


INTRODUCTION
To date, malaria remains one of the top infectious diseases of public health importance. According to the World Health Organization, there were 219 million cases of malaria with an estimated 660 000 deaths in 2010 (WHO, 2013). Of all Plasmodium strains that infect humans, Plasmodium falciparum causes the majority of severe form of malaria (Beeson and Brown, Reference Beeson and Brown2002).
In order to complete their life cycle, parasites such as Plasmodium, Leishmania and Trypanosoma rely on insect vectors and mammalian hosts. Hence, they are exposed to a drastic change in temperature of more than 10 °C. In order to adapt to the abrupt change in temperature, the parasites are endowed with a variety of molecular chaperone proteins. For instance, about 2% of P. falciparum genes code for proteins that function as molecular chaperones (Acharya et al. Reference Acharya, Kumar and Tatu2007).
HEAT SHOCK PROTEINS
Heat shock proteins (Hsp) are molecular chaperones that are expressed under both normal physiological conditions and as a result of environmental stress. Thus, Hsp are both constitutively expressed and inducible. They are abundant proteins in eukaryotic cells and play a crucial role in proper functioning of cells under physiological conditions and also in withstanding environmental stress. Under stressful conditions, the expression of Hsp increases dramatically. Environmental conditions that trigger increased Hsp response include abrupt change in temperature and nutrient deprivation, as well as exposure to heavy metal ions (Bonnefoy et al. Reference Bonnefoy, Attal, Langsley, Tekaia and Mercereau-Puijalon1994; Scheibel and Buchner, Reference Scheibel and Buchner1998).
Heat shock proteins are ubiquitous, evolutionarily conserved proteins that exist in all life forms, ranging from prokaryotic cells to higher organisms such as plants and mammals. For instance, Hsp90 from different levels of eukaryotes such as unicellular protozoa, insects and mammals show greater than 40% homology (Lindquist and Craig, Reference Lindquist and Craig1988; Bonnefoy et al. Reference Bonnefoy, Attal, Langsley, Tekaia and Mercereau-Puijalon1994). Due to their chaperoning activities, Hsp are involved in a variety of cell functions. In addition to helping newly synthesized proteins to fold properly, Hsp are also involved in intracellular protein trafficking, gene expression and the cell cycle, as well as differentiation and development of cells (Acharya et al. Reference Acharya, Kumar and Tatu2007; Neckers and Tatu, Reference Neckers and Tatu2008; Shonhai, Reference Shonhai2010). Moreover, Hsp prevent aggregation of proteins by inhibiting intermolecular interactions (Wiech et al. Reference Wiech, Buchner, Zimmermann and Jakob1992).
Based on molecular size, the major Hsp families are grouped into small Hsp (sHsp), Hsp40, Hsp60, Hsp70, Hsp90 and Hsp110. Hsp90 is one of the most abundant cytosolic proteins of a cell (Shonhai, Reference Shonhai2010). Hsp90 is a dimeric protein and has three major domains: N-terminal domain, middle domain, and C-terminal domain. The N-terminal domain is characterized by having a deep pocket that is used for ATP binding. The C-terminal domain is used for dimerization of the protein. Located in between the N- and C-terminal domains is the highly charged middle domain. The middle domain of Hsp90 protein is involved in binding client proteins and co-chaperones as well as in regulating the ATPase activity of the N-terminal domain. While the N- and C-terminal domains are highly conserved regions, the middle domain is variable in different organisms. Hsp90 exists as a multiprotein chaperone comprising a dimeric Hsp90, Hsp70 and different co-chaperones (Csermely et al. Reference Csermely, Schnaider, Soti, Prohaszka and Nardai1998; Scheibel and Buchner, Reference Scheibel and Buchner1998; Bracher and Hartl, Reference Bracher and Hartl2006; Pallavi et al. Reference Pallavi, Roy, Nageshan, Talukdar, Pavithra, Reddy, Venketesh, Kumar, Gupta, Singh, Yadav and Tatu2010).
