Background
Post-traumatic stress disorder (PTSD) is a mental disorder that develops in up to a third of individuals who are exposed to extreme stressors, with significant work and social impairment, increased risk of suicide, higher medical and social costs and higher psychiatric comorbidity (Norris & Sloane, Reference Norris, Sloane, Friedman, Keane and Resick2007; Steel et al. Reference Steel, Chey, Silove, Marnane, Bryant and van Ommeren2009; Krysinska & Lester, Reference Krysinska and Lester2010). The estimated lifetime prevalence of PTSD among adults in the general population is around 6.8% and current (12-month) prevalence about 3.6% (Kessler et al. Reference Kessler, Aguilar-Gaxiola, Alonso, Angermeyer, Anthony, Brugha, Kessler and Ustun2011). However, recent surveys of military personnel have yielded higher estimates, ranging from 6.2% for US service members who fought in Afghanistan to 12.6% for those who fought in Iraq and 15.4% in conflict-affected populations (Dohrenwend et al. Reference Dohrenwend, Turner, Turse, Adams, Koenen and Marshall2006; Seal et al. Reference Seal, Bertenthal, Miner, Sen and Marmar2007). Treatments available for PTSD span a variety of psychological and pharmacological interventions (Jonas et al. Reference Jonas, Cusack, Forneris, Wilkins, Sonis and Middleton2013). Numerous organizations have issued guidance for the treatment of patients with PTSD (APA, 2004; NICE, 2005; PA-CPMH, 2013), supporting trauma-focused psychological interventions as first-line treatments for PTSD. These guidelines have also recognised some benefit of pharmacological interventions; however, they have arrived at different conclusions about the value of such interventions, and have limited themselves to recommendations about broad categories of treatments. From a clinical point of view, though, it is very important to know what pharmacological intervention is more efficacious and acceptable than others among the various treatment options available for PTSD. Notwithstanding the recent publication of two systematic reviews (Watts et al. Reference Watts, Schnurr, Mayo, Young-Xu, Weeks and Friedman2013; Hoskins et al. Reference Hoskins, Pearce, Bethell, Dankova, Barbui and Tol2015), clinical uncertainty still remains about what pharmacological treatment to select among all available compounds because traditional pair-wise meta-analyses do not allow the simultaneous integration of direct and indirect evidence (Cipriani et al. Reference Cipriani, Higgins, Geddes and Salanti2013). The aim of this paper is to assess efficacy and acceptability of different pharmacological treatments against one another, to provide a clinically useful summary of the comparative evidence that can be used to guide decisions about acute treatment of PTSD in adults.
Methods
The study protocol was drafted and made available on our institutional website (http://cebmh.warne.ox.ac.uk/cebmh/research_other_reviews.htm) (Appendix 1). Furthermore, with the publication of this paper, the overall dataset will be in the public domain for anyone who would be interested to use it.
Study eligibility criteria
We identified all double-blind randomised controlled trials (RCTs) comparing any psychotropic agent at a therapeutic dose with another psychotropic drug or placebo as oral therapy in the treatment of adults with PTSD, diagnosed according to operationalised criteria. We included both monotherapy and add-on studies, and assumed that add-on interventions had an additive effect on the treatments of interest. The dose ranges for the medication included in the network meta-analysis were defined according to the FDA, whenever possible (https://dailymed.nlm.nih.gov/dailymed/ or http://www.accessdata.fda.gov/). If information was not available, we used the British National Formulary (https://www.bnf.org/). We included all the following pharmacological interventions: amitriptyline, clomipramine, desipramine, imipramine, maprotiline, mianserin, tianeptine, trazodone, nefazodone, citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline, bupropion, desvenlafaxine, venlafaxine, duloxetine, mirtazapine, reboxetine, brofaromine, moclobemide, phenelzine (among antidepressants); haloperidol, chlorpromazine, olanzapine, risperidone, quetiapine (among antipsychotics); lithium, lamotrigine, valproate, tiagabine, topiramate (among mood stabilisers/anti-epileptic drugs); guanfacine, prazosin and selective NK1R antagonists (among the new drugs with different mechanisms of action). The synthesis comparator set consists of all the interventions listed above, their combinations and placebo (for all details about Methods, see Appendix 1).
Identification and selection of studies
We searched Cochrane Depression, Anxiety and Neurosis Group specialised register and the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, EMBASE, PsycINFO, the National PTSD Center Pilots database and PubMed (last update: 15 February 2016). We also searched the databases of the most important regulatory agencies worldwide and online trial registers for published and unpublished RCTs (see Appendix 2 for full details on the search strategy). No language restrictions were applied. All relevant authors and principal manufacturers were contacted to supplement the incomplete report of the original papers.
Data collection and study appraisal
Acute treatment was defined as 8-week treatment in all analyses. Mean change scores on the Clinician-Administered PTSD Scale (CAPS) and dropout rates (treatment discontinuation) were chosen as primary outcomes to represent the most sensible and sensitive estimate of acute treatment efficacy and acceptability, respectively. As secondary analyses, we also estimated the proportion of patients who responded to treatment (according to original study authors’ definition) and the proportion of patients who left the study early due to adverse events (tolerability). The risk of bias tool was used to assess study quality (Appendix 3) (Higgins & Green, Reference Higgins and Green2011). Additionally, we assessed the quality of evidence contributing to each network estimate using the GRADE framework, which characterises the quality of a body of evidence on the basis of the study limitations, imprecision, inconsistency, indirectness, and publication bias for the primary outcomes (Salanti et al. Reference Salanti, Del Giovane, Chaimani, Caldwell and Higgins2014).
Data synthesis and statistical analysis
For continuous outcome, where different measures were used to assess the same outcome, standardised mean difference (SMD) (Hedges's adjusted g) were used (Altman & Bland, Reference Altman and Bland1996; Higgins & Green, Reference Higgins and Green2011). Dichotomous outcomes were analysed by calculating the odds ratio (OR).
Assessment of clinical and methodological heterogeneity and transitivity
To evaluate the presence of heterogeneity we generated descriptive statistics for trial and study population characteristics across all eligible trials (Furukawa et al. Reference Furukawa, Barbui, Cipriani, Brambilla and Watanabe2006). If the distributions were balanced across comparisons, we concluded against evidence of intransitivity (Salanti, Reference Salanti2012; Jansen & Naci, Reference Jansen and Naci2013).
