot, in which there is already a more guarded cardiac prognosis regardless of extracardiac abnormality. It is in the anticipated good cardiac outcome category of tetralogy of Fallot with pulmonary stenosis that decisions about the relative risks and benefits of invasive foetal investigation for chromosomal analysis can be the most difficult. Aberrant origin of a subclavian artery is detectable using colour flow or power Doppler assessment during foetal echocardiography. The additional prognostic information provided by knowledge of aortic arch branching when tetralogy of Fallot with pulmonary stenosis is diagnosed prenatally can usefully inform the often difficult decision regarding invasive foetal chromosomal investigation.