HEAT SHOCK PROTEIN 90 – A TARGET FOR ANTIMALARIAL DRUGS
The crucial role of Hsp90 in the folding of proteins that are involved in important cellular functions such as signalling and the cell cycle makes it an important target for anti-tumour drugs (Scheibel and Buchner, Reference Scheibel and Buchner1998). In protozoan parasites such as Plasmodium, Leishmania and Trypanosoma, Hsp90 plays a pivotal role in the differentiation of the parasites from one stage to another and in adapting to environmental stress conditions such as abrupt increases in temperature. Using small molecular inhibitors, Banumathy et al. (Reference Banumathy, Singh, Pavithra and Tatu2003) have shown that Hsp90 is important for the development of P. falciparum from ring to trophozoite stage.
The discovery of the antibiotic geldanamycin (DeBoer et al. Reference DeBoer, Meulman, Wnuk and Peterson1970) and demonstration of its antitumour activity (Sasaki et al. Reference Sasaki, Yasuda and Onodera1979) opened a new avenue for using Hsp90 as an anti-tumour drug target. A study on the mechanism of reversal of oncogenic transformation of certain cell types by geldanamycin identified the specific interaction between the drug and Hsp90. The study demonstrated that geldanamycin inhibits the formation of the Hsp90-v-src kinase heteroprotein complex without significantly inhibiting kinase activity. It also showed that geldanamycin binds to Hsp90 in a stable and pharmacologically specific manner (Whitesell et al. Reference Whitesell, Mimnaugh, De Costa, Myers and Neckers1994). Later, Prodromou et al. (Reference Prodromou, Roe, O'Brien, Ladbury, Piper and Pearl1997) demonstrated the crystal structure of the N-terminal domain of Hsp90 and its specific interaction with ATP. This finding showed that geldanamycin inhibits the ATPase activity of Hsp90 by competing with ATP for binding to the specific N-terminal domain of Hsp90. Since then, several small molecules have been tested as anti-tumour agents targeting Hsp90. These include geldanamycin and its derivatives, such as 17AAG (17-(allylamino)-17-demethoxygeldanamycin).
The demonstration of the anti-tumour activity of geldanamycin and its specific interaction with Hsp90 laid the ground-work for the testing of various Hsp90-inhibiting small molecules as possible drugs for the treatment of cancer. Although geldanamycin shows anti-tumour activity, it has never been tested in clinical trials. This is due to its high degree of hepatotoxicity in animals as well as poor solubility and limited in vivo stability. Structural modification of geldanamycin, however, has resulted in the development of Hsp90-inhibiting molecules with improved quality to be used in clinical trials. One of these molecules is 17AAG. In addition to geldanamycin derivatives, synthetic purine and purine-like analogues such as PU-H71 are being tested in patients as Hsp90-inhibitor anti-cancer drugs. To date, several Hsp90 inhibitors have reached the phase I, phase II and even phase III stages of clinical trials as candidate drugs for the treatment of a variety of human cancers (Jhaveri et al. Reference Jhaveri, Taldone, Modi and Chiosis2012).
Apart from their anti-tumour activity, small-molecule inhibitors of Hsp90 have also been tested as candidate drugs for infectious diseases such as malaria. The inhibitory effects of these molecules on P. falciparum Hsp90 makes them feasible antimalarial candidate drugs. The fact that many human Hsp90 inhibitor small molecules have reached human clinical trials has provided an impetus for considering these or similar molecules as candidate antimalarial drugs.
Several in vitro and in vivo antimalarial drug studies that specifically target P. falciparum Hsp90 (PfHsp90) have shown promising results. These molecules include geldanamycin (GA), 17AAG, 17-PEG-Alkyn-GA (Pallavi et al. Reference Pallavi, Roy, Nageshan, Talukdar, Pavithra, Reddy, Venketesh, Kumar, Gupta, Singh, Yadav and Tatu2010), harmine (Shahinas et al. Reference Shahinas, Macmullin, Benedict, Crandall and Pillai2012) and PU-H71 (Shahinas et al. Reference Shahinas, Folefoc, Taldone, Chiosis, Crandall and Pillai2013).
