



Isomerism, also referred to as heterotaxy syndrome, is a rare congenital disorder characterised by abnormal symmetry and malposition of the thoracoabdominal organs and vessels.Reference Loomba, Hlavacek, Spicer and Anderson1 Consistent nomenclature has been controversial and is best summarized in a recent review by Jacobs et al.Reference Jacobs, Anderson and Weinberg2 It is believed to be underpinned by disorders of left-right axis determination during early embryonic development and failure of the developing embryo to establish normal asymmetry.Reference Catana and Apostu3 Most cases of isomerism are the result of sporadic mutations, though recent evidence suggests some are due to familial inheritance with variable penetrance.Reference Aylsworth4,Reference Sempou and Khokha5 A variety of genetic mutations have been found to be associated with isomerism.Reference Loomba, Frommelt and Anderson6 The heterogeneity of the isomerism phenotype and genotype makes gene discovery challenging.Reference Sempou and Khokha5
Any organ system can be impacted in the setting of isomerism, although the number of systems and the extent to which they are impacted is highly variable. Manifestations can be anatomic and/or functional.Reference Loomba, Ahmed, Spicer, Backer and Anderson7,Reference Loomba, Shah, Anderson and Arora8 While hepatic abnormalities are often present in heterotaxy patients, congenital extrahepatic portosystemic shunts, also known as Abernethy malformation, are infrequent.Reference Newman, Feinstein and Cohen9 We describe one such case of isomerism diagnosed later in life and presenting with pulmonary vascular disease secondary to Abernethy malformation. We also review the multiple cardiac and extracardiac manifestations of isomerism.
Case Presentation
A previously healthy 14-year-old female presented to the emergency room solely for evaluation of pharyngitis, but upon further questioning was found to have exercise intolerance and significant dyspnea on exertion. Past medical history was otherwise unremarkable and family history was significant for a brother with a cerebrovascular accident at the age of 13 years. Her blood pressure, temperature, and respiratory rate were within normal limits; however, bradycardia was noted with a heart rate between 40 and 50 beats per minute. Physical exam was remarkable for a thin, prepubescent girl without distress at rest but with noticeable tachypnea when walking. She had multiple retained primary teeth. Her cardiac exam revealed a normally located cardiac impulse, a loud single second heart sound, and no murmur. There was no oedema or hepatomegaly.
Routine labs revealed no abnormalities. Her electrocardiogram was significant for left atrial rhythm alternating with sinus node dysfunction with a junctional escape rhythm and multiform ventricular bigeminy (Fig 1). A chest X-ray (Fig 2) revealed significantly enlarged central pulmonary arteries with distal pruning and bilateral anatomically left pulmonary arteries. The echocardiogram showed a dilated right atrium, right ventricle, and pulmonary arteries. There was no tricuspid regurgitation to estimate right ventricular pressure, but the interventricular septal position was flat consistent with elevated right ventricular pressure. Biventricular function was normal. She underwent a 6-minute walk test of 488 m and she did not experience oxygen desaturations with exertion. Cardiac catheterisation revealed a normal right atrial pressure of 8 mmHg, a significantly elevated right ventricular pressure of 70/8 mmHg, as well as a significantly elevated main pulmonary artery pressure of 70/14 with a mean of 40 mmHg. The pulmonary capillary wedge pressure was normal at 9 mmHg and the pulmonary vascular resistance was elevated at 8.4 Wood Units. Her oxygen saturations were 95% in room air, and her mixed venous saturation was 77% as measured in the branch pulmonary artery, consistent with normal cardiac output. There were no shunts. Angiography demonstrated a dilated right ventricle, main pulmonary artery, and branched pulmonary arteries. With administration of the pulmonary vasodilators nitric oxide at 40 parts per million and 100% oxygen, the pulmonary artery pressure decreased to 40/22 mmHg with a mean of 30 mmHg. Her mixed venous saturation was 86% and her Qp:Qs remained 1:1. Though still abnormal, her pulmonary vascular resistance was reactive to pulmonary vasodilators and decreased to 3.9 Wood Units.

Figure 1. Sinus node dysfunction with junctional escape rhythm and multiform ventricular bigeminy.

Figure 2. Chest X-ray with blue arrows demonstrating left-sided morphology of both pulmonary arteries.
High-resolution CT revealed clear lungs with no masses or arteriovenous malformations, but with marked enlargement of the main pulmonary arteries (Fig 3). Other findings were notable for left-sided morphology of both bronchi and pulmonary arteries, and lungs with absence of a right-sided middle lobe consistent with left-sided isomerism, as also depicted on chest X-ray (Fig 2). An abdominal and pelvic CT supported the above diagnosis with multiple spleens of varying size (Fig 4) each with their own arterial and venous supply, a small truncated pancreas, and two lobulated liver masses within the hypertrophied right lobe, consistent with focal nodular hyperplasia (Fig 5). Additionally, there was absence of the hepatic segment of the inferior vena cava with azygos continuation above the renal veins and the hepatic lobes drained into the underside of the right atrium. The small intestine was located entirely in the right hemiabdomen with an ill-defined ileocecal valve. CT of the abdomen also revealed complex vascular malformations with multiple tortuous collaterals and anatomic variants of the superior mesenteric artery and its branches (Fig 6). The portal vein was not defined on any of her studies corroborating the diagnosis of Abernethy malformation. A diagnosis of left isomerism with Abernethy malformation was established and the patient’s pulmonary hypertension and pulmonary vascular disease was secondary to portopulmonary hypertension and led to the initiation of pulmonary vasodilator therapy.
