INTRODUCTION
Blastocystis spp. are common intestinal parasites found in several animal groups with a widespread geographic distribution (Tan, Reference Tan2008). They are the most prevalent protozoa found in human fecal samples. Although the involvement of Blastocystis spp. in human diseases remains unclear, a potential link with the irritable bowel syndrome was recently reported (Poirier et al. Reference Poirier, Wawrzyniak, Vivares, Delbac and El Alaoui2012). These anaerobic parasites were first described by Alexeieff at the beginning of the 20th century and classified as yeasts by Brumpt (Alexeieff, Reference Alexeieff1911; Brumpt, Reference Brumpt1912). Blastocystis spp. were then classified in the heterogeneous group of the Stramenopiles on the basis of the sequence of the gene encoding the 18S rRNA (Silberman et al. Reference Silberman, Sogin, Leipe and Clark1996). Recently, the 18·8 Mb genome sequence of a Blastocystis subtype 7 isolate (strain B) has been reported (Denoeud et al. Reference Denoeud, Roussel, Noel, Wawrzyniak, Da Silva, Diogon, Viscogliosi, Brochier-Armanet, Couloux, Poulain, Segurens, Anthouard, Texier, Blot, Poirier, Ng, Tan, Artiguenave, Jaillon, Aury, Delbac, Wincker, Vivares and El Alaoui2011). Genome sequencing has also revealed the presence of a small circular genome of ∼30 kb in size located within double-membrane surrounded-compartments known as mitochondria-like organelles (MLOs) (Wawrzyniak et al. Reference Wawrzyniak, Roussel, Diogon, Couloux, Texier, Tan, Vivares, Delbac, Wincker and El Alaoui2008). This circular genome consists of 45 genes including 27 ORFs, 16 tRNAs and two rRNA genes (the small (srRNA) and large (lrRNA) subunit ribosomal RNAs). The entire annotated sequence of the MLO genome with a high gene synteny conservation has been also reported for 3 other subtypes including ST1, ST3 and ST4 (Perez-Brocal and Clark, Reference Perez-Brocal and Clark2008; Wawrzyniak et al. Reference Wawrzyniak, Roussel, Diogon, Couloux, Texier, Tan, Vivares, Delbac, Wincker and El Alaoui2008; Stensvold et al. Reference Stensvold, Alfellani and Clark2011).
Molecular studies revealed a high genetic diversity within the Blastocystis genus. At least 17 different subtypes (STs) referred to as ST1 to ST17 have been identified, with 9 of them (ST1 to ST9) having been recovered from human stool samples (Alfellani et al. Reference Alfellani, Taner-Mulla, Jacob, Imeede, Yoshikawa, Stensvold and Clark2013). In humans, the most frequently encountered ST is ST3, followed by ST1, ST2 and ST4 whereas ST5 to ST9 are more rarely found (Stensvold et al. Reference Stensvold, Alfellani, Norskov-Lauritsen, Prip, Victory, Maddox, Nielsen and Clark2009; Alfellani et al. Reference Alfellani, Taner-Mulla, Jacob, Imeede, Yoshikawa, Stensvold and Clark2013). For subtyping, most epidemiological studies used a 600 bp barcoding region within the 18S small subunit ribosomal DNA (18S rDNA) (Scicluna et al. Reference Scicluna, Tawari and Clark2006). The amplification of this 18S rDNA region is followed by sequencing of the PCR product to assign isolates to specific STs. The sequencing of several clones is required to detect potential co-infections by different STs. Indeed, co-infections have been shown to occur at different frequencies between 3·6 and 17·2% of positive Blastocystis spp. patients and are probably underestimated (Tan, Reference Tan2008; Meloni et al. Reference Meloni, Poirier, Mantini, Noel, Gantois, Wawrzyniak, Delbac, Chabe, Delhaes, Dei-Cas, Fiori, El Alaoui and Viscogliosi2012). A case of co-infection by 3 different STs and probably by different strains belonging to the same ST was recently described (Meloni et al. Reference Meloni, Poirier, Mantini, Noel, Gantois, Wawrzyniak, Delbac, Chabe, Delhaes, Dei-Cas, Fiori, El Alaoui and Viscogliosi2012). Among the 50 clones that had been sequenced, 6 corresponded to ST2, 6 to ST4 and 38 to ST3. The suspicion of co-infections by different strains of ST3 was supported by the polymorphism observed between the 38 sequences belonging to ST3 with an identity ranging from 86·9% to 100%. It was hypothesized that these differences could be due to co-infections by different strains from the same ST (Souppart et al. Reference Souppart, Sanciu, Cian, Wawrzyniak, Delbac, Capron, Dei-Cas, Boorom, Delhaes and Viscogliosi2009, Reference Souppart, Moussa, Cian, Sanciu, Poirier, El Alaoui, Delbac, Boorom, Delhaes, Dei-Cas and Viscogliosi2010).