Using the antibiotic geldanamycin (GA), Banumathy et al. (Reference Banumathy, Singh, Pavithra and Tatu2003) clearly demonstrated the essential role of Hsp90 in the growth of P. falciparum. The study showed that treatment of parasite cultures with GA significantly inhibited growth by preventing development from the ring to trophozoite stage. It was also shown that GA treatment appreciably increases the synthesis of PfHsp90. Modelling studies confirmed that the overall folding of the N-terminal domain of PfHsp90 is similar to that of human Hsp90. In addition, this domain showed 69% identity with the N-terminal domain of human Hsp90. Another publication that appeared at almost the same time corroborated those results (Kumar et al. Reference Kumar, Musiyenko and Barik2003). Among other things, the study showed that GA effectively inhibits the in vitro growth of P. falciparum with IC50 value comparable to that of chloroquine. The effect of GA was similar in both chloroquine-sensitive and -resistant strains of P. falciparum. Moreover, the study demonstrated that GA and chloroquine exert a synergistic antimalarial effect (Kumar et al. Reference Kumar, Musiyenko and Barik2003). Both studies clearly demonstrated the importance of using Hsp90 inhibitors as potential antimalarial drugs.
A more recent study has further strengthened the rationale of using Hsp90 as a drug target against unicellular parasitic infections (Pallavi et al. Reference Pallavi, Roy, Nageshan, Talukdar, Pavithra, Reddy, Venketesh, Kumar, Gupta, Singh, Yadav and Tatu2010). A detailed study showed that PfHsp90 has the highest ATPase activity of all known Hsp90s, this being six times higher than that of human Hsp90. Moreover, it was shown that geldanamycin effectively inhibits ATPase activity of PfHsp90. In vivo study using Plasmodium berghei infection in mice showed that 17AAG significantly inhibited parasite growth and increased survival of infected mice. A similar result was seen in Trypanosoma evansi infection in mice (Pallavi et al. Reference Pallavi, Roy, Nageshan, Talukdar, Pavithra, Reddy, Venketesh, Kumar, Gupta, Singh, Yadav and Tatu2010). In light of what has been done so far, the use of natural and synthetic small molecules that inhibit PfHsp90 as antimalarial drugs is a promising endeavour. However, the effectiveness of these antimalarial candidate drugs will ultimately require passage through well-designed human clinical trials.
ANIMAL STUDIES ON P. FALCIPARUM HSP90 INHIBITORS
Plasmodium species that cause malaria in humans do not infect non-primate animal models that are often used to test drug efficacy in vivo. Moreover, none of the Plasmodium species that are currently used for in vivo experiments is a natural pathogen of laboratory mice. In spite of this, in vivo experiments using rodent malaria such as P. berghei have long been employed to test the efficacy of new antimalarial drugs. Rodent models of Plasmodium infection has been developed and validated through identification of anti-malarials such as mefloquine and halofantrine. These models are being more widely used and are proven to be good indicators of treatment outcomes for human malaria (Peters et al. Reference Peters, Howells, Portus, Robinson, Thomas and Warhurst1977, Reference Peters, Robinson and Ellis1987; Sanni et al. Reference Sanni, Fonseca and Langhorne2002; Fidock et al. Reference Fidock, Rosenthal, Croft, Brun and Nwaka2004). Recently, a SCID (Severe Combined Immunodeficiency) mouse model was tested for in vivo drug efficacy studies against P. falciparum infection. A strain of SCID mice was engrafted with human erythrocytes by continuously injecting the mice with human erythrocytes. These mice have been found to establish infection with a clone of P. falciparum 3D7 (Angulo-Barturen et al. Reference Angulo-Barturen, Jimenez-Diaz, Mulet, Rullas, Herreros, Ferrer, Jimenez, Mendoza, Regadera, Rosenthal, Bathurst, Pompliano, Gomez de las Heras and Gargallo-Viola2008; Jimenez-Diaz et al. Reference Jimenez-Diaz, Mulet, Viera, Gomez, Garuti, Ibanez, Alvarez-Doval, Shultz, Martinez, Gargallo-Viola and Angulo-Barturen2009). The feasibility of using this strain of mice to test different classes of antimalarials against P. falciparum remains to be seen.