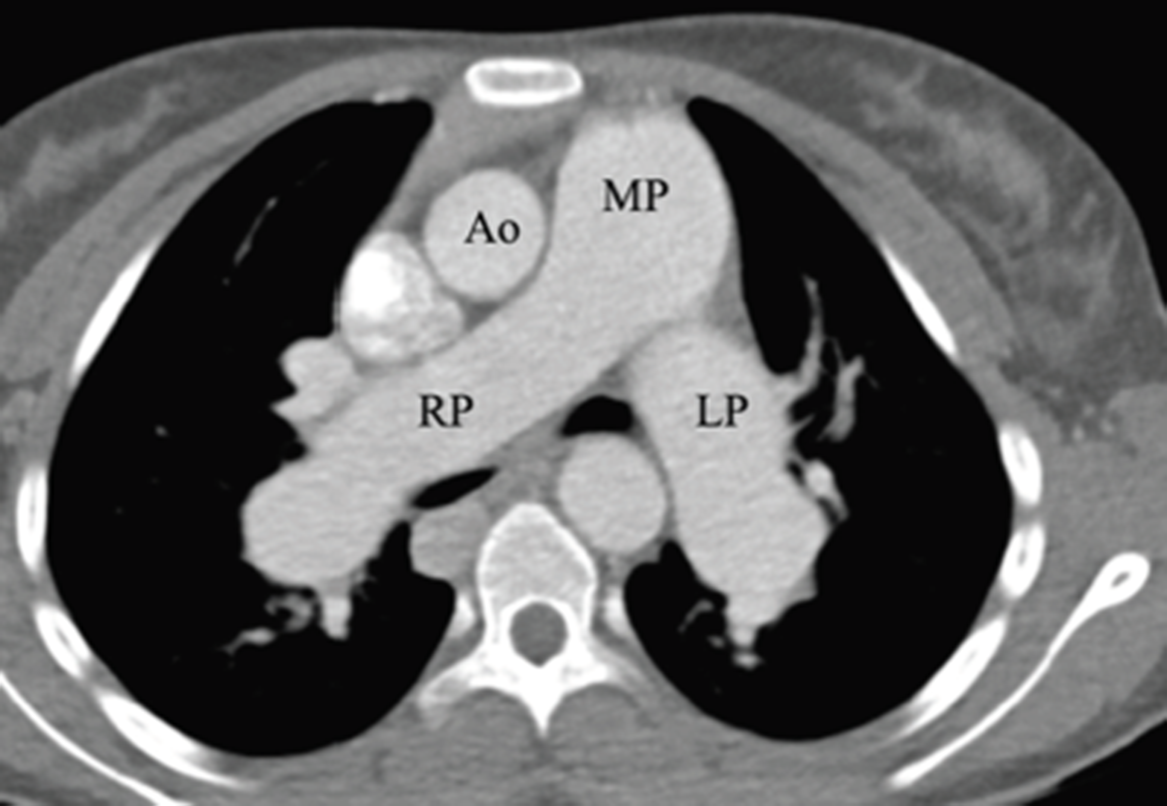
Figure 3. CT axial image demonstrating marked enlargement of the main pulmonary arteries (MP) as well as right and left branch pulmonary arteries (RP and LP, respectively). Note that each branch pulmonary artery is larger than the aorta (Ao).

Figure 4. CT demonstrating multiple splenic lobes lateral to the stomach (blue arrows).

Figure 5. CT abdomen demonstrating hepatomegaly with two lobulated liver masses (blue arrows) that appear to be multifocal nodular hyperplasia.

Figure 6. CT abdomen shows superior mesenteric vein (green arrow) draining via a collateral vein (blue arrow) which eventually drains into the inferior vena cava. Superior mesenteric vein does not continue superiorly and no portal vein is formed.
Genetic testing was performed and a karyotype demonstrated no abnormalities and 46XX. A microarray revealed a 451-kb deletion in 7q11.23 which impacted the following genes: GTF2I, NCF1, GTFIRD2, and GTFIRD2B. A FBN mutation was also identified in this patient; all of which are variants of unknown significance.