In the present study, we first analysed the sequence polymorphism of the multiple copies coding for the 18S rRNA reported in the genome of a BlastocystisST7 isolate (Denoeud et al. Reference Denoeud, Roussel, Noel, Wawrzyniak, Da Silva, Diogon, Viscogliosi, Brochier-Armanet, Couloux, Poulain, Segurens, Anthouard, Texier, Blot, Poirier, Ng, Tan, Artiguenave, Jaillon, Aury, Delbac, Wincker, Vivares and El Alaoui2011). We then demonstrated that sequence variation between intragenomic copies also concerns the 600 bp barcoding region which is commonly used as the method of choice for Blastocystis subtyping. Finally, we tested and validated a new subtyping rDNA marker from the circular mitochondrial-like genome of Blastocystis spp. A partial sequence of this new marker was successfully used to discriminate ST1 to ST10 among 66 isolates of Blastocystis spp.
MATERIALS AND METHODS
Analysis of the multiple copies of the 18S rDNA gene within the genome of Blastocystis ST7 (strain B)
The identification of the 17 complete copies of the 18S rDNA gene from the genome of Blastocystis ST7 (strain B) was done using Genome Browser 1.68. Alignment of these copies (accession numbers are mentioned in Fig. 1) was performed with Geneious Pro 4.8.3 software. The percentage of identity was determined between both the 17 complete rDNA copies and the sequences corresponding to the 600 bp barcoding region.

Fig. 1. Full-length sequence alignment of the 17 complete copies of the 18S rDNA gene identified in the genome of the Blastocystis ST7 strain B. Alignment was performed using the Geneious Pro 4.8.3 software. The names of the 18S rDNA sequences include the scaffold (scf) number followed by the number of nucleotides. Some scaffolds (scf18, scf11) contain multiple 18S rDNA copies (a, b). The 600 bp barcoding region commonly used for Blastocystis subtyping is located in the first part of the gene. Polymorphic sites between the 17 copies are summarized in the consensus: homology of 100% in green, from 100 to 60% yellow and less than 60% in red. Some sites of polymorphism were located in the barcoding region.
Identification of a new subtyping rDNA marker within the MLO genome
The MLO genome full sequences of ST1, ST4 and ST7 were uploaded from databases and aligned using ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html). A region located in the second part of the gene encoding the rRNA of the small ribosomal subunit of the MLOs (MLO-rDNA) was selected and amplified using MLOsrDNA-D1 (5′-GACATTGATAGACGAAAG-3′) and MLOsrDNA-R1 (5′-GTAGCACATGTGTAGCCC3′) primers. This region presented variations in both nucleotide sequence (at least 7% of nucleotide differences between the three STs) and length (from 440 nt for ST2 to 536 nt for ST7). The amplification of this MLO-rDNA region was then performed for isolates representatives of ST2, ST3, ST5, ST6, ST8, ST9 and ST10. All these STs were previously subtyped using the 18S nuclear rDNA marker.
