Introduction
Cryptosporidium is an apicomplexan protist parasite that primarily infects the gastrointestinal epithelium of a broad range of vertebrate species including humans (Lv et al., Reference Lv, Zhang, Wang, Jian, Zhang, Ning, Wang, Feng, Wang, Ren, Qi and Xiao2009). Infections can be asymptomatic or can result in diarrhoea ranging from mild to severe. Disease severity depends mainly on the age and immune status of the host (Checkley et al., Reference Checkley, White, Jaganath, Arrowood, Chalmers, Chen, Fayer, Griffiths, Guerrant, Hedstrom, Huston, Kotloff, Kang, Mead, Miller, Petri, Priest, Roos, Striepen, Thompson, Ward, Van Voorhis, Xiao, Zhu and Houpt2015; Baneth et al., Reference Baneth, Thamsborg, Otranto, Guillot, Blaga, Deplazes and Solano-Gallego2016). Field studies have shown that genus Cryptosporidium is genetically diverse, with much of that diversity found in wildlife. Rodents are ubiquitous mammals comprising about 40% of mammalian diversity and occupying a wide range of habitats. Studies to date have shown that rodent species are predominantly parasitized with host-specific Cryptosporidium species and genotypes (Feng et al., Reference Feng, Alderisio, Yang, Blancero, Kuhne, Nadareski, Reid and Xiao2007; Foo et al., Reference Foo, Farrell, Boxell, Robertson and Ryan2007; Ziegler et al., Reference Ziegler, Wade, Schaaf, Chang and Mohammed2007a; Kváč et al., Reference Kváč, Hofmannová, Bertolino, Wauters, Tosi and Modrý2008, Reference Kváč, McEvoy, Loudová, Stenger, Sak, Květoňová, Ditrich, Rašková, Moriarty, Rost, Macholán and Piálek2013; Feng, Reference Feng2010; Ng-Hublin et al., Reference Ng-Hublin, Singleton and Ryan2013; Stenger et al., Reference Stenger, Clark, Kváč, Khan, Giddings, Dyer, Schultz and McEvoy2015a, Reference Stenger, Clark, Kváč, Khan, Giddings, Prediger and McEvoy2015b, Reference Stenger, Horčičková, Clark, Kváč, Čondlová, Khan, Widmer, Xiao, Giddings, Pennil, Stanko, Sak and McEvoy2018), although zoonotic species such as C. parvum and C. ubiquitum (Hajdušek et al., Reference Hajdušek, Ditrich and Šlapeta2004; Rašková et al., Reference Rašková, Květoňová, Sak, McEvoy, Edwinson, Stenger and Kváč2013; Li et al., Reference Li, Xiao, Alderisio, Elwin, Cebelinski, Chalmers, Santin, Fayer, Kváč, Ryan, Sak, Stanko, Guo, Wang, Zhang, Cai, Roellig and Feng2014; Perec-Matysiak et al., Reference Perec-Matysiak, Bunkowska-Gawlik, Zalesny and Hildebrand2015) and livestock-specific species such as C. scrofarum, C. andersoni and C. baileyi (Ziegler et al., Reference Ziegler, Wade, Schaaf, Chang and Mohammed2007a; Lv et al., Reference Lv, Zhang, Wang, Jian, Zhang, Ning, Wang, Feng, Wang, Ren, Qi and Xiao2009; Ng-Hublin et al., Reference Ng-Hublin, Singleton and Ryan2013; Danišová et al., Reference Danišová, Valenčáková, Stanko, Luptaková, Hatalová and Canady2017) have been reported. Despite a large number of studies, the diversity and biology of Cryptosporidium in several rodent hosts, including voles, have not been thoroughly characterized (Kváč et al., Reference Kváč, McEvoy, Stenger, Clark, Cacciò and Widmer2014; Stenger et al., Reference Stenger, Horčičková, Clark, Kváč, Čondlová, Khan, Widmer, Xiao, Giddings, Pennil, Stanko, Sak and McEvoy2018).