Discussion
Isomerism is a multisystem disease which may be particularly challenging when complicated by the complex interactions between the liver, lungs, and cardiovascular systems. We describe a case of left isomerism that was diagnosed in adolescence rather than the typical infancy or early childhood period. Although she had an interrupted inferior caval vein and a persistent left superior caval vein, the presence of an otherwise structurally normal heart undoubtedly facilitated her later presentation. Her diagnosis was ultimately made due to shortness of breath and exercise intolerance from a combination of a slow heart rate and portopulmonary hypertension, a direct result of Abernethy malformation, which is a rare finding in patients with isomerism. This case is unique in that it highlights the variability in how patients with isomerism may present as well as the association of an additional rare congenital anomaly. Two important manifestations resulting from hepatic, pulmonary, and cardiovascular interactions are hepatopulmonary syndrome and portopulmonary hypertension. Hepatopulmonary syndrome is seen more commonly following superior cavopulmonary anastomoses for patients with a functionally univentricular heart.Reference Loomba10,Reference Krowka, Fallon and Kawut11 Portopulmonary hypertension describes the presence of pulmonary arterial hypertension in the setting of portal hypertensionReference Krowka, Fallon and Kawut11,Reference Simonneau, Gatzoulis and Adatia12 and is diagnosed by right heart catheterisation measurements demonstrating a mean pulmonary artery pressure >25 mmHg, pulmonary vascular resistance >3 Wood Units, and a pulmonary artery wedge pressure <15 mmHg.Reference Krowka, Fallon and Kawut11
Abernethy malformation and portopulmonary hypertension
Abernethy malformation is a rare vascular anomaly in which the splanchnic venous blood bypasses the liver, secondary to congenital absence or hypoplasia of the portal vein, and drains directly into the systemic veins. This rare association can be seen with left isomerism as exhibited by our patient. Type I is characterised by complete absence of a portal vein and type II by reduced portal flow through a hypoplastic portal vein.Reference Lisovsky, Konstas and Misdraji13 Abernethy malformation can have a wide variety of clinical manifestations, ranging from asymptomatic to severe hepatopulmonary manifestations. Portopulmonary hypertension is defined as the concomitant presence of pulmonary arterial hypertension and portal hypertension with or without cirrhosisReference Savale, Guimas and Ebstein14–Reference Sitbon, Bosch and Cottreel16; it is associated with progressive shortness of breath and exercise limitation and results from complex pathophysiologic interactions between the portal and pulmonary circulations.Reference Savale, Guimas and Ebstein14,Reference Savale, Watherald and Sitbon15 Aetiologies of portal hypertension include cirrhosis, alcohol, hepatitis, autoimmune, extrahepatic portal hypertension, and congenital portosystemic shunts, among others.Reference Savale, Guimas and Ebstein14 Portopulmonary hypertension is characterised by constricted pulmonary vasculatureReference Law, Mack, Sokol, Rice, Parsley and Ivy17; in contrast to hepatopulmonary syndrome where the pulmonary vascular abnormality consists of diffuse arteriocapillary dilation with rapid transit though the lung, and intrapulmonary shunt, and systemic hypoxemia.Reference Newman, Feinstein and Cohen9,Reference Law, Mack, Sokol, Rice, Parsley and Ivy17,Reference Whitworth, Ivy, Gralla, Narkewicz and Sokol18 In cases of Abernethy malformation, there is an imbalance leading towards pulmonary vasoconstriction and subsequent pulmonary vascular disease, right ventricular failure, and in some cases, death.
The ultimate goal is reversal of the portal hypertension, often requiring a liver transplantation; however, patients with severe portopulmonary hypertension have a higher mortality with transplant unless the pulmonary arterial pressure can be lowered into a safe range, and aggressive management of the pulmonary arterial hypertension to ensure patients remain ideal candidates for liver transplantation is crucial. Such therapies include supplemental oxygen to maintain saturations greater than 92%, diuretics to control volume overload, and calcium channel blockers and vasodilators.Reference Savale, Guimas and Ebstein14,Reference Condino, Ivy and O’Connor19 In a large French registry of patients with portopulmonary hypertension, Savale et al noted improved survival in a subgroup of patients who underwent liver transplant compared with those who were medically managed.Reference Savale, Guimas and Ebstein14
In cases of congenital absence of the portal vein and pulmonary arterial hypertension, liver transplantation remains the primary option and should be considered early in the disease course so as to prevent the development of severe pulmonary arterial hypertension and subsequent right ventricular dysfunction at the time of transplant. Because pulmonary arterial hypertension is progressive in this disease, minimisation of pulmonary vascular remodelling is crucial and treated with pulmonary arterial vasodilators. In a case report of two young children with heterotaxy polysplenia anatomy and congenital absence of the portal vein, a significant clinical improvement in pulmonary artery pressures were noted after the treatment was initiated with sildenafil.Reference Law, Mack, Sokol, Rice, Parsley and Ivy17 In another patient in whom a liver transplant was required secondary to complete absence of the portal vein, a clinical response was seen when portal venous flow was reestablished, indicating that the disease process is reversible.Reference Condino, Ivy and O’Connor19 For Abernethy type II cases involving a hypoplastic portal vein and extrahepatic shunts, occlusion of the abnormal shunt can be attempted and in some may obviate or delay the need for transplant. Newman et al described an identifiable hypoplastic portal vein in three children with Abernethy type II; all three underwent percutaneous shunt occlusion with short-term improvement in hypoxemia and pulmonary hypertension obviating the need for immediate liver transplant.Reference Newman, Feinstein and Cohen9