Data synthesis
Initially, we performed standard pairwise meta-analyses using a random effects model (DerSimonian & Lair, Reference DerSimonian and Laird1986) in STATA (StataCorp, 2015). We then carried out a network meta-analysis (NMA) to synthesise the available evidence from the entire network of trials by integrating direct and indirect estimates and using the methodology of multivariate meta-analysis (different treatment comparisons were treated as different outcomes, assuming a common heterogeneity parameter) (White et al. Reference White, Barrett, Jackson and Higgins2012). We also estimated the ranking probabilities using the surface under the cumulative ranking curve (SUCRA) (Salanti et al. Reference Salanti, Ades and Ioannidis2011) and mean ranks. To perform NMA we used the mvmeta command in Stata (White, Reference White2011) while checking the model assumptions and presenting the results we used a suite of previously published Stata commands (Chaimani et al. Reference Chaimani, Higgins, Mavridis, Spyridonos and Salanti2013a).
Statistical heterogeneity and inconsistency
To measure the percentage of variability that cannot be attributed to random error, we assessed statistically the presence of heterogeneity in all pairwise comparisons by calculating the heterogeneity standard deviation parameter (τ) and within each comparison using the I-squared (I2) statistic (Higgins et al. Reference Higgins, Thompson, Deeks and Altman2003) and its 95% confidence interval (CI). For all outcomes τ was compared with its empirical distribution (Turner et al. Reference Turner, Davey, Clarke, Thompson and Higgins2012; Rhodes et al. Reference Rhodes, Turner and Higgins2015). We also estimated a total I2 value for heterogeneity in the network (Jackson et al. Reference Jackson, Barrett, Stephen, White and Higgins2014). We evaluated the presence of inconsistency locally (local tests) using the loop-specific approach (Song et al. Reference Song, Altman, Glenny and Deeks2003; Chaimani et al. Reference Chaimani, Vasiliadis, Pandis, Schmid, Welton and Salanti2013b; Veroniki et al. Reference Veroniki, Vasiliadis, Higgins and Salanti2013). To check the assumption of consistency in the entire network (global test) we used the design-by-treatment model (White, Reference White2011; Higgins et al. Reference Higgins, Jackson, Barrett, Lu, Ades and White2012). To distinguish between inconsistency and heterogeneity, we employed the I2 for inconsistency and measured the percentage of variability that cannot be attributed to random error or heterogeneity (Turner et al. Reference Turner, Davey, Clarke, Thompson and Higgins2012). In case of important heterogeneity or/and inconsistency, we carried out meta-regression or subgroup analyses by using the following effect modifiers: (a) variance of the observed relative effects; (b) baseline severity; (c) publication year and (d) sponsorship.
Sensitivity analysis
To check the robustness of study findings, we carried out a sensitivity analysis excluding studies rated as at high or unclear risk of bias (Wood et al. Reference Wood, Egger, Gluud, Schulz, Jüni and Altman2008).
Results
Description of the evidence base
The systematic review included overall 51 studies with 6189 patients (see Appendices 4 and 5), randomised to 25 different interventions (Fig. 1). The mean duration of primary studies was 10.3 weeks (s.d. 2.1) and the mean age of trial participants was 42.9 years (s.d. 5.9). The great majority of studies were 2-arm (45 studies), but the search retrieved also six 3-arm studies. Thirty-six studies (71.0%) used CAPS (or CAPS-2) as the rating scale for assessing efficacy, with a mean baseline severity of 74.1 (s.d. 16.7). Only 11 trials (22.0%) had an add-on design and overall 36 studies (71.0%) were sponsored by pharmaceutical industry. In terms of study quality, the reports often did not provide full details about randomisation procedures (28 out of 51, 54.9%) but the reporting about allocation concealment was much worse, as only 19 studies out of 51 (37.2%) were judged at low risk of bias (Appendix 3). The networks of eligible comparisons by outcome are shown in Fig. 2 and Appendix 6. The results of the direct pairwise comparisons for all outcomes are reported in Appendix 7. There was considerable variability in the placebo response rates among studies, which varied between 3% and 60% and resulted in a τ = 0.1. However, there was no evidence of a time trend in placebo response (p value 0.93). Heterogeneity was not low only for the dichotomous response rate and results did not materially change when we assumed comparison-specific heterogeneity parameters.