PCR reactions
The MLO-rDNA marker amplifications were run in a final volume of 50 μL according to standard conditions for the Platinum Taq High-Fidelity DNA polymerase (Invitrogen). 5 μL of genomic DNA was added to 1 μ m of each primer MLOsrDNA-D1 (5′-GACATTGATAGACGAAAG-3′) and MLOsrDNA-R1 (5′-GTAGCACATGTGTAGCCC3′). After denaturation at 95 °C for 10 min, 45 cycles of amplification were performed as follows: 30 s at 95 °C, 45 s at 56 °C, and 45 s at 68 °C. The final extension step was continued for 20 min. The PCR products were separated by agarose gel electrophoresis, and bands of the expected size were purified using the Wizard SV Gel and PCR clean-up system (Promega, Madison, WI, USA). Purified PCR products were cloned in the T-vector, pCR 2.1-TOPO (Invitrogen) and amplified in Escherichia coli One Shot TOP10 competent cells. Plasmid DNA was purified using the QIAprep Spin Miniprep kit (Qiagen). Two clones were randomly sequenced for each isolate by MWG eurofins (Germany).
Subtyping of isolates from human and animal stool samples
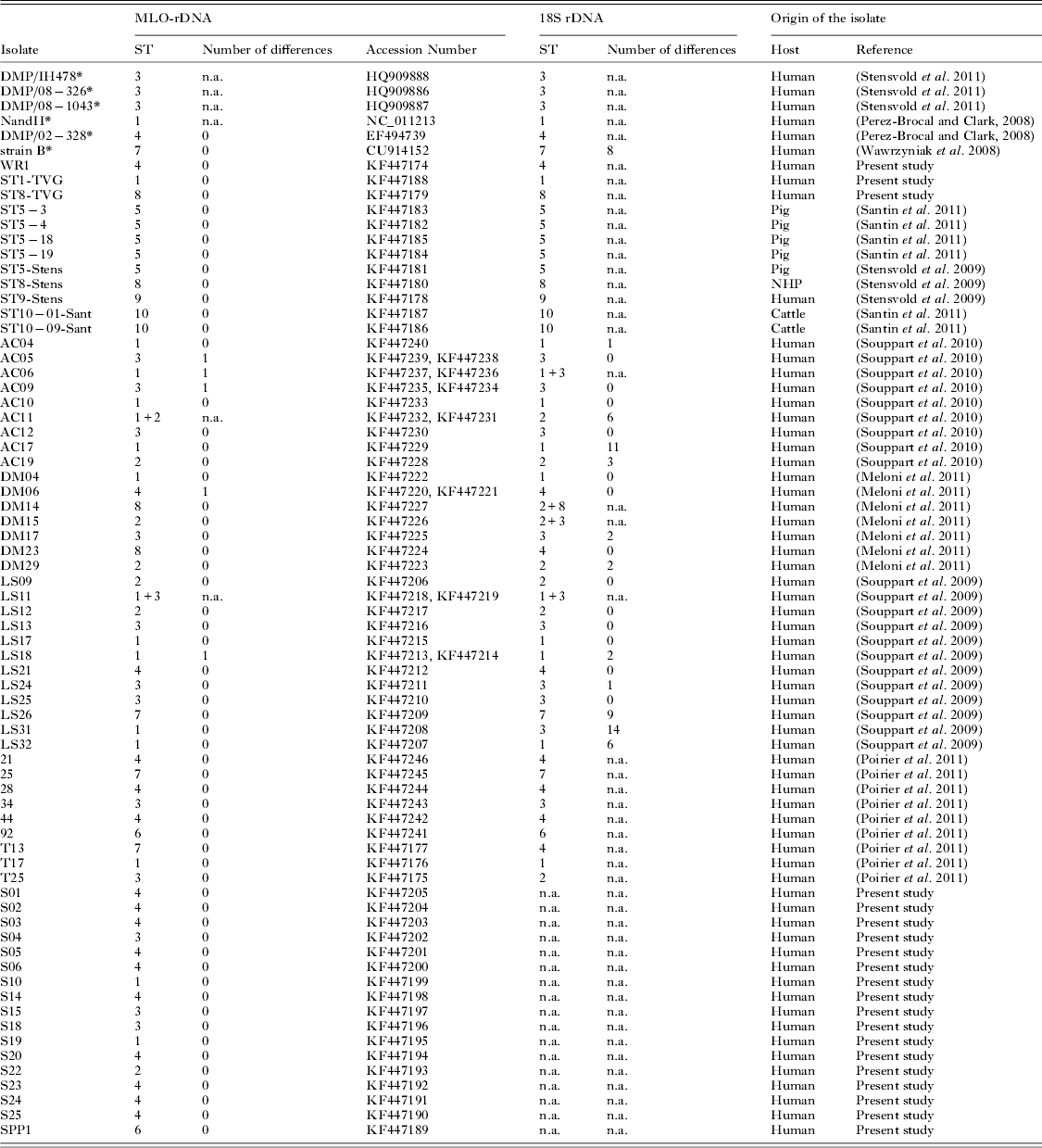
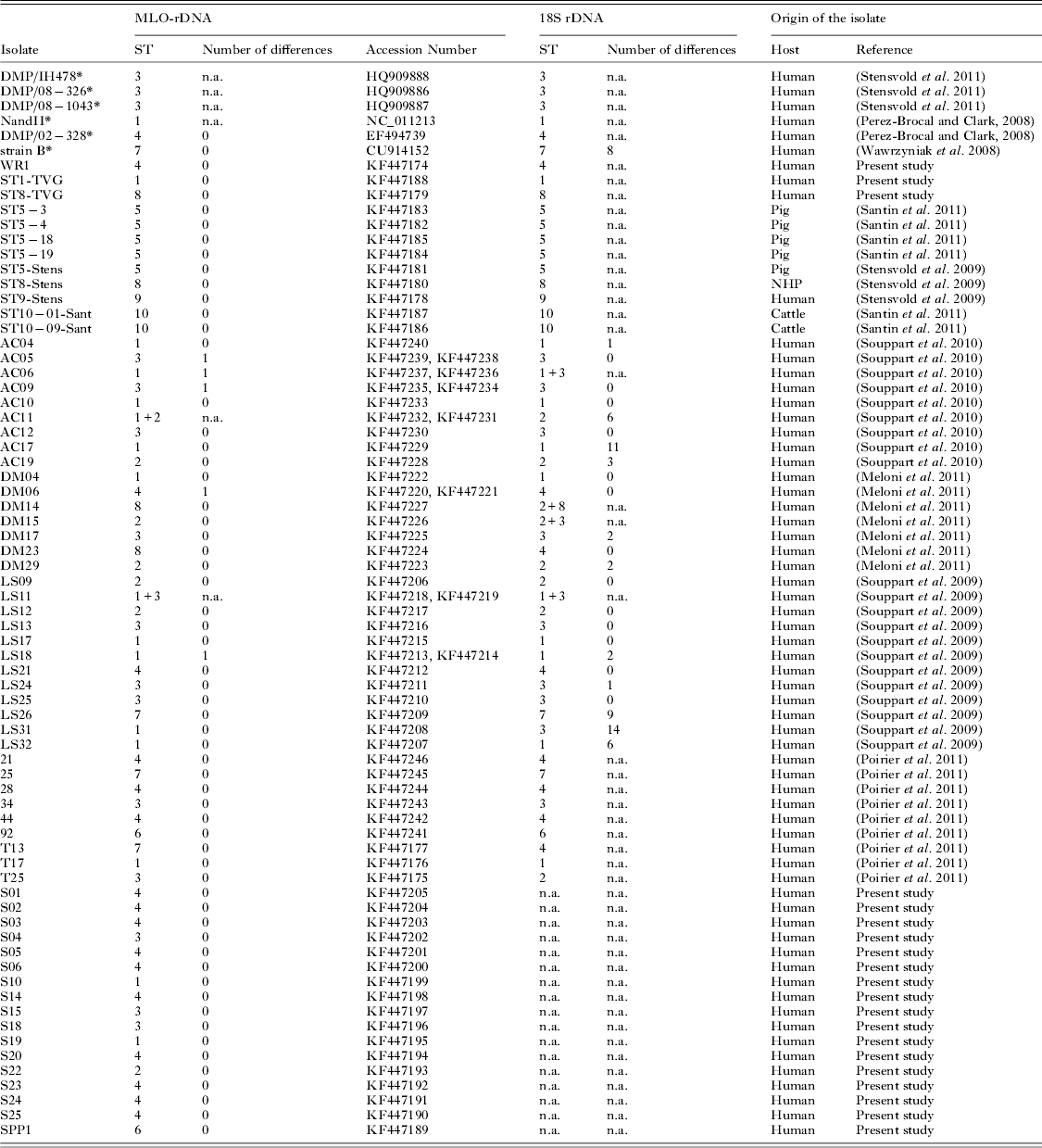
The MLO-rDNA marker was amplified, cloned and sequenced for 66 Blastocystis spp. isolates from both human and animals (accession numbers are mentioned in Table 2). Among the 66 isolates, 49 were also subtyped in previous studies with the 18S rDNA barcoding region and 28 by sequencing 2 clones of the 18S rDNA barcoding region (isolates of the ‘AC’, ‘DM’ and ‘LS’ sets). The MLO-rDNA marker was also used to subtype 17 isolates prospectively diagnosed by fecal smear observation in the French Medical Laboratory of Parasitology of the teaching hospital of Clermont-Ferrand, France (isolates S01 to S25, and SPP1). DNA of the 17 isolates was extracted using the QIAamp DNA stool mini kit (Qiagen, France) according to the manufacturer's recommendations.
Phylogenetic analysis of the MLO-rDNA marker
MLO-rDNA sequences from DMP/IH478 (ST3), DMP/08-1043 (ST3), DMP/08-326 (ST3), NandII (ST1), DMP/02-328 (ST4) and strain B (ST7) were uploaded (http://www.ncbi.nlm.nih.gov/genbank/) (Perez-Brocal and Clark, Reference Perez-Brocal and Clark2008; Wawrzyniak et al. Reference Wawrzyniak, Roussel, Diogon, Couloux, Texier, Tan, Vivares, Delbac, Wincker and El Alaoui2008; Stensvold et al. Reference Stensvold, Alfellani and Clark2011). These 6 sequences were added for the analysis to the 66 sequences from isolates subtyped in the present study. Tree was generated using Mega 5.05 software (Fig. 3).
RESULTS
Multiple variable copies of the 18S rDNA gene within the Blastocystis ST7 genome