Returning to our patient, later in her course, macitentan was added to her vasodilatory therapy. Macitentan is an endothelin receptor antagonist indicated for the treatment of pulmonary arterial hypertension and has demonstrated efficacy in both the SERAPHIN and PORTICO trials, addressing pharmacologic treatment for pulmonary arterial hypertension in patients with and without portopulmonary hypertension.Reference Sitbon, Bosch and Cottreel16,Reference Galiè, Jansa and Pulido20
Isomerism
Isomerism affects approximately 1 in 10,000 live births and can impact any combination of the organ systems in anatomic and/or functional manner.Reference Loomba21–Reference Gabriel and Lo24 Due to the multisystem nature of isomerism, it highly impacts morbidity and mortality in these patients.Reference Loomba and Frommelt25–Reference Zou, Wang and Cui35
Cardiac anatomy
Isomerism itself is best segregated into the subsets of left and right based on the morphology of the atrial appendages. While isomerism has previously been described based on subsets of splenic anatomy, the atrial appendage method offers a more robust means by which to sort isomerism. This method also eliminates the issue that arises with patients who have a normal spleen.Reference Loomba, Hlavacek, Spicer and Anderson1,Reference Uemura, Ho, Devine and Anderson36,Reference Uemura, Ho, Devine, Kilpatrick and Anderson37 Within the heart, only the atrial appendages are truly isomeric, and isomerism within the atrial component of the heart points to a left-right patterning abnormality during embryonic development.Reference Anderson, Spicer and Loomba38 The atrial appendages in the setting of isomerism no longer remain lateralised; both the left and right appendage possess the same morphology. In the usual arrangement, the right atrial appendage is broad and pyramidal and has pectinate muscles that spill outside the appendage and extend to the atrioventricular junction. The left atrial appendage is finger-like and has pectinate muscles that are contained within the appendage and do not extend to the atrioventricular junction. It is the internal components of the atrial appendages which are of most importance as these are not influenced by surgery or haemodynamics, while the external appearance of the atrial appendages might be influenced.Reference Uemura, Ho, Devine, Kilpatrick and Anderson37 Echocardiographic assessment of the atrial appendages is extremely difficult, but recent advances in CT allow for detailed imaging with relatively low radiation doses.Reference Mori, Anderson, Nishii, Matsumoto and Loomba39 It must also be noted that the atria themselves are not isomeric but simply the appendages, and thus the term atrial isomerism is not accurate.
In regard to the cardiac anatomy, common atrioventricular junctions are present in a majority of patients with both left and right isomerism. A tongue of tissue connecting the superior and inferior bridging leaflets and thus creating two atrioventricular orifices is more common in left isomerism. Analysis of post-mortem specimens with isomerism found that approximately 60% of those with left isomerism had two orifices compared to 17% of those with right isomerism.
Pulmonary atresia was more prevalent in those with right isomerism with 45% of those patients having pulmonary atresia compared to 9% of those with left isomerism. The ventriculoarterial connections were more likely to be concordant in those with left isomerism and consequently more likely to be discordant in those with right isomerism. In regard to the position of the aorta and the pulmonary artery, those with left isomerism were more likely to have usual or mirror-imaged spiraling of the arterial trunks, while those with right isomerism almost never had spiraling arterial trunks. A posterior, rightward aorta with usual spiraling was the most common relationship in those with left isomerism and found in 40% of such patients. For those with right isomerism, the most common relationship of the arterial trunks was with a more anterior, rightward aorta that is present in 33%.Reference Tremblay, Loomba and Frommelt40
Ventricular topology did not differ between those with left and right isomerism and right-handed topology was present in the majority. Aortic arch sidedness also did not differ nor did rates of aortic coarctation or interruption. Abnormal coronary artery distribution was present in 66% of patients with either left or right isomerism with single coronary arteries being the most common variant. This multi-centre analysis published by Trembaly found that a single coronary artery was present in 31% of those with left isomerism and 37% of those with right isomerism. The direction of the cardiac apex did not differ between those with right and left isomerism. In both instances, a leftward pointing apex was noted in approximately 60% and a rightward apex in the remainder. Some patients had a midline pointing apex.Reference Tremblay, Loomba and Frommelt40
Systemic Venous Connection Abnormalities