Although a few non-human primates can be used as models for testing the efficacy of drugs for falciparum malaria, their widespread use is hampered by high cost and ethical issues (Angulo-Barturen et al. Reference Angulo-Barturen, Jimenez-Diaz, Mulet, Rullas, Herreros, Ferrer, Jimenez, Mendoza, Regadera, Rosenthal, Bathurst, Pompliano, Gomez de las Heras and Gargallo-Viola2008).
Several species and strains of rodent Plasmodium have been used for in vivo anti-malarial efficacy tests. The choice of the right Plasmodium and mouse strain is of paramount importance in the interpretation of the experimental results. Rodent Plasmodium species differ from each other in their degree of infection, the severity of the disease they cause as well as in sensitivity to certain classes of compounds (Sanni et al. Reference Sanni, Fonseca and Langhorne2002; Fidock et al. Reference Fidock, Rosenthal, Croft, Brun and Nwaka2004). For example, P. berghei ANKA, P. berghei K173, P. yoelii YM and P. vinckei vinckei strains cause lethal infection in BALB/c mice whereas other strains, such as P. chabaudi chabaudi, P. chabaudi adami and P. vinckei petteri, cause non-lethal infection. Plasmodium strains that cause lethal infection are used to test the efficacy of new drugs and vaccines while the non-lethal infection-causing strains are used to study the mechanisms of immunity and immune regulation (Sanni et al. Reference Sanni, Fonseca and Langhorne2002).
In addition to the type of rodent malaria and strain of mouse used, there have been differences in the drug-efficacy test procedure followed in different studies. As reviewed in Fidock et al. (Reference Fidock, Rosenthal, Croft, Brun and Nwaka2004), most antimalarial drug tests use a ‘4-day suppressive test’ followed by a ‘dose-ranging test’ using P. berghei ANKA infection. The former is used to determine the efficacy of four daily injections of a candidate drug by comparing parasitaemia on day 4 after infection and survival of treated mice with untreated controls. The dose-ranging test is used to further evaluate the effectiveness of a candidate drug that shows a good in vivo activity in a 4-day suppressive test. The candidate drug is given at a minimum of four different doses (Fidock et al. Reference Fidock, Rosenthal, Croft, Brun and Nwaka2004).
Mouse studies that have been performed so far to evaluate candidate Hsp90 inhibitors as antimalarial drugs have followed this general procedure (see Fig. 1). The procedure includes infection of mice with the Plasmodium parasite, confirmation of infection by peripheral smear, administration of candidate drugs and evaluation of the efficacy of the drugs. The latter involves quantification of the parasitaemia and determination of the survival rate of treated mice as compared with untreated ones. However, the studies appears to differ from each other in the details of the procedure. For instance, in a study of the derivatives of geldanamycin in mice, Pallavi et al. (Reference Pallavi, Roy, Nageshan, Talukdar, Pavithra, Reddy, Venketesh, Kumar, Gupta, Singh, Yadav and Tatu2010) injected mice with the drug after confirmation of infection by microscopy. However, Mout et al. (Reference Mout, Xu, Wolf, Jo Davisson and Jarori2012) waited for the establishment of clinical malaria before administering the candidate drugs, while Shahinas et al. (Reference Shahinas, Macmullin, Benedict, Crandall and Pillai2012, Reference Shahinas, Folefoc, Taldone, Chiosis, Crandall and Pillai2013) used a parasitaemia of 1% as an indicator to start the drug injection. Uniformity of methodology in regards to mouse efficacy trials will greatly aid in interpretation. In addition, there is a marked difference between different studies with regard to the dose of the drug administered in each injection. Table 1 shows a summary of the procedures used in different studies of PfHsp90 inhibitor candidate drugs. It appears that the choice of the detailed procedure in these studies is random and focuses mostly on convenience. Only a few studies have been conducted to test the efficacy of PfHsp90 inhibitor drugs in mice. These studies have generally supported the results seen in in vitro studies regarding the efficacy PfHsp90 inhibitor molecules as potential antimalarial drugs. This review tries to summarise the studies that have been done on PfHsp90 inhibitor molecules in mice so far.