Genome analysis of the Blastocystis ST7 strain B revealed the presence of 31 sequences encoding the 18S rRNA, with 17 completely assembled copies. The total length of these 17 non-identical copies ranged from 1798 to 1819 bp and the sequence identity between copies was shown to vary from 99·9% to 98·1% (i.e. 1 to 38 nucleotide differences between copies) (Fig. 1). As mentioned above, most epidemiological studies used a 600 bp barcoding region (Scicluna et al. Reference Scicluna, Tawari and Clark2006), which is located within the 18S rDNA gene (Fig. 1). Comparison of this barcoding region from the 17 copies of the 18S rDNA gene indicated a sequence identity ranging from 100% to 98·4% (i.e. 0 to 10 nucleotide differences between copies). As shown in Table 1, these 17 sequences could be classified into 6 clusters (C1 to C6), each cluster including sequences with 100% identity. One sequence of each cluster was then used for a Blastn analysis. As expected, all clusters corresponded to ST7, but maximal homology with isolate sequences deposited in GenBank varied among clusters. Indeed, sequences from C1 and C2 clusters had respectively 100% and 99% identity with strain B corresponding to the sequenced ST7 genome isolate (Table 1), whereas clusters C3 to C6 had the highest homology with sequences assigned to two other ST7 strains. C3 presented 100% identity with a sequence of the strain QQ98-4. C4 and C5 also presented the highest homology with the strain QQ98-4. C6 showed a 100% identity with the ST7 isolate H (Table 1).
Table 1. Analysis of the 6 clusters from the 600 bp barcoding region of Blastocystis ST7 (strain B). For each cluster, a Blastn analysis was performed. We reported the maximal homology results from the GenBank database for each cluster

Identification of a new subtyping rDNA marker from the MLO circular genome
We determined a new barcoding region named MLO-rDNA located within the gene encoding the small ribosomal subunit rRNA of the MLOs. This sequence was selected on the basis of the alignment between MLOs genomes from Blastocystis ST1 (NandII), ST4 (DMP/02-328) and ST7 (strain B). Even though the mitochondria rDNA gene is known to be present as a single copy within the circular genome, we sequenced 20 clones of this marker from axenic cultures of both ST7 (strain B) and ST4 (WR1) isolates. All clones were 100% identical for each ST. This mitochondrial marker was then successfully amplified and sequenced for different Blastocystis spp. isolates belonging to ST1 to ST10. Fragment length of the MLO-rDNA marker ranged from 440 bp for ST2 to 536 bp for ST7 (Fig. 2), and sequence identity between the 10 STs ranged from 83% to 93%. The difference between each ST was at least 7%, allowing easy discrimination between STs. We then used this MLO-rDNA marker to subtype 66 isolates from human or animal stools (Table 2). Among these isolates, we identified 16 ST1, 7 ST2, 15 ST3, 16 ST4, 3 ST5, 2 ST6, 3 ST7, 2 ST8 and 2 ST10.