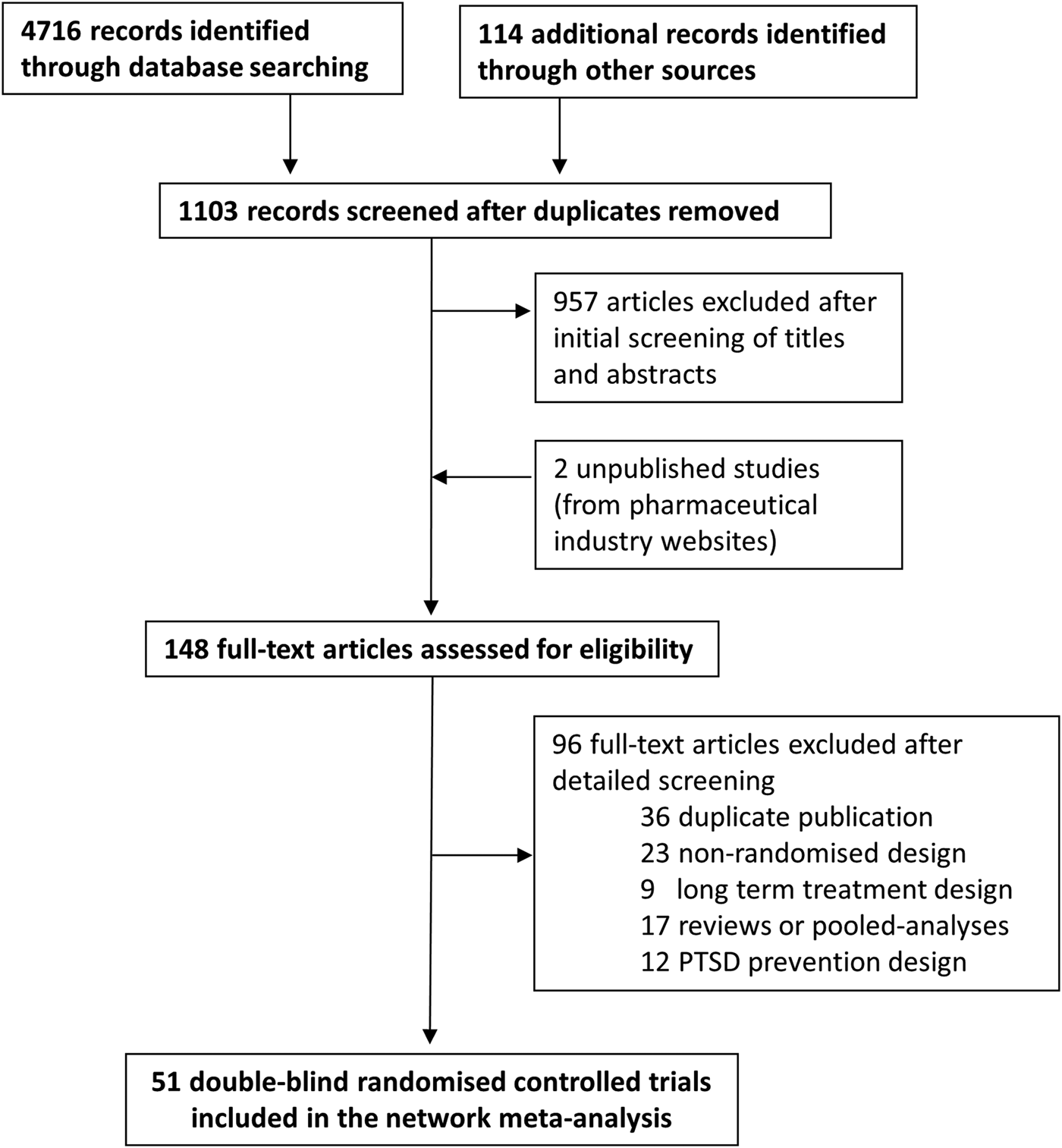
Fig. 1. PRISMA flow diagram. Included and excluded studies.
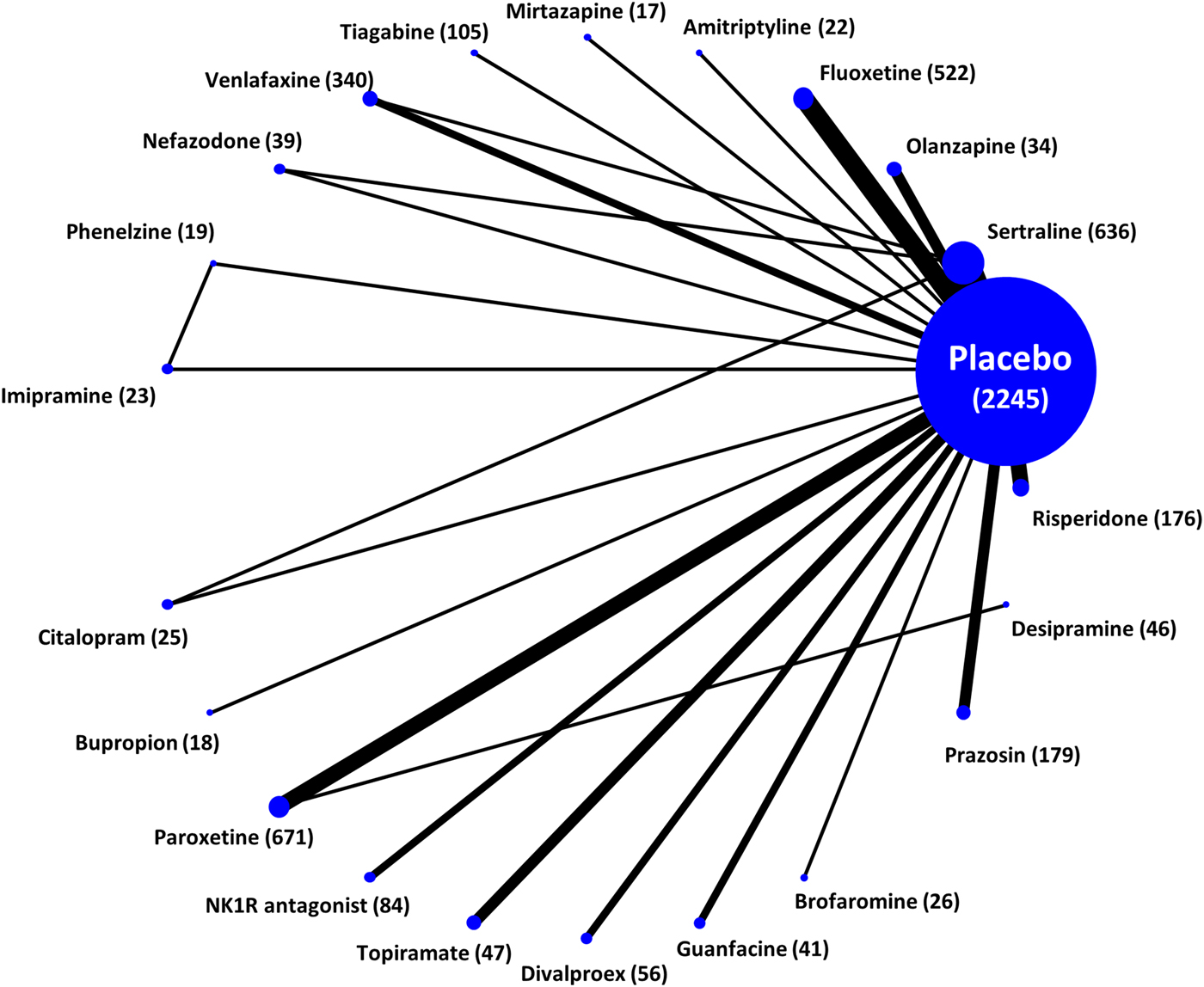
Fig. 2. Network plot for the primary efficacy outcome. Nodes and edges are weighted according to the number of studies involved in each treatment comparison.