Fig. 1. The general procedure used to study the efficacy of PfHsp90 inhibitors.
Table 1. Summary of the procedure used in in vivo mouse studies on inhibitors of PfHsp90

As described above, Pallavi et al. (Reference Pallavi, Roy, Nageshan, Talukdar, Pavithra, Reddy, Venketesh, Kumar, Gupta, Singh, Yadav and Tatu2010) studied the interaction of PfHsp90 with its inhibitor, geldanamycin, in vitro. Moreover, the in vivo antimalarial activity of one of the derivatives of GA (17AAG) was evaluated in P. berghei infection of Swiss mice. The study followed a modified ‘4-day suppressive’ test procedure. Female Swiss mice were infected with P. berghei intraperitoneally. After confirmation of infection, the mice were treated with intraperitoneal injection of 50 mg kg−1 body weight of 17AAG for 4 consecutive days. Negative control mice were not treated with the drug. The efficacy of the drug was then evaluated based on the difference in per cent parasitaemia and survival rate of drug-treated mice and controls over a period of 3 weeks (Pallavi et al. Reference Pallavi, Roy, Nageshan, Talukdar, Pavithra, Reddy, Venketesh, Kumar, Gupta, Singh, Yadav and Tatu2010).
The result clearly showed that treatment of Swiss mice with four doses of 17AAG significantly reduced the parasitaemia and increased survival of P. berghei-infected mice. Control mice that did not receive the drug showed a parasitaemia of 80–90% at day 6 post-infection and the majority died less than one week after infection. The drug-treated mice showed a significantly lower parasitaemia than the controls. Twenty-one days after infection, 40–50% of the treated mice were alive while all of the control mice succumbed to the infection (Pallavi et al. Reference Pallavi, Roy, Nageshan, Talukdar, Pavithra, Reddy, Venketesh, Kumar, Gupta, Singh, Yadav and Tatu2010).
A study by Mout et al. (Reference Mout, Xu, Wolf, Jo Davisson and Jarori2012) evaluated the efficacy of two derivatives of geldanamycin as potential antimalarials using an in vivo model of P. yoelii infection in Swiss mice. The drugs studied were 17AAG and a new derivative of GA, 17-PEG-Alkyn-GA (17-N-(3-(2-(2-(3-aminopropoxy) ethoxy) ethoxy) propyl) pent-4-ynamide – 17demethoxygeldanamycin). Swiss mice were infected with 106 P. yoelii 17XL parasites intraperitoneally. The first dose of the drugs was given when the mice showed characteristic symptoms of malaria with a parasitaemia of 8–12%. The second dose was given at day 12 post-infection. Each mouse in the experimental groups received 300 nm of either 17AAG or 17-PEG-Alkyn-GA. Positive control mice were given chloroquine phosphate at a dose of 300 nm per mouse. In order to study the level of protection against re-infection, drug-treated mice were allowed to recuperate for one month and were re-challenged with same dose of P. yoelii 17XL. Two control groups were included for the re-challenge experiment; one group that had received chloroquine and another group consisting of naïve Swiss mice (Mout et al. Reference Mout, Xu, Wolf, Jo Davisson and Jarori2012).