Fig. 2. Alignment of the MLO-rDNA marker sequence from Blastocystis ST1 to ST10 using ClustalW2 software. The sequences of the primers are not included in the alignment. Fragment length of the amplified region varied from 440 bp for ST2 to 536 bp for ST7 with a homology between STs ranging from 83 to 93%. Nucleotides are numbered on the top of the alignment.
Table 2. Comparison of nuclear 18S rDNA and MLO-rDNA markers for the subtyping of Blastocystis isolates. 66 isolates subtyped with the MLO-rDNA marker were sequenced in our work (2 clones per isolate). Among them, 49 were also subtyped in other studies with the 18S rDNA marker (sequencing of 2 clones per isolates for 28 samples ‘AC’, ‘DM’, ‘LS’). The number of nucleotide differences between the two clones was reported when available. The table also includes 6 sequences (*) available in databases. (n.a. : not available; NHP : Non-Human Primate)
Comparison of subtyping methods using the nuclear 18S rDNA and the MLO-rDNA markers
Among the 66 isolates we subtyped with the MLO-rDNA marker, 49 had been previously subtyped with the 18S rDNA barcoding region, and among them 28 with a cloning/sequencing approach that is commonly used to detect co-infections. As shown in Table 2, some discrepancies were observed between the 2 markers for 8 isolates (AC06, AC11, DM14, DM15, DM23, LS31, T13 and T25) suggesting potential co-infections by at least 2 subtypes.
Subtyping of unknown isolates using the MLO-rDNA marker
We then subtyped 17 human isolates in a ‘blind-test’ to assess the feasibility of using only the MLO-rDNA marker. These isolates (S01 to S25 and SPP1) were prospectively collected in the Medical Laboratory of Parasitology of the teaching hospital of Clermont-Ferrand (France). Subtyping with the MLO-rDNA marker revealed the presence of 2 ST1, 1 ST2, 3 ST3, 10 ST4 and 1 ST6 isolates (Table 2).