Network meta-analysis results
Table 1 and Fig. 3 show the relative treatment effects from NMA for the two primary outcomes (change in symptom's severity and all-cause dropouts). In terms of efficacy, phenelzine, desipramine, paroxetine, venlafaxine, fluoxetine, risperidone, and sertraline were more effective than placebo (Fig. 3). Phenelzine appeared to be statistically significantly more effective than nearly half of the other active treatments included in the network (9 out of 21), while both citalopram and divalproex were statistically significantly less efficacious than phenelzine, desipramine, and paroxetine (citalopram was also inferior to mirtazapine, olanzapine, venlafaxine, and fluoxetine) (Table 1). In terms of dropout rate, phenelzine was the only drug which was significantly better than placebo (OR 7.50, 95% CI 1.72–32.80) and most of the other active treatments (Table 1). No other statistically significant differences were found between the rest of the treatments and placebo (Fig. 1). Notwithstanding fewer events, response rate data did match with our efficacy findings using continuous data (Appendix 8); in terms of dropouts due to adverse events, no drug was statistically significantly better tolerated than placebo, but sertraline and paroxetine performed worse than placebo (OR 0.59, 95% CI 0.38–0.91 and 0.63, 95% CI 0.42–0.94, respectively), and brofaromine better than paroxetine (OR 2.78, 95% CI 1.04–7.42), topiramate (OR 4.77, 95% CI 1.04–21.75), and sertraline (OR 2.99, 95% CI 1.10–8.09) (Appendix 8).
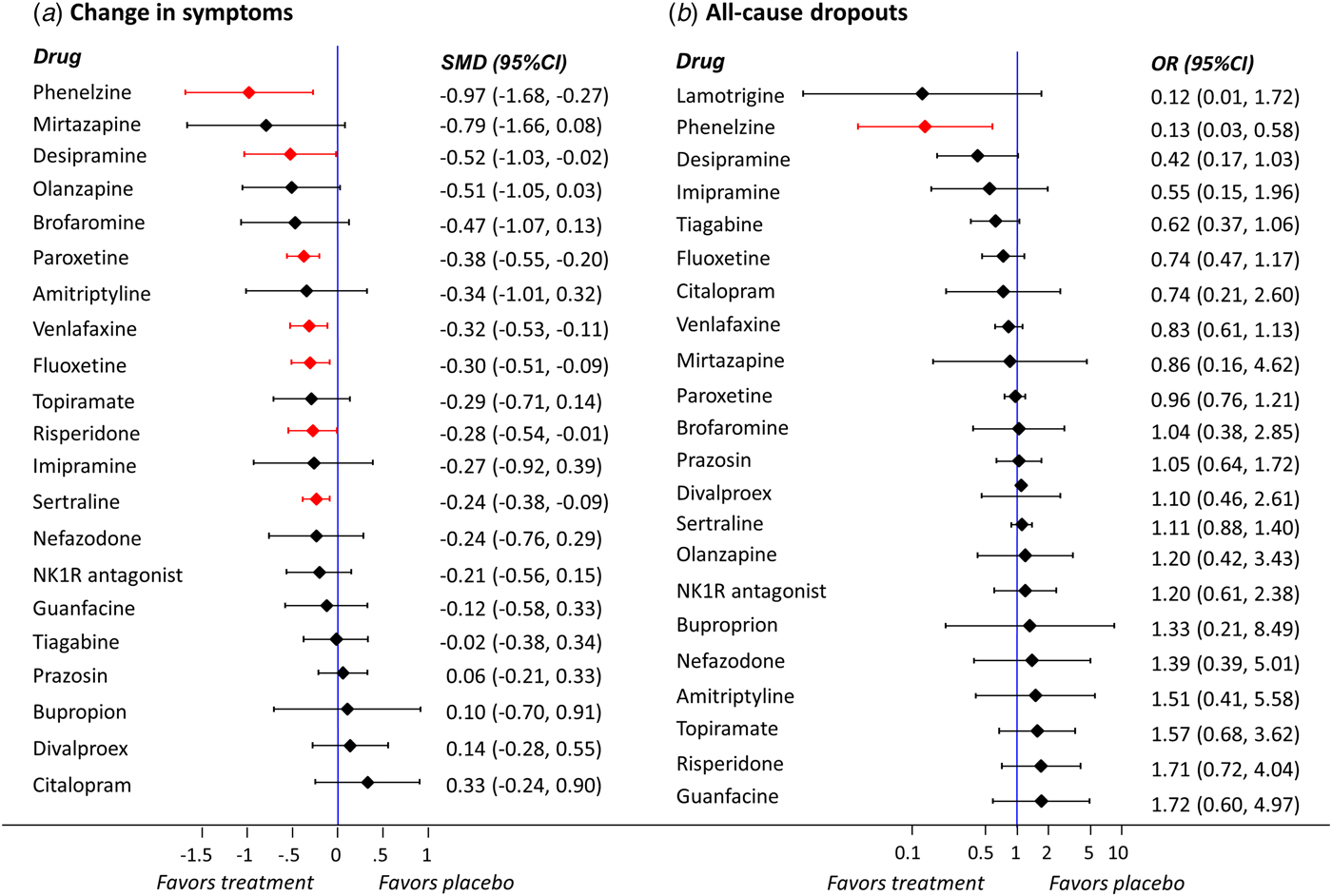
Fig. 3. Network meta-analysis results for the comparisons of active treatments v. placebo for the primary outcomes (a) change in symptoms and (b) all-cause dropouts (reference treatment: placebo). Legend: SMD: standardised mean difference; OR: odds ratio; CI: confidence interval. Effect sizes are listed on the basis of SUCRA rankings. Statistically significant results are highlighted in red.