Systemic venous abnormalities were also exceedingly common in those with isomerism. A right-sided superior caval vein was present in approximately 80% of those with either left or right isomerism, while a left-sided superior caval vein was present in approximately 65% of those with either right or left isomerism. When present, the superior caval vein drained into the roof of the left-sided atrium in the setting of right isomerism. This was also the most common drainage of a left-sided superior caval vein in the setting of left isomerism, although the left-sided superior caval vein may also drained into the coronary sinus in patients with left isomerism. While a coronary sinus was never present in those with right isomerism, it was present in 80% of those with left isomerism. Thus, the presence of a coronary sinus can rule out a diagnosis of right isomerism, but its absence cannot rule out left isomerism.Reference Tremblay, Loomba and Frommelt40
An interrupted inferior caval vein was present in 78% of those with left isomerism but was not noted in any patient with right isomerism. For those with left isomerism but without interruption of the inferior caval vein, the drainage was most commonly to the right-sided atrium although drainage to the left-sided atrium was also noted. For those with right isomerism the inferior caval vein drained to the right-sided atrium in 73% of patients and to the left-sided atrium in the remainder.Reference Tremblay, Loomba and Frommelt40
Drainage of the hepatic veins did not significantly differ between those with left and right isomerism. In approximately 67% of patients with either left or right isomerism, the hepatic veins drained directly into the inferior caval vein. Hepatic veins may drain into either atrium or to both atria through multiple hepatic veins returning to the heart.Reference Tremblay, Loomba and Frommelt40 Pulmonary vein drainage is also important to note. The presence of infradiaphragmatic, supracardiac, or mixed pulmonary venous connection is almost always indicative of right isomerism. Supracardiac pulmonary venous connections was the most common variant in those with right isomerism and was noted in 28%. In those with left isomerism connection of the pulmonary veins directly to the right-sided atrium was most frequently found and was the case in 39% of these patients. The second most frequent variant in these patients was ipsilateral connection of the pulmonary veins, in 36%.Reference Tremblay, Loomba and Frommelt40
Functional cardiac abnormalities
While the anatomic cardiac considerations are outlined above, it is also important to note the functional considerations in these patients. Arrhythmias are commonly noted in those with both left and right isomerism. The median age of onset in either subset is approximately 4 years of age, with reports of left atrial isomerism having an earlier age of onset.Reference Ozawa, Asakai and Shiraga41 Those with right isomerism are more likely to have atrial flutter, atrial tachycardia, twin atrioventricular nodes, junctional tachycardia, supraventricular tachycardia, and ventricular tachycardia and those with left isomerism are more likely to have atrioventricular block, bradyarrhythmias, or sick sinus syndrome.Reference Loomba, Willes, Kovach and Anderson42–Reference Niu, Dickerson and Moore44 A single-centre retrospective review on arrhythmias in paediatric patients with heterotaxy syndrome found that tachyarrhythmias occurred in 30% of patients with nearly twice the frequency of bradyarrhythmias and were independently associated with an increase in death or need for transplant. The reported incidence of tachyarrhythmias in right atrial isomerism is higher than in patients with left atrial isomerism and ranges from 18 to 34%.Reference Niu, Dickerson and Moore44–Reference Cheung, Cheng, Yung and Chau47 Tachyarrhythmia has been reported as a survival disadvantage.Reference Broda, Salciccioli, Lopez, Ermis, Moodie and Dickerson48 Pacemaker insertion is common in heterotaxy patients with most frequent aetiologies of AV block, severe sinus node dysfunction, and post-operative complications. Left atrial isomerism is an independent predictor for pacemaker insertion.Reference Baban, Cantarutti and Adorisio49
Our patient has left isomerism with normal cardiac anatomy and less severe functional manifestation of sinus node dysfunction with junctional escape rhythm and multiform ventricular bigeminy, undoubtedly facilitating her late presentation and diagnosis. At present, she reports feeling well without symptomatic interference in daily activities. Her echocardiograms have remained unchanged and her most recent left ventricular ejection fraction was 50%. Her right ventricular pressure remains elevated with Doppler estimated at 50 mmHg above right atrial pressure. Her 6-minute walk test was notable for 390 m and she does not have oxygen desaturations with exercise. Current medications include tadalafil and macitentan.
Extracardiac Manifestations
Extracardiac manifestations have not been examined as extensively as the cardiac defects but are present in a majority of patients with isomerism and pose significant problems with many clinical symptoms.
Pulmonary abnormalities