Variability of the MLO-rDNA sequence within STs
The AC17, AC19, DM17 and DM29 isolates presented respectively 11, 3, 2 and 2 nucleotide differences between the 2 sequenced clones of the 18S rDNA (Table 2) whereas no intra-isolate polymorphism was observed with the MLO-rDNA marker. In contrast, isolates AC05, AC06, AC09, DM06 and LS18 presented 1 nucleotide polymorphism between the 2 clones for the MLO-rDNA marker, suggesting a co-infection by 2 different isolates of the same ST.
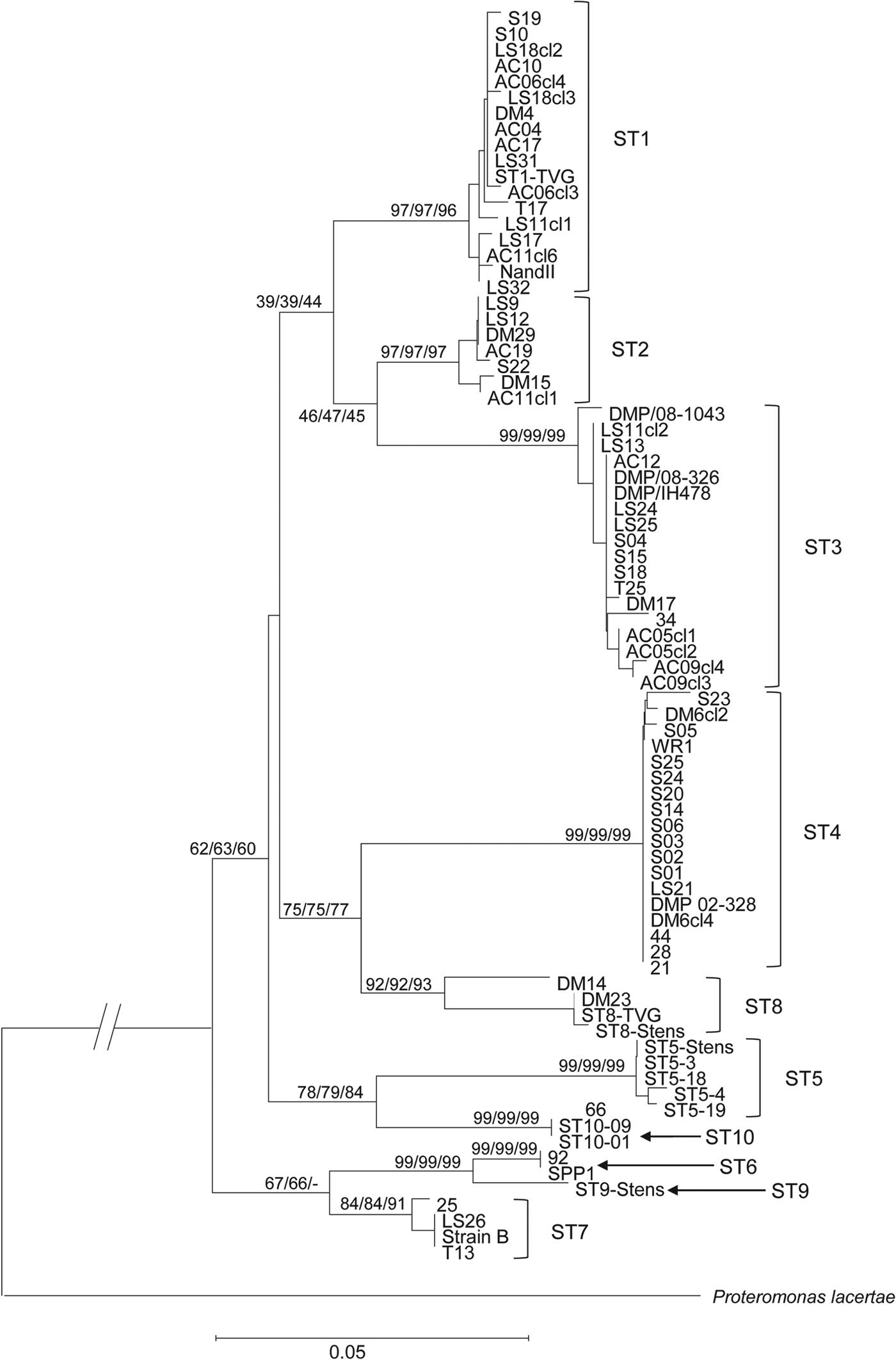
Phylogenetic analysis of all the MLO-rDNA sequences obtained in our study revealed different groups of sequences within the different STs (Fig. 3). Looking at the ST1, ST3 and ST4 for which we had the highest number of isolates, ST3 was the most diversified ST. This analysis also identified 4 main clades, most of them being consistent with those already described in previous phylogenetic studies based on 18S rRNA sequences (Alfellani et al. Reference Alfellani, Taner-Mulla, Jacob, Imeede, Yoshikawa, Stensvold and Clark2013). A first not well-resolved clade included the STs 1, 2 and 3. A second one grouped together the STs 4 and 8. A next clade united the STs 5 and 10 whereas the last clade included the STs 6, 7 and 9.
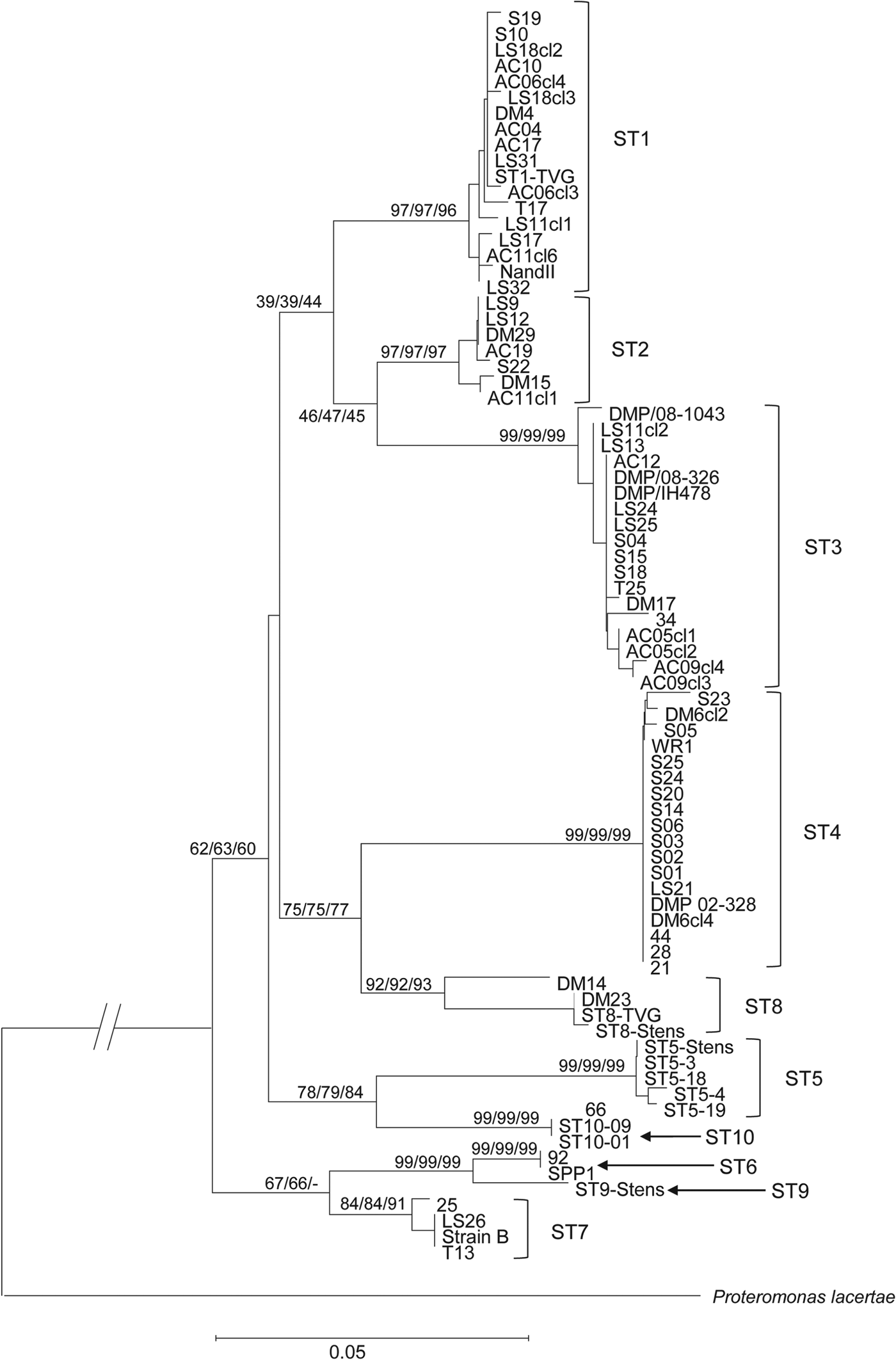
Fig. 3. Minimum evolution (10 000 replicates) tree showing the phylogenetic relationships among the MLO-rDNA sequences of the 66 Blastocystis isolates using the proteromonad P. lacertae as outgroups. The relative frequency from Maximum likelihood (10 000 replicates) and from Neighbour-joining methods (10 000 replicates) are indicated close to nodes.