Table 1. NMA results for the two primary outcomes
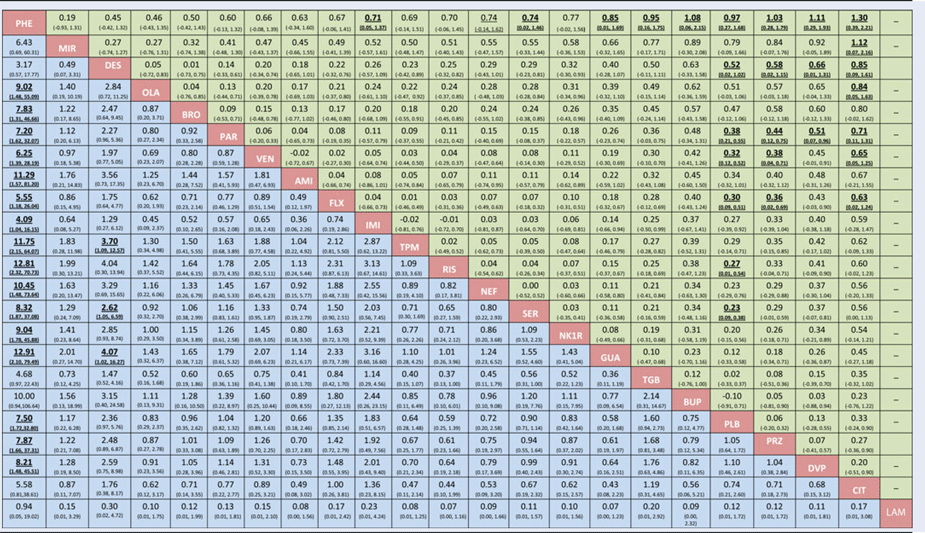
Legend: Mean change in symptoms severity (green) and all-cause dropout rate (blue). For efficacy (green), standardized mean difference values greater than 0 indicated that the treatment specified in the row is more efficacious. For acceptability, odds ratios greater than 1 indicated that the treatment specified in the column is better (fewer dropouts). Bold underlined results indicate statistical significance. The overall heterogeneity (τ) is equal to 0.1 for efficacy and equal to 0 for acceptability. For efficacy, I2 was 22.4% for heterogeneity and 22.5% for inconsistency; for acceptability, I2 was 0% for heterogeneity and 8% for inconsistency. AMI, amitriptyline; BRO, bromipramine; BUP, bupropion; CIT, citalopram; DES, desipramine; DVP, divalproex; FLX, fluoxetine; GUA, guanfacine; IMI, imipramine; LAM, lamotrigine; MIR, mirtazapine; NEF, nefazodone; NK1R, NK1 receptor antagonist; OLA, olanzapine; PAR, paroxetine; PHE, phenelzine; PLB, placebo; PRZ, prazosin; RIS, risperidone; SER, sertraline; TGB, tiagabine; TPM, topiramate; VEN, venlafaxine.
We were not able to assess statistically the transitivity assumption by comparing the distributions of potential effect modifiers across comparisons as most comparisons were informed by a small number of studies. We also evaluated inconsistency in each loop of available evidence (Appendix 9). Although we did not find any statistical evidence of inconsistency assuming either network-specific or loop-specific heterogeneity, there are a few closed loops per outcome and few studies involved, so we cannot exclude the possibility of potentially important inconsistency to be present. The design × treatment interaction model did not provide evidence that any of the networks present as a whole, statistically significant inconsistency (Appendix 10); again the small number of closed loops and studies limits the power of these tests. The I2 for both heterogeneity and inconsistency in NMA were calculated to distinguish between these two sources of variability. For efficacy as a continuous outcome, I2 was 22.4% for heterogeneity and 22.5% for inconsistency; for acceptability, I2 was 0% for heterogeneity and 8% for inconsistency. The percentage of variability attributed to heterogeneity was 43.2% for the dichotomous efficacy outcome (we could not derive the respective measure for inconsistency as inconsistency could not be assessed for this outcome – see above). For dropout rate due to adverse events, I2 was 0% for heterogeneity and 13% for inconsistency. Overall, all these measures were in general low but it should be acknowledged that the uncertainty around them might be high (see below).
For a continuous outcome in a ‘drug versus placebo’ comparison in mental health the suggested median value for τ is 0.22 and for a ‘drug versus drug’ comparison it is 0.20. Both values are greater than our estimation of heterogeneity for the efficacy as a continuous outcome using the restricted maximum likelihood (REML) estimator (τ = 0.1). For a binary mental health outcome the suggested median values for τ are 0.35 and 0.31 for a ‘drug versus placebo’ and a ‘drug versus drug’ comparison, respectively. Our estimation of τ for the dichotomous efficacy outcome was 0.4, whereas the values of τ were 0 for all the remaining dichotomous outcomes, dropouts due to any cause and dropouts due to adverse events.
In Fig. 4 we used the hierarchical cluster analysis to group treatments according to their ranking for the two primary outcomes. Different colours represent different groups of treatments by considering jointly their relative ranking for two outcomes (treatments that belong to the same group may be considered as being of comparable performance with respect to both outcomes). According to all possible cluster rankings, phenelzine performed better than the rest of the treatments both in terms of efficacy and dropouts (Appendix 11); this was also supported by considering the actual effect sizes presented in Table 1 and Fig. 3. Although mirtazapine yielded a relatively high rank in the efficacy outcomes, the respective SUCRA value for the dropout outcome was 51.5% (Appendix 12). Brofaromine performed well in terms of change in symptoms and adverse events, whereas divalproex had the worst ranking considering this particular pair of outcomes. Risperidone was the best treatment in terms of response rate whereas it is among the worst for the outcome of adverse events. See Appendix 12 for all details about rankings and SUCRA values. We could not perform the pre-planned sensitivity analyses because only five studies were rated as low risk of bias for allocation concealment (Appendix 3) and because we did not impute any outcome data (all information we needed was retrieved by checking for unpublished data and contacting the original study authors). According to GRADE, the quality of evidence for primary outcomes was rated as low or very low for most comparisons.