Pulmonary manifestations in right isomerism are characterised by bilaterally trilobed lungs and bilaterally eparterial bronchi; although this is typical, cases of bilaterally unilobed or bilaterally multilobed lungs have been described.Reference Bartram, Wirbelauer and Speer50–Reference Phoon and Neill54 In patients with left isomerism, a high frequency of bilaterally bilobed lungs and bilateral hyparterial bronchi are found, as was identified in our patient.Reference Gabriel and Lo24,Reference Bartram, Wirbelauer and Speer50–Reference Ticho, Goldstein and Van Praagh52,Reference Peoples, Moller and Edwards55 There is emerging evidence that two-fifths of those with isomerism lie on the ciliopathy spectrum, although it is abnormal motion and not ultrastructure that represents the underlying defect.Reference Loomba, Ahmed, Spicer, Backer and Anderson7,Reference Gabriel and Lo24,Reference Nakhleh, Francis and Giese56 This predisposes to recurrent sinopulmonary infections, which, along with bronchiectasis, are well recognised in Kartageners syndrome due to primary ciliary dyskinesia or immotile ciliary dysfunction. Ciliary dysfunction also results in post-operative lung complications, and these are well described in heterotaxy patients.Reference Gabriel and Lo24,Reference Nakhleh, Francis and Giese56,Reference Kothari57 Unrecognised ciliary dysfunction may contribute to poor secretion clearance, atelectasis, and recurrent chest infections. Patients often need vigorous chest physiotherapy and beta-agonists.Reference Kothari57,Reference Harden, Tian and Giese58 Unrecognised ciliary dysfunction might be the cause for respiratory distress in some neonates with undiagnosed heterotaxy.Reference Kothari57
Central nervous system abnormalities
Central nervous system abnormalities are noted in many case reports and meta-analyses undertaking the description of isomerism. Affecting around 10%, anomalies include porencephalic cysts,Reference Ticho, Goldstein and Van Praagh52,Reference Freedom59 hydrocephalus,Reference Ozawa, Asakai and Shiraga41,Reference Kothari57,Reference Freedom59 hypoplasia of the third ventricle,Reference Freedom59 partial absence of the septum pellucidum,Reference Freedom59 partial or complete absence of the corpus callosum,Reference Kothari57 cerebellar dysplasia or agenesis,Reference Ticho, Goldstein and Van Praagh52 microphthalmia,Reference Freedom59 meningeomyelocele,Reference Ticho, Goldstein and Van Praagh52,Reference Kothari57 encephalocele,Reference Ticho, Goldstein and Van Praagh52 Dandy–Walker malformation,Reference Ticho, Goldstein and Van Praagh52 holoprosencephaly,Reference Ticho, Goldstein and Van Praagh52,Reference Kothari57 diplomelia and hydromelia.Reference Ticho, Goldstein and Van Praagh52 Craniofacial anomalies including cleft lip and palate, agnathia or micrognathia, choanal atresia, high-arched palate, laryngeal cleft, or cyclopia have also been reported.Reference Ticho, Goldstein and Van Praagh52 Ciliopathies such as Joubert, Meckel–Gruber, and Bardet–Beidel syndromes further underscore how abnormal ciliopathies can lead to alterations in cortical formation and neurodevelopmental impairments.Reference Loomba, Ahmed, Spicer, Backer and Anderson7,Reference Hildebrandt, Benzing and Katsanis60–Reference Marley and von Zastrow62 The clinical implications of brain anomalies in heterotaxy has not been well defined.Reference Kothari57
Renal and genitourinary abnormalities
Renal and genitourinary malformations and anomalies are seen approximately in 14–25% of patients. The most common renal anomalies include horseshoe kidney,Reference Ticho, Goldstein and Van Praagh52,Reference Kothari57,Reference Freedom59 hydronephrosis both in isolation or secondary to posterior urethral valves,Reference Freedom59 congenital renal hypoplasia,Reference Kothari57,Reference Freedom59 unilateral “pancake” kidney, unilateral duplicating collecting system, severe obstructive uropathy,Reference Loomba, Aggarwal and Gupta43 polycystic kidney disease,Reference Loomba, Ahmed, Spicer, Backer and Anderson7 nephronophthisis,Reference Loomba, Ahmed, Spicer, Backer and Anderson7 hypoplastic kidneys,Reference Ticho, Goldstein and Van Praagh52,Reference Kothari57 oral-facial-digital syndrome,Reference Loomba, Ahmed, Spicer, Backer and Anderson7 and absent kidney.Reference Kothari57 Genitourinary anomalies include hypospadias,Reference Ticho, Goldstein and Van Praagh52 bilateral cryptorchidism,Reference Van Praagh, Santini and Sanders51,Reference Kothari57 urethral duplications,Reference Kothari57 vaginal duplications or atresia, duplicated uterus, and unicornate or bicornate uterus.Reference Kothari57 These abnormalities predispose to urinary tract infections, pelviureteral obstruction, or nephrolithiasis. The unilateral hypoplastic kidney can also cause hypertension or decreased renal function in the future.Reference Kothari57,Reference Westland, Schreuder, van Goudoever, Sanna-Cherchi and van Wijk63
Gastrointestinal and hepatobiliary abnormalities
Intestinal obstructions and mesenteric abnormalities including a common mesentery, abnormal mesenteric attachments, and non-rotation or malrotation of the intestine are common in both forms of isomerism.Reference Bartram, Wirbelauer and Speer50,Reference Ticho, Goldstein and Van Praagh52,Reference Gayer, Apter and Jonas64–Reference Ryerson, Pharis and Pockett70 There is controversy in the literature regarding the investigation for intestinal rotation abnormalities in patients with isomerism, and if present, whether an elective LADD procedure should be undertaken.Reference Ryerson, Pharis and Pockett70 In most reports, 80% of patients with symptomatic malrotation present within the first month of life, and 90% present within the first year.Reference Ryerson, Pharis and Pockett70–Reference Filston and Kirks72 A recent study examined intestinal rotational abnormalities in a multicentre cohort of infants with heterotaxy syndrome and found a 72% incidence of intestinal rotational abnormalities in their cohort of 34 infants; there was no failure of expectant management in the asymptomatic infants and none of the infants developed a volvulus, and there was no difference in the presentation or incidence of intestinal rotational abnormalities in infants with right- versus left-sided isomerism.Reference Ryerson, Pharis and Pockett70 The decision to do a LADD procedure in patients with complex CHD must be made on a case-by-case basis.