DISCUSSION
Analysis of the 18S rDNA marker in the Blastocystis ST7 genome
At least 17 different STs of Blastocystis spp. have been described on the basis of a 600 bp barcoding region from the 18S rDNA gene (Alfellani et al. Reference Alfellani, Taner-Mulla, Jacob, Imeede, Yoshikawa, Stensvold and Clark2013). Recently, we highlighted the difficulty to demonstrate the co-infection by different strains of the same ST due to the high intra-genomic variability of the 18S rDNA gene copies (Meloni et al. Reference Meloni, Poirier, Mantini, Noel, Gantois, Wawrzyniak, Delbac, Chabe, Delhaes, Dei-Cas, Fiori, El Alaoui and Viscogliosi2012). In the present study, we analysed the 17 complete copies of this gene in the sequenced genome of Blastocystis ST7 (strain B). All these copies were polymorphic in size and sequence. When considering the only 600 bp barcoding region, the 17 copies could be classified in 6 different clusters (C1 to C6) of identical sequences (Fig. 1). C1 and C2 were identical to the sequences deposited in GenBank database for the strain B. Surprisingly, C3 to C6 were more closely related to 2 other isolates of Blastocystis ST7 named QQ98-4 and H. These results suggest that the genetic diversity of Blastocystis ST7 could be overestimated by the use of the 600 bp region and confirm that this barcoding region cannot be used to discriminate between ST7 isolates. However, the existence of a similar intra-genomic polymorphism of the 18S rDNA gene in the other Blastocystis STs remains to be determined. Even though the intra-genomic polymorphism of the 600 bp in Blastocystis spp. ST7 is above 1%, we cannot confirm that it is the same in the other STs.
A new rDNA marker located in the MLO genome
In the present study, we validated a new DNA barcoding sequence to subtype Blastocystis spp. isolates. This barcoding region is located within the gene encoding the srRNA of the MLOs of Blastocystis spp. Mitochondrial genes evolve at higher rates than nuclear genes, thus becoming advantageous for studies of closely related taxa (DeSalle et al. Reference DeSalle, Freedman, Prager and Wilson1987; Moriyama and Powell, Reference Moriyama and Powell1997; Monteiro and Pierce, Reference Monteiro and Pierce2001). Then, cytochrome b mitochondrial gene was successfully used to recognize close lineages among Babesia species and the srRNA gene demonstrated its usefulness in the molecular phylogeny of the filarial genus Onchocerca (Krueger et al. Reference Krueger, Fischer and Morales-Hojas2007; Tian et al. Reference Tian, Luo, Zheng, Xie, Shen, Yin, Luo, Tian, Yuan, Wang and Liu2013).The MLO-rDNA gene of Blastocystis spp. is present as a single copy in the circular genome and the sequence divergence reaches at least 7% between STs. We confirmed the absence of variability between the genomes of the multiple MLOs present per parasite by sequencing 20 clones of this marker from axenic cultures of both ST7 (strain B) and ST4 (strain WR1). Even though this marker is present as a single copy, Wawrzyniak et al. reported that one MLO enclosed multiple copies of the circular genome (Wawrzyniak et al. Reference Wawrzyniak, Roussel, Diogon, Couloux, Texier, Tan, Vivares, Delbac, Wincker and El Alaoui2008). These ‘multiple identical copies’ facilitated the subtyping when the samples enclosed poor parasite load. Indeed, we were able to subtype some isolates that were only detected by qPCR in the study of Poirier et al. (Reference Poirier, Wawrzyniak, Albert, El Alaoui, Delbac and Livrelli2011). One limitation of our study is that we did not include rare STs (i.e. ST11–ST17). Nevertheless, the main objective was to provide a new tool to detect potential co-infections by different strains of a same ST. This question mainly concerns human studies and only ST1 to ST9 have been so far recovered from human stool samples. Obviously, characterization of the MLO-rDNA marker for these other Blastocystis STs could be of great interest in the field of phylogenetic analysis.