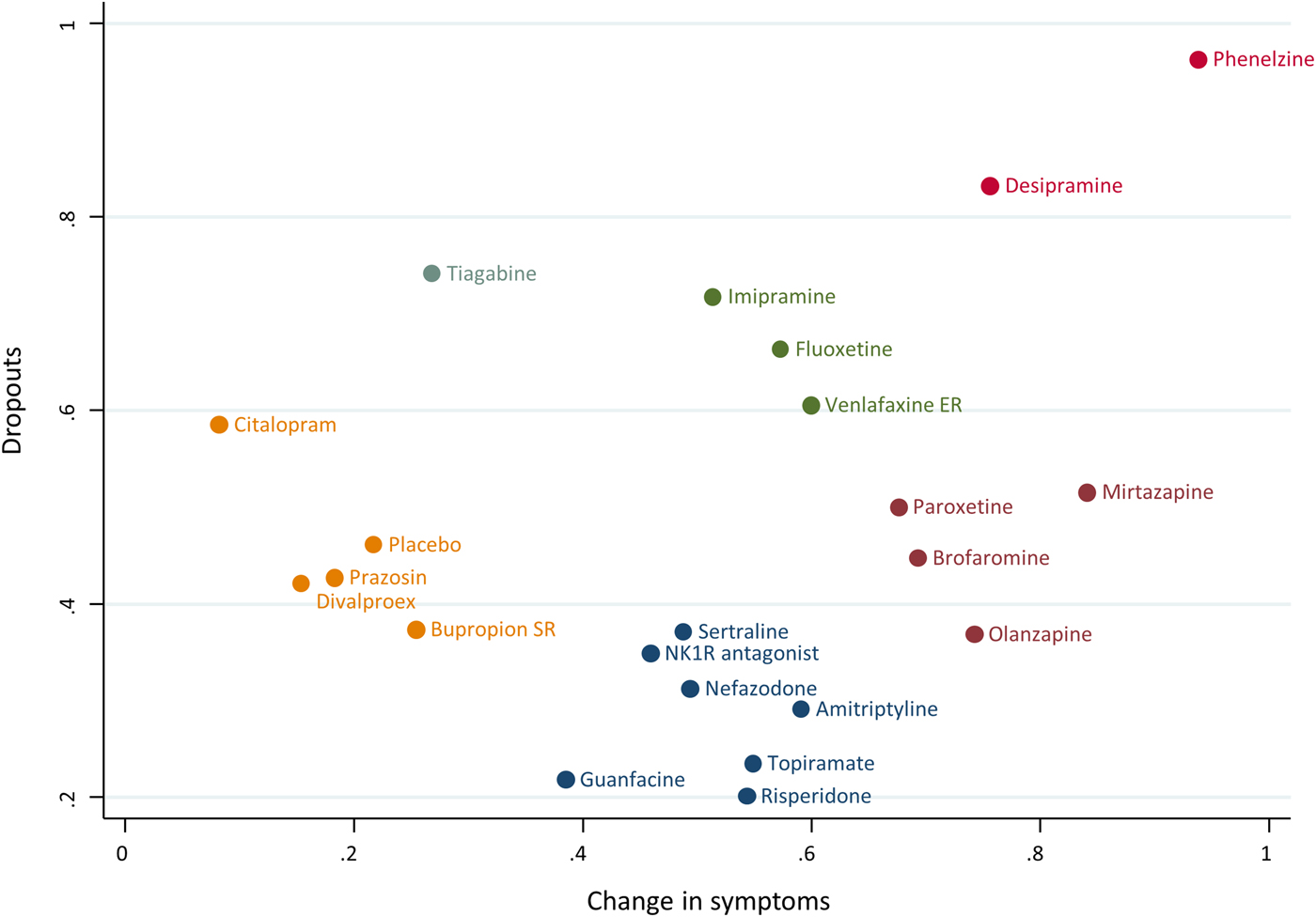
Fig. 4. Clustered ranking based on SUCRA values for the outcomes ‘Change in symptoms’ (unadjusted model) and ‘Any cause dropouts’. Hierarchical cluster analysis is performed to group the competing treatments. Different colours represent different groups according to treatments’ similarity regarding the two outcomes.
Impact of effect modifiers
Because of concerns about power to detect potentially important heterogeneity and inconsistency, we applied network meta-regression models to explore the impact on effect modifiers on heterogeneity and inconsistency. We included the pre-specified covariates in separate network meta-regression models. All results are presented in Appendix 13. For the primary efficacy outcome, the meta-regression model that accounted for baseline severity resulted in a marginally non-significant coefficient 0.11 (95% CI −0.01 to 0.22) implying that the active treatments tended to be more effective (compared with placebo) in studies with more severe patients. The estimation of heterogeneity (τ) was of 0.1 for both the meta-regression model accounting for baseline severity and the model without covariates, showing that accounting for baseline severity did not explain heterogeneity. Appendix 14 presents the effect sizes and the treatment hierarchy (using SUCRAs) of the standard analysis and those from the meta-regression model at the mean centralised baseline severity. Accounting for differences in baseline severity across studies had a minimal impact on the effect sizes and the treatment ranking.
The funnel plot analyses suggested a possible association between study precision and dropout due to adverse events (Appendix 15). However, the relative effect estimates derived from the network meta-regression model that accounted for small study effects did not converge and drawing conclusions based on these findings would be invalid. We performed a random-effects pairwise meta-analysis for the comparison active v. placebo which resulted in a summary effect OR of 1.42 (95% CI 1.15–1.75) indicating that placebo is associated with fewer dropouts due to adverse events than active treatments. Accounting for study precision in a meta-regression model resulted in a statistically significant coefficient 2.07 (95% CI 1.10–3.88). This result suggests that less precise studies tended to show placebo to be better tolerated than it would have been in larger studies (Appendix 15).
Discussion
Our network meta-analysis provides an evidence-based hierarchy for the efficacy and acceptability of pharmacological treatments for PTSD, using all available comparative data and thus overcoming the major limitation of previous pairwise meta-analyses (NICE, 2005; Watts et al. Reference Watts, Schnurr, Mayo, Young-Xu, Weeks and Friedman2013; Hoskins et al. Reference Hoskins, Pearce, Bethell, Dankova, Barbui and Tol2015). Interestingly, results from this study challenge the previous dogma that the efficacy of psychopharmacological agents within the same drug class is the same. Similarly, clinically important differences in efficacy were found among antipsychotics for schizophrenia (Leucht et al. Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter2013) and also among antidepressants, where differences were reported not only in major depression (Cipriani et al. Reference Cipriani, Furukawa, Salanti, Geddes, Higgins and Churchill2009), but also for anxiety disorders, like generalised anxiety disorder (NICE, 2011) and social anxiety disorder (NICE, 2013). The efficacy and acceptability hierarchies generated by our study were robust against many sources of bias, as various supplementary analyses related to severity at baseline, year of publication, small study effect, and sponsorship did not change the final ranking to an important extent.