The liver is large and bilaterally symmetric in right isomerism,Reference Bartram, Wirbelauer and Speer50,Reference Van Praagh, Santini and Sanders51,Reference Applegate, Goske, Pierce and Murphy65 and conversely is abnormally symmetric in left isomerism.Reference Bartram, Wirbelauer and Speer50,Reference Van Praagh, Santini and Sanders51,Reference Gayer, Apter and Jonas64,Reference Applegate, Goske, Pierce and Murphy65 Absence of the gallbladder or extrahepatic biliary atresia has been seen in some patients with left isomerism.Reference Bartram, Wirbelauer and Speer50,Reference Van Praagh, Santini and Sanders51,Reference Kothari57,Reference Freedom59 The portal vein runs ventral to the duodenum,Reference Gayer, Apter and Jonas64 and biliary atresia has been found to be associated with hypoplasia of the portal vein.Reference Falchetti, de Carvalho and Clapuyt73 Biliary atresia and liver fibrosis have been seen in ciliopathy syndromes,Reference Loomba, Ahmed, Spicer, Backer and Anderson7,Reference Rock and McLin74,Reference Chu, Russo and Wells75 and so their association with isomerism is not surprising. A short pancreasReference Gayer, Apter and Jonas64 and annular and semiannular pancreasReference Freedom59 have been noted in patients with left isomerism. Less frequently seen but reported in necropsied patients with isomerism are midline defects such trachea-oesophageal fistula, oesophageal atresia, omphalocele, and congenital rectal stenosis or atresiaReference Ticho, Goldstein and Van Praagh52,Reference Kothari57,Reference Freedom59 Interestingly, rectal stenosis and atresia have been seen exclusively in right isomerism and biliary atresia and extrahepatic portosystemic anastomoses, also known as Abernethy malformation, have been seen exclusively in left isomerism.Reference Ticho, Goldstein and Van Praagh52,Reference Kothari57 These extrahepatic portosystemic anastomoses are thought to be responsible for idiopathic pulmonary arterial hypertension or diffuse pulmonary arteriovenous fistulas causing cyanosis.Reference Newman, Feinstein and Cohen9,Reference Kothari57,Reference Murray, Yoo and Babyn76,Reference McElhinney, Marx and Newburger77 Structural gastrointestinal defects may also play a role in feeding difficulties, failure to thrive, recurrent aspirations, atypical abdominal pain, and other symptoms.Reference Kothari57 Propensity for gallstones, pancreatitis, diabetes mellitus, or intestinal obstruction can occur from structural abnormalities. Rare case reports of preportal duodenal vein causing obstructive jaundiceReference Kothari57,Reference Low, Williams and Chaganti78 or appendicitis resulting in epigastric or right hypochondrial pain because of an undescended appendix emphasise the importance of awareness of gastrointestinal symptom involvement in these patients.Reference Kothari57,Reference Odabasi, Arslan and Abuoglu79,Reference Ely, Gorelik and Cohen-Sivan80
The gastrointestinal and hepatobiliary manifestations in our patient, including polysplenia, interrupted inferior vena cava with azygos continuation, and intestinal malrotation are consistent with left isomerism. In addition, she has Abernethy malformation with concomitant portopulmonary hypertension for which she is prescribed vasodilator medication. She is also prescribed amoxicillin for functional asplenia prophylaxis.
Lympho-Reticulo-Endothelial abnormalities
The situs of abdominal organs can readily be diagnosed by all imaging modalities and particular attention should be paid to the spleen. Originally, the organ used to assign isomerism, now replaced by cardiovascular manifestations, the presence of absent, single, or multiple spleens remains important as even in the presence of a single, normally located spleen function can be abnormal. Although the amount of splenic tissue needed for adequate immunologic function is not known, it is generally agreed that the presence of Howell–Jowell bodies or pitted red blood cells in the peripheral blood smear is indicative of hypo- or asplenia and represents a risk for overwhelming infection.Reference Loomba, Ahmed, Spicer, Backer and Anderson7,Reference Bartram, Wirbelauer and Speer50,Reference Brigden81 A pitted red blood cell count of >3.8% is indicative of splenic hypofunction; normal is <2.0%.Reference Kothari57,Reference Melles and de Marie82 The risk of overwhelming sepsis is greatest in the young infants and perhaps decreases with age. Infants less than 6 months of age are more susceptible to gram-negative organisms, and older children are susceptible to unusual organisms like Babesia and Capnocytophaga in addition to known capsular microbes.Reference Kothari57,Reference Melles and de Marie82 It is recommended to treat asplenic patients with daily prophylactic penicillin, though duration has varied from 5 to 15 years with some reports recommending lifelong prophylaxis.Reference Kothari57,Reference Melles and de Marie82,Reference Yamamura, Joo and Ohga83 Vaccination with pneumococcal polysaccharide PCV 23 is given after 2 years of age as the antibody response in children under 2 years is not adequate, but the PCV 7 can be given in the first 2 years of life. One dose of H influenza B vaccine is given at 2 months of life. Seasonal influenza, varicella, salmonella, and meningeococcemia vaccines can be considered.Reference Kothari57 Whether thromboembolism in isomerism patients is more common is unclear and has not been systematically studied, although a study by Yamamura et al looked at the incidence of thrombocystosis and thromboembolisms in single-ventricle patients with asplenia (group A) and single-ventricle patients without asplenia (group B). The incidence of thromboembolic events was higher in group A (28% versus 10%) and was associated with increased incidence of thromboembolic events in BT shunt malfunction, cerebral infarcts, venous thromboembolisms, and Fontan route thrombosis. Single-ventricle patients with asplenia were found to have a poorer outcome than other single-ventricle patients; reasons cited were pulmonary vein obstruction, arrhythmias secondary to twin atrioventricular nodes, and susceptibility to pneumococcal infections.Reference Yamamura, Joo and Ohga83,Reference Knott-Craig, Danielson, Schaff, Puga, Weaver and Driscoll84 The spleen plays a major role in platelet aggregation and is the primary site of platelet destruction. Platelet adhesion and aggregation are key events in the development of thromboembolism. Antithrombotic therapy should be optimised in patients with asplenia showing high platelet counts.Reference Yamamura, Joo and Ohga83,Reference Khan, Nair, Olivares, Tingle and Li85