Early studies, relying on oocyst morphology to distinguish species, reported C. parvum, C. muris and Cryptosporidium sp. in voles (Chalmers et al., Reference Chalmers, Sturdee, Bull, Miller and Wright1997; Torres et al., Reference Torres, Gracenea, Gomez, Arrizabalaga and Gonzalez-Moreno2000; Sinski et al., Reference Sinski, Hlebowicz and Bednarska1993, Reference Sinski, Bednarska and Bajer1998; Bull et al., Reference Bull, Chalmers, Sturdee and Healing1998; Bajer et al., Reference Bajer, Bednarska, Pawelczyk, Behnke, Gilbert and Sinski2002; Bednarska et al., Reference Bednarska, Bajer, Sinski, Girouard, Tamang and Graczyk2007). In more recent studies of voles, using more discriminatory genotyping tools to distinguish species, the prevalence of C. parvum was much lower than previously reported and C. muris was not detected. Additionally, common voles were not susceptible to C. muris, C. proliferans or C. andersoni under experimental conditions (Modrý et al., Reference Modrý, Hofmannová, Antalová, Sak and Kváč2012). In contrast, Cryptosporidium muskrat genotypes I and II and Cryptosporidium isolates closely related to muskrat genotypes I and II have been reported frequently (online Supplementary Table S1). In the most recent study, the largest to date, Stenger et al. (Reference Stenger, Horčičková, Clark, Kváč, Čondlová, Khan, Widmer, Xiao, Giddings, Pennil, Stanko, Sak and McEvoy2018) reported greater diversity of Cryptosporidium spp. infecting North American and European voles than previously known. They identified at least 18 different Cryptosporidium spp. by sequencing of the partial sequence of the small ribosomal subunit rRNA and actin genes in European and North American voles, and most of these were identified for the first time. Phylogenetic analyses indicated the Cryptosporidium spp. infecting voles from the different continents remained closely related (Stenger et al., Reference Stenger, Horčičková, Clark, Kváč, Čondlová, Khan, Widmer, Xiao, Giddings, Pennil, Stanko, Sak and McEvoy2018). Collectively, data from studies on voles show that they are host to at least 20 Cryptosporidium species and genotypes (see online Supplementary Table S1). Most of the genotypes lack biological data such as course of infection and host range.
We undertook the present study to extend knowledge of the occurrence and diversity of Cryptosporidium spp. infecting the common vole (Microtus arvalis). We selected two isolates from wild-caught common voles and, in accordance with ICZN nomenclature rules and criteria established by the scientific community studying Cryptosporidium (Xiao et al., Reference Xiao, Fayer, Ryan and Upton2004; Jirků et al., Reference Jirků, Valigurová, Koudela, Křížek, Modrý and Šlapeta2008; Fayer, Reference Fayer2010), we describe the morphometry of oocysts, determine phylogenetic relatedness at multiple genetic loci and report on the infectivity for several hosts (voles, laboratory and yellow-necked mice, laboratory rats and chickens) under natural and experimental conditions. Outcomes from the study support the conclusion that the Cryptosporidium isolates are genetically and biologically distinct from previously described species. We therefore propose them as new species named Cryptosporidium alticolis sp. n. and Cryptosporidium microti sp. n.