We emphasise that the statistically significant differences in efficacy between drugs and placebo were small, with the only exception of phenelzine (according to Cohen, Reference Cohen1988, an effect size of 0.2 is small, 0.5 is medium, and 0.8 is large (Cohen, Reference Cohen1988)). It is probably misleading clinically to dismiss the effects of desipramine, fluoxetine, paroxetine, risperidone, sertraline, and venlafaxine as being small. Their point estimates of SMD were around 0.3, which is the same as antidepressants for depression, and indeed for many standard treatments in psychiatry and medicine (Leucht et al. Reference Leucht, Hierl, Kissling, Dold and Davis2012). Phenelzine had a standardised mean difference of 0.97 when compared with placebo and ranged between 0.19 and 1.30 with other active drugs. These differences in efficacy between active treatments were possibly substantial enough to be clinically important and pose the question whether phenelzine should be used as a first line drug treatment for PTSD. However, the overall evidence about this drug is based only on one trial with 60 participants. We assessed the contribution of this single study to all ‘phenelzine versus other’ network estimates for the primary outcome ‘change in symptoms’ (Appendix 16 - Fig. 1). The only study comparing phenelzine contributes more than 35% to the relative effects of all treatments against this specific intervention. Given that the contribution of this study to the relative effects of the rest of the treatment comparisons is very low (results not shown but available from the authors), it is therefore unlikely that the retrieved evidence can currently be considered robust enough to suggest phenelzine as a drug of choice, particularly in view of the need for concomitant dietary restrictions and the risk of drug interactions. This is in line with the interpretation of recent clinical practice guidelines (Katzman et al. Reference Katzman, Bleau, Blier, Chokka, Kjernisted and Van Ameringen2014), however, our analysis had higher statistical power than previous analyses, because of the comprehensive search of published and unpublished data and because we used the network meta-analytical method.
Phenelzine is a potent inhibitor of monoamine oxidase (MAOI), licensed in the USA and UK for depressed patients clinically characterised as ‘atypical,’ ‘non-endogenous,’ or ‘neurotic’, who often have mixed anxiety and depression and phobic or hypochondriacal features (http://www.accessdata.fda.gov/drugsatfda_docs/label/2007/011909s038lbl.pdf). Even though approved for depression only, phenelzine is also effective in the treatment of anxiety disorders such as panic disorder and social phobia (Sheehan et al. Reference Sheehan, Ballenger and Jacobsen1980; Johnson et al. Reference Johnson, Lydiard, Ballenger and Kennedy1994). Of course, phenelzine may owe its apparently striking efficacy in PTSD to the expected increases in brain levels of biogenic amines (serotonin, noradrenaline, and dopamine) produced by MAO inhibition. However, the reversible MAOI-A inhibitor, brofaromine, was of only modest efficacy in the current analysis. It may, therefore, be relevant that unlike other MAOIs, phenelzine also possesses the ability (at least in animal studies) to increase brain levels of the inhibitory neurotransmitter, γ-aminobutyric acid (GABA), through blocking the GABA metabolising enzyme, GABA transaminase. Studies with magnetic resonance spectroscopy (MRS) indicate that cortical GABA levels may be lower in patients with PTSD (Rosso et al. Reference Rosso, Weiner, Crowley, Silveri, Rauch and Jensen2014). Hence the efficacy of phenelzine in PTSD might result from a combination of monoamine and GABA potentiation. However, it is also possible that GABA transaminase inhibition by itself, for example with the anticonvulsant drug vigabatrin, could be a viable therapeutic strategy in PTSD.
Interestingly, treatment with phenelzine reduces dream recall frequency (Landolt et al. Reference Landolt, Raimo, Schnierow, Kelsoe, Rapaport and Gillin2001) and induces almost total REM sleep suppression in anxious-depressed patients (Wyatt et al. Reference Wyatt, Fram, Kupfer and Snyder1971). Although objective sleep findings in PTSD are mixed, there is some evidence that REM sleep fragmentation and REM sleep autonomic imbalance are associated with an increased risk for the development and persistence of PTSD (Mellman et al. Reference Mellman, Pigeon, Nowell and Nolan2007). Sleep disturbances are an indicator of heightened risk for poor psychiatric outcomes following trauma exposure (Germain, Reference Germain2013). To date, prazosin (an alpha-1 antagonist) is the recommended pharmacological treatment options for PTSD-related nightmares, as it is associated with improvements in nightmares and insomnia (Aurora et al. Reference Aurora, Zak, Auerbach, Casey, Chowdhuri and Karippot2010). In our analysis prazosin was not among the most efficacious treatments for relieving acute symptoms of PTSD, so the fact that phenelzine may reduce the REM sleep disturbances associated with PTSD could also give it additional benefit over drugs like SSRIs and SNRIs which also lower REM sleep, but to a lesser extent than phenelzine (Sharpley & Cowen, Reference Sharpley and Cowen1995).