Phylogenetic tree based on the MLO-rDNA marker was congruent with trees built using the 18S rDNA (Alfellani et al. Reference Alfellani, Taner-Mulla, Jacob, Imeede, Yoshikawa, Stensvold and Clark2013). Indeed, ST1 and ST2 were closely related, as well as ST4 and ST8. Avian STs (ST6 and ST7) were also closely related to ST9. The major differences concern the position of the ST3 that was related to ST1 and ST2 using the MLO-rRNA marker whereas this ST is related to ST4/ST8 on the basis of the 18S rDNA (Stensvold et al. Reference Stensvold, Suresh, Tan, Thompson, Traub, Viscogliosi, Yoshikawa and Clark2007). Using the MLO-rDNA marker, we showed that the 3 major STs (i.e. ST1–ST3) in terms of frequency in the human population are more closely related.
Using the MLO-rDNA marker, 66 isolates from both human and animals were successfully subtyped. Among them, 49 were also subtyped with the 18S rDNA marker in other previously reported studies. The use of both methods for subtyping was globally congruent. However, differences between both markers were observed for 8 isolates. These differences could be due to the poor number of clones sequenced and consequently the lack of detection of co-infections with both methods. For 4 isolates (AC06, AC11, DM14 and DM15), discrepancies corresponded to co-infection alternatively detected by either the 18S rDNA marker or the MLO-rDNA marker. DM23 and LS31 isolates were subtyped as ST4 and ST3, respectively, with the 18S rDNA barcoding region, whereas the MLO-rDNA marker identified ST8 and ST1, respectively. T13 (ST7) and T25 (ST3) were previously subtyped by qPCR product sequencing (Poirier et al. Reference Poirier, Wawrzyniak, Albert, El Alaoui, Delbac and Livrelli2011), but the MLO-rDNA sequences identified both isolates as ST4 and ST2, respectively. A possible explanation for these discordances is also that isolates DM23, LS31, T13 and T25 could be co-infected by different STs as illustrated for isolates AC06, AC11, DM14 and DM15. This reinforces the importance of sequencing a sufficient number of clones to study the repartition of STs in the population. Indeed, we recently sequenced 50 clones from 1 isolate and highlighted the presence of 3 different STs (ST2, ST3 and ST4), with a quantitative dominance of ST3 compared with ST4 and ST2 (Meloni et al. Reference Meloni, Poirier, Mantini, Noel, Gantois, Wawrzyniak, Delbac, Chabe, Delhaes, Dei-Cas, Fiori, El Alaoui and Viscogliosi2012). Thus, it will be valuable to establish a consensus about methods and number of clones to sequence with the aim of performing comparisons between studies.
Sequence variability of the MLO-rDNA marker
Authors from different studies have pointed out the variability of the 18S rDNA marker (Souppart et al. Reference Souppart, Sanciu, Cian, Wawrzyniak, Delbac, Capron, Dei-Cas, Boorom, Delhaes and Viscogliosi2009, Reference Souppart, Moussa, Cian, Sanciu, Poirier, El Alaoui, Delbac, Boorom, Delhaes, Dei-Cas and Viscogliosi2010; Meloni et al. Reference Meloni, Sanciu, Poirier, El Alaoui, Chabe, Delhaes, Dei-Cas, Delbac, Luigi Fiori, Di Cave and Viscogliosi2011, Reference Meloni, Poirier, Mantini, Noel, Gantois, Wawrzyniak, Delbac, Chabe, Delhaes, Dei-Cas, Fiori, El Alaoui and Viscogliosi2012). Our results clearly confirmed that the 18S rDNA gene is not the marker of choice to detect co-infection by different strains of the same ST. The MLO-rDNA marker used in our study is present as a single gene copy and its intra-ST polymorphism enables the detection of co-infection by different strains of a same ST as shown in the present study for isolates AC05 (ST3), AC06 (ST1), AC09 (ST3) and LS18 (ST1). The fact that ST1 and ST3 are the most prevalent STs in the human population could explain the co-infection by different strains of these STs. It could also be explained by differences in the genetic diversity between the different STs infecting humans. Indeed, Stensvold et al. demonstrated that ST3 from humans was more genetically diversified than ST4 (Stensvold et al. Reference Stensvold, Alfellani and Clark2011). Our results seem to confirm a lower genetic diversity for ST4. Indeed, only 4 different types of sequences were found among the 18 isolates studied belonging to ST4.
CONCLUSION
The involvement of Blastocystis spp. in human health as well as the potential correlation between certain STs and symptoms and/or gastrointestinal illnesses remains unclear, hence the need to conduct epidemiological studies. In these studies, authors commonly used a 600 bp barcoding region of the 18S rDNA gene (Scicluna et al. Reference Scicluna, Tawari and Clark2006). However, our results highlighted the intra-genomic polymorphism of this marker. We thus proposed a new barcoding region allowing the detection of co-infection by different strains of the same ST. We subtyped 66 isolates with this new marker and detected 4 co-infections by different strains of ST1 and ST3.
ACKNOWLEDGEMENTS
We are grateful to Rune Stensvold, Monica Santin and Tom Van Gool for providing DNA from rare STs (i.e. ST5, ST8 and ST9) of Blastocystis.
FINANCIAL SUPPORT
This work was supported by grants from the Programme Orientations Stratégiques from the University of Lille 2, the Centre National de la Recherche Scientifique, and the Institut Pasteur of Lille. DM was supported by fellowships from the Regione Autonoma della Sardegna and Japan Society for the Promotion of Science (JSPS).