The results showed that treatment of P. yoelii 17XL-infected mice with two doses of the GA derivatives resulted in parasite clearance and about 80% survival at day 15 post-infection. However, only a single injection of either of the drugs did not clear the parasite. Treatment of positive control mice with a single dose of chloroquine given at day 6 was sufficient to clear the parasite and cure all the mice. Control mice that did not receive any antimalarial drug showed a parasitaemia of up to 60%. All of these mice died by day 14 post-infection (Mout et al. Reference Mout, Xu, Wolf, Jo Davisson and Jarori2012). This study also assessed, for the first time, the stage specificity of 17AAG and 17-PEG-Alkyn-GA in vivo. Differential counting of the three stages of the parasite, ring, trophozoite and schizont showed that the drugs inhibit the transition of ring stage parasites to trophozoite stage (Mout et al. Reference Mout, Xu, Wolf, Jo Davisson and Jarori2012).
Re-challenge infection with the same dose of P. yoelii 17XL one month after cure demonstrated that GA-derivative drug-treated mice developed milder disease and cleared the infection faster than those that received chloroquine or the control naïve mice that did not receive any drug. The parasitaemia in both of the GA-derivative drug-treated groups peaked at day 2 post-re-challenge infection and never went higher than 4%. The parasites were cleared at day 9. The chloroquine-treated mice showed a parasitaemia of 8–13% and cleared by day 16. The negative control group that did not receive any drug showed 40–60% parasitaemia and succumbed to the infection between 5–8 days after re-challenge (Mout et al. Reference Mout, Xu, Wolf, Jo Davisson and Jarori2012).
Our group has been actively working on the evaluation of different PfHsp90 inhibiting compounds as potential drugs for malaria. Both in vitro and in vivo studies have demonstrated that the PfHsp90 inhibitors tested synergize with chloroquine and exert better anti-parasitic effect. Animal studies have been conducted using infection of BALB/c mice with the P. berghei ANKA strain. Two PfHsp90 inhibitor molecules were tested in vivo: (1) a beta-carboline alkaloid, harmine (Shahinas et al. Reference Shahinas, Macmullin, Benedict, Crandall and Pillai2012), and (2) a purine analogue, PU-H71 (Shahinas et al. Reference Shahinas, Folefoc, Taldone, Chiosis, Crandall and Pillai2013). Harmine belongs to a beta-carboline alkaloid family of compounds that is found in different plant extracts and is used as a traditional antimalarial medicine (Ancolio et al. Reference Ancolio, Azas, Mahiou, Ollivier, Di Giorgio, Keita, Timon-David and Balansard2002; Azas et al. Reference Azas, Laurencin, Delmas, Di, Gasquet, Laget and Timon-David2002). The chemical structure of harmine is composed of an indole skeleton bound to a pyridine ring (Fig. 2A). A study on the mechanism of antimalarial action of harmine showed that the molecule competitively inhibits ATP binding to PfHsp90. As compared with its derivative, methoxy-6-harmalan, harmine preferentially binds to PfHsp90 rather than human Hsp90 (Shahinas et al. Reference Shahinas, Liang, Datti and Pillai2010).