Our study has several limitations. The validity of the above findings might be limited due to the lack of sufficient data to properly evaluate the required assumptions. Results from network meta-analysis may be unreliable when there is a high risk of intransitivity in the network (Salanti, Reference Salanti2012; Cipriani et al. Reference Cipriani, Higgins, Geddes and Salanti2013). Statistical assessment of the transitivity assumption (i.e. one can learn indirectly for a pairwise comparison via one or more anchor treatments) considers whether the distribution of the potential effect modifiers is balanced across the available direct comparisons. In our network, we were unable to judge the plausibility of transitivity since most comparisons included very few studies. Thus, we could only rely on our clinical understanding of the studied condition and outcomes to assume that transitivity was likely to hold in the identified data. Important intransitivity might be reflected in the data in the form of statistical inconsistency. Our network included only a few closed loops of evidence, for which inconsistency could be assessed (three loops for efficacy as a continuous outcome, two for all-cause dropout, none for response rate and two for dropouts due to adverse events). Using the loop-specific approach and the design-by-treatment interaction model we did not find any statistically significant inconsistency (Song et al. Reference Song, Altman, Glenny and Deeks2003; Higgins et al. Reference Higgins, Jackson, Barrett, Lu, Ades and White2012). However, the tests for inconsistency very often have low power and may fail to detect inconsistency as statistically significant even when it is present (Veroniki et al. Reference Veroniki, Vasiliadis, Higgins and Salanti2013). Inconsistency is closely related with heterogeneity and the two notions should be considered jointly. The small number of studies in the majority of the available direct comparisons renders the assessment of comparison-specific heterogeneities impractical (when it is estimable). The common heterogeneity across all comparisons from the network meta-analysis was estimated close to zero and the I2 measure for heterogeneity suggested that heterogeneity was low in the network. This finding slightly mitigates our concerns for the reliability of our results. However, all statements comparing the drugs in the network must be tempered by the potential limitations of the methodology, the complexity of patients with PTSD and the uncertainties that may result from the choice of dose or treatment setting.
We explored the association of several characteristics and their impact on the relative effects using network meta-regression models. The model accounting for baseline severity for the efficacy primary outcome resulted in a marginally non-statistically significant regression coefficient suggesting that the active treatments have a tendency to appear more effective (when compared with placebo) in studies with more severe populations. The meta-regression model that accounted for small-study effects on the outcome of adverse events suggested that small studies showed the placebo safer than did larger studies. This finding might be due to publication bias or selective reporting bias, or might be the manifestation of other characteristics such as study quality (Chaimani & Salanti, Reference Chaimani and Salanti2012; Chaimani et al. Reference Chaimani, Vasiliadis, Pandis, Schmid, Welton and Salanti2013b). Despite this finding, we were unable to use the results from this model for drawing conclusions with respect to the relative safety of the treatments due to the lack of convergence for the estimated relative effect.
Most trials included in our analysis did not report adequate information about randomisation and allocation concealment, and this might undermine the validity of overall findings. However, all the studies were double-blind and the scant information in terms of quality assessment could be more an issue of reporting in the text than real defects in study design, as it has been commonly found in other systematic reviews (Huwiler-Müntener et al. Reference Huwiler-Müntener, Jüni, Junker and Egger2002). Finally, findings from this analysis apply only to acute-phase treatment of PTSD. Clinically, the assessment of efficacy after 4 weeks of treatment or after 16 weeks or more might lead to wide differences in treatment outcome, and this may limit the ability to provide valid estimates of treatment effect if different durations of follow-up are combined. To overcome this problem, we employed a common definition of acute response that included a pre-defined follow-up duration (8 weeks). Moreover, because network meta-analysis requires reasonably homogeneous studies, in this project we had to restrict ourselves to short-term trials and decided to address the issue of long-term relapse prevention treatment in an ad hoc network meta-analysis, as we previously did for bipolar disorder (Cipriani et al. Reference Cipriani, Barbui, Salanti, Rendell, Brown and Stockton2011; Miura et al. Reference Miura, Noma, Furukawa, Mitsuyasu, Tanaka and Stockton2014).
All-cause discontinuation has been used in other NMAs as a measure of the acceptability of treatments because it encompasses at the same time efficacy and tolerability. Response rate and dropout rate were chosen as primary outcomes to have the best estimate of acute treatment effectiveness (Cipriani et al. Reference Cipriani, Furukawa, Salanti, Geddes, Higgins and Churchill2009; Leucht et al. Reference Leucht, Cipriani, Spineli, Mavridis, Orey and Richter2013). In our analysis, the results paralleled the efficacy findings in that phenelzine, the most effective drug also had the lowest discontinuation rates. We used the neutral term all-cause discontinuation because clinicians might intuitively associate the word acceptability more with tolerability than with efficacy. However, it is worth noting that phenelzine, even though not statistically significant, showed a trend of better tolerability in terms of dropout rate due to adverse events when compared with placebo. Even if in clinical practice MAOIs are used much less frequently than other antidepressant agents because of the dangers of dietary and drug interactions (http://www.bnf.org/bnf/index.htm), findings from this review reinforce the idea that phenelzine should be prioritised in future trials in PTSD, particularly in the significant proportion of patients who remain symptomatic despite first-line psychological and pharmacological treatments. It would also be of interest to use magnetic resonance spectroscopy to assess the effect of phenelzine treatment on GABA levels in the human brain and to relate symptomatic improvement in PTSD symptomatology to changes in GABA concentration.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S003329171700349X
Acknowledgements
We thank the following authors for providing us with additional data: Prof Rothbaum, Prof Killeen, Dr Lindley, Dr Saygin and Ms Suliman. We would also like to thank Prof Davis, Prof Baker, Prof Zohar, Dr Davidson, Dr Reich and Dr Tucker for responding to our request even though they were unable to provide us with missing data (most of these studies were published more than 10 years ago). We also thank Sarah Dawson (Cochrane Depression, Anxiety and Neurosis Group) for her help in the search strategy. Andrea Cipriani is supported by the NIHR Oxford Cognitive Health Clinical Research Facility. John Geddes is an NIHR Senior Investigator.
Declaration of Interest
Taryn Amos, Adriani Nikolakopoulou, Georgia Salanti, Anna Chaimani, Jonathan Ipser, John R. Geddes: none. Andrea Cipriani: In the past 3 years, he was an expert witness for Accord Healthcare for a patent issue about quetiapine extended release. Phil Cowen: In the past 3 years, he has been a member of an advisory board for Lundbeck. Dan Stein: In the past 3 years, he has received research grants and/or consultancy honoraria from AMBRF, Biocodex, Cipla, Lundbeck, National Responsible Gambling Foundation, Novartis, Servier, and Sun. Previously, he received research grants and/or consultancy honoraria from Abbott, ABMRF, Astrazeneca, Biocodex, Eli-Lilly, GlaxoSmithKline, Jazz Pharmaceuticals, Johnson & Johnson, Lundbeck, National Responsible Gambling Foundation, Novartis, Orion, Pfizer, Pharmacia, Roche, Servier, Solvay, Sumitomo, Sun, Takeda, Tikvah, and Wyeth.