Endocrine abnormalities
Endocrine manifestations in isomerism patients are not as prevalent as other system manifestations but have been noted in autopsied patients. A single institution review of autopsies in 29 patients with asplenia found 4 patients to have notable manifestations including horseshoe adrenal glands fused across the midline in 2 patients, congenital testicular hypoplasia in 1, and bilateral cystic ovaries and congenital adrenal hyperplasia in 1 patient.Reference Freedom59 Similarly, Ticho et al reviewed medical records and autopsy reports of 160 paediatric cases with an isomerism diagnosis and found only 1 endocrine abnormality, which was fusion of adrenal glands across midline and was seen in 10% of patients with asplenia.Reference Ticho, Goldstein and Van Praagh52
Musculoskeletal abnormalities
As with endocrine manifestations, musculoskeletal abnormalities are reported, though not with high prevalence. Several retrospective reports have found sporadic defects. Freedom et al found one patient who had left equinovarus deformity and bilaterally overlapping toes. Another was found to have clubbing of both hands and bilateral absence of the radii, and the last patient had macrodactyly as an isolated skeletal anomaly.Reference Freedom59 Ticho et al reported musculoskeletal manifestations in 13% including severe kyphosis or scoliosis, hemivertebrae, fused vertebrae, pectus deformity, vertebral anomalies, bifid sacrum, sacral agenesis, and three patients with caudal regression.Reference Ticho, Goldstein and Van Praagh52
Adult Outcomes
Adult CHD patients now outnumber children with CHD, and the adult CHD population continues to grow.Reference Broda, Salciccioli, Lopez, Ermis, Moodie and Dickerson48,Reference Diller, Kempny and Alonso-Gonzalez86 Data regarding the long-term survival and morbidity of adults with CHD and isomerism are lacking, but the few available in the literature report a heavy burden of morbidity, with wide variations in mortality. In a review of 62 patients with CHD and isomerism, 38% required interventions in adulthood, the median age for death or heart transplant was 45.3 (95%CI 34.0-56.1 years). Twenty-six per cent had an arrhythmia, 12.7% were diagnosed with pulmonary hypertension, 22.4% had a cerebrovascular incident, with seven patients before 18 years of age. The median age of cerebrovascular incident-free survival for the entire cohort was 33.5 years (95%CI 26.1–40.8). Heart failure was diagnosed in 29.8% of patients, with seven of these patients diagnosed prior to 18 years of age. The median age of heart failure-free survival for the entire cohort was 32.6 years (95% CI 27.0–38.1). Heart failure was associated with a worse prognosis for adult patients, and there was a high proportion of patients with early-onset heart failure in this cohort, >40% by 30 years of age. An interesting find was that no specific congenital anatomic factor was associated with decreased survival, and a confounder may be that patients with severe anatomic (cardiovascular or other) lesions may have auto-selected out of this adult-only cohort. The authors of the review note that despite this increased morbidity, the adults with CHD and isomerism were able to live a fulfilling life and accomplish important milestones such as attending college, full-time employment, marriage, and having children.Reference Broda, Salciccioli, Lopez, Ermis, Moodie and Dickerson48
An Italian review of 136 patients over a 26-year follow-up found a reduction in mortalityReference Baban, Cantarutti and Adorisio49 compared with prior studies reaching 50% mortality.Reference Chen, Ma and Cui28,Reference Bhaskar, Galati and Brooks87–Reference Anderson, Baker, Redington, Rigby, Penny and Wernovsky89 The overall survival and freedom from heart transplant was 69.8% for patients with right atrial isomerism and was 87.8% for patients with left atrial isomerism at 40 years of age. In the right atrial isomerism subset, the mortality rate was 12% and the heart transplant rate was 7%, while in the left atrial isomerism subset, the mortality rate was 4% and the heart transplant rate was 4%. The authors concluded that right atrial isomerism was a major risk factor for mortality, but that were no other significant independent predictors for mortality.Reference Baban, Cantarutti and Adorisio49
Conclusion
This case highlights the complexities and varied clinical manifestations of isomerism, including the uncommon association of Abernethy syndrome and portopulmonary hypertension. Our patient’s late presentation and delayed diagnosis with complicated portopulmonary hypertension have put her at risk for significant morbidity and mortality, but her clinical course thus far has been reassuring. In any patient with a diagnosis of isomerism or with unexplained pulmonary hypertension, hypoxemia, or pulmonary arteriovenous shunting, we recommend detailed imaging of the liver including CT or MRI, hepatic Doppler, and a venous phase of the liver,Reference Loomba, Shah, Anderson and Arora8 as well as upper abdominal venous vasculature to assess for this malformation. The complexity of these conditions further highlights the importance of a multidisciplinary approach with cardiologists and intensivists, pulmonary hypertension experts, hepatologists, and primary care physicians for optimal diagnosis and management in the paediatric population.
Author contributions
Megan L. Ringle, MD, contributed to this manuscript in the form of concept, data collection and interpretation, drafting of article, revision of article, and approval of article.
Rohit Loomba, MD, contributed to this manuscript in the form of concept and design, data interpretation, drafting of article, revision of article, and approval of article.
John Dykes, MD, contributed to this manuscript in the form of concept and design, data analysis and interpretation, data collection, drafting of the article, revision of the article, and in approval of the article.
Danyal Khan, MD, contributed to this manuscript in the form of data analysis, collection, and interpretation, revision of the article, and approval of the article.
David Schidlow, MD, contributed to this manuscript in the form of data analysis and interpretation, revision of the article, and approval of the article.
Gil Wernovsky, MD, contributed to this manuscript in the form of design and concept, data analysis and interpretation, drafting of the article, critical revision of the article, and approval of the article.
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflicts of Interest
None.