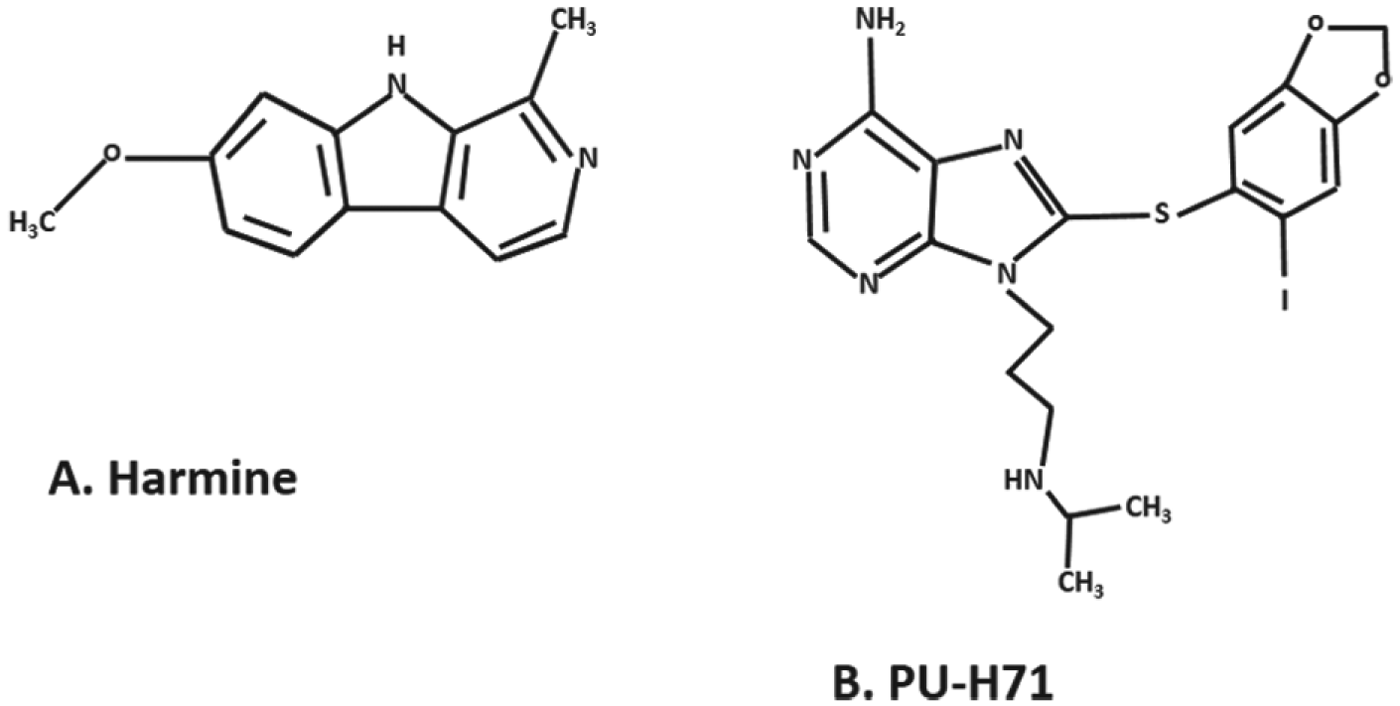
Fig. 2. The chemical structure of harmine (A) and PU-H71 (B).
An in vivo efficacy study on female BALB/c mice showed that harmine potentiates the activity of chloroquine in a combination treatment regimen. Mice were infected with a virulent P. berghei ANKA strain intraperitoneally. Once parasitaemia reached 1%, two groups of mice in the experimental category were given three doses of harmine, each group with a different concentration of the drug (i.e. one group with 75 mg kg−1 and the other with 100 mg kg−1). The positive and negative control groups were given three doses of 30 mg kg−1 chloroquine and PBS, respectively. A combination treatment study was conducted by treating mice with a combination of 5 mg kg−1 chloroquine with either 75 mg kg−1 or 100 mg kg−1 harmine. Control mice were given 5 mg kg−1 chloroquine alone, 75 mg kg−1 harmine alone or 100 mg kg−1 harmine alone. In both combination experiments, a group of mice treated with 30 mg kg−1 chloroquine was included as a positive control. After three consecutive drug injections, the mice were examined daily for signs of clinical malaria. Moreover, the parasitaemia was assessed using Giemsa-stained smears of blood samples daily (Shahinas et al. Reference Shahinas, Macmullin, Benedict, Crandall and Pillai2012).
The result showed that compared with control injection with PBS, treatment with 100 mg kg−1 body weight of harmine induced a statistically significant reduction in parasite load at day 8 post-infection. Combination of 5 mg kg−1 chloroquine with 75 mg kg−1 or 100 mg kg−1 harmine resulted in an average of 96·4%±1·5% and 97·8%±1·5% reduction of parasitaemia, respectively. However, no significant difference was seen between drug-treated and untreated groups with respect to per cent survival of mice. No significant toxicity was observed in treated mice as assessed by weight loss, cage activity and grooming behaviour (Shahinas et al. Reference Shahinas, Macmullin, Benedict, Crandall and Pillai2012).
Recently, our group has tested the efficacy of another inhibitor of PfHsp90, PU-H71 (Shahinas et al. Reference Shahinas, Folefoc, Taldone, Chiosis, Crandall and Pillai2013). As shown in Fig. 2B, the PU-H71 structure consists of a purine backbone, a sulphur linker and aryliodide. As with other purine analogues, the PU-H71 molecule is chemically synthesized using the adenine skeleton. The modification gives the molecule improved pharmaceutical properties without changing its specific interaction with Hsp90 (He et al. Reference He, Zatorska, Kim, Aguirre, Llauger, She, Wu, Immormino, Gewirth and Chiosis2006). PU-H71 is being tested in a phase 1 clinical trial in patients with advanced malignancies (ClinicalTrials.gov, 2013). Interaction studies between PU-H71 and PfHsp90 have shown that the molecule binds with the ATP-binding domain of PfHsp90 with high affinity and inhibits its ATPase activity. An in vitro study on P. falciparum also demonstrated that PU-H71 exerts an antimalarial effect and acts synergistically with known antimalarials such as chloroquine (Shahinas et al. Reference Shahinas, Folefoc, Taldone, Chiosis, Crandall and Pillai2013).
A mouse study on the in vivo efficacy of PU-H71 was done using infection of BALB/c mice with the P. berghei ANKA strain following the procedure described with harmine. Mice were injected with different concentrations of the drug alone or in combination with 0·25 or 0·5 mg kg−1 chloroquine intraperitoneally three times starting day 5 post-infection. The efficacy of the drug in vivo was then assessed based on the per cent parasitaemia and degree of survival of treated mice relative to untreated controls.
The results demonstrated that upon injection with three doses of 75 or 100 mg kg−1, PU-H71 exerts antimalarial activity, with parasitaemia significantly lower than that of vehicle control at day 8 post-infection. As with harmine, PU-H71 was also found to be synergistic with chloroquine in vivo. Compared with treatment with 0·25 mg kg−1 chloroquine alone, injection of a combination of 25 mg kg−1 PU-H71 and 0·25 mg kg−1 chloroquine resulted in a significantly higher reduction in parasitaemia and also improved survival by 3 days. Moreover, the drug did not show a significant toxic effect in both infected mice and uninfected controls. Both the in vitro and in vivo results suggest that PU-H71 is a good antimalarial candidate drug in combination with established drugs such as chloroquine (Shahinas et al. Reference Shahinas, Folefoc, Taldone, Chiosis, Crandall and Pillai2013).
CHALLENGES IN ANIMAL STUDIES OF P. FALCIPARUM HSP90 INHIBITORS
As discussed above, few studies have so far been done to evaluate the in vivo antimalarial efficacy of PfHsp90 inhibitor molecules. These studies have shown different level of efficacy of the candidate drugs in vivo. Although the studies separately show excellent results, comparing the results of the studies is a very challenging endeavour. This is partly because the procedures used in the studies differ. The differences include the strain of rodent Plasmodium used, dose of the drugs, time of drug administration and number of drug injections. Table 1 summarizes some of the differences in the procedures followed in different in vivo studies in mice. A consensus is needed on the design of mouse trials that seek to establish new Hsp90 inhibitors to enter human clinical trials, the design of which is yet to be established. One possibility is the use of Hsp90 inhibitors as an adjunctive drug in concert with current antimalarials used in the region concerned.
ACKNOWLEDGEMENTS
We thank Dr Dea Shahinas for assistance in preparing Figure 1.