Material and methods
Area and specimens studied
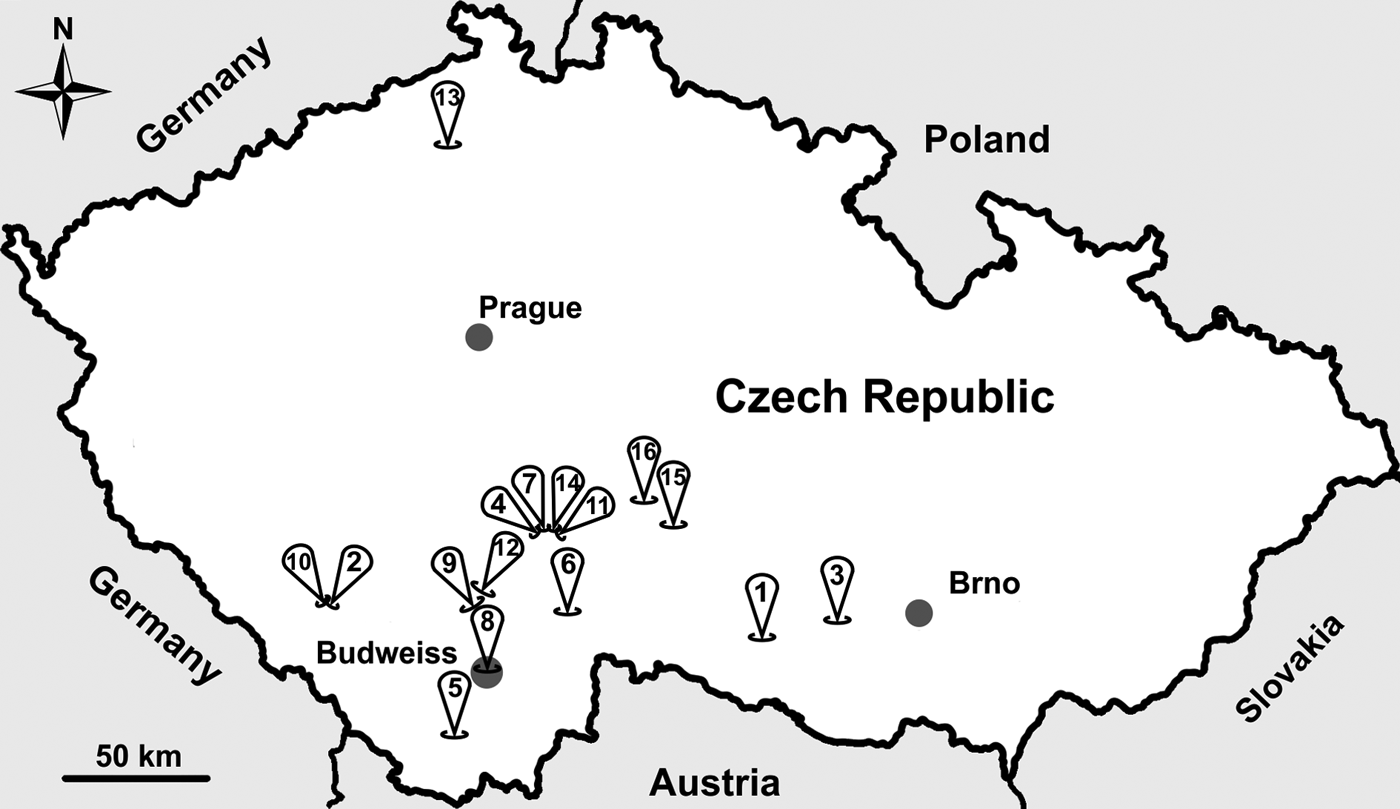
From 2014 to 2017 (May to September each year), wild-caught common voles were trapped using snap traps baited with apple and peanut at 16 locations in the Czech Republic (Fig. 1). After trapping, we identified the species, measured body mass (±1 g) and determined the sex of each individual. We estimated the age of each individual using body mass, such that an individual weighing <15 g was considered a juvenile and all other animals were considered adults. Following collection, we dissected each individual and collected a fecal sample from the colon. Fecal samples were stored at 4 °C without fixation. All fecal samples were screened for the presence of Cryptosporidium oocysts using the aniline–carbol–methyl violet (ACMV) staining (Miláček and Vítovec, Reference Miláček and Vítovec1985) followed by microscopic examination at 1000× magnification (light microscope Olympus BX51, Tokyo, Japan). During microscopic examination, we counted oocysts and we quantified the infection intensity as number of oocysts per gram of feces (OPG) according to Kváč et al. (Reference Kváč, Ondráčková, Květoňová, Sak and Vítovec2007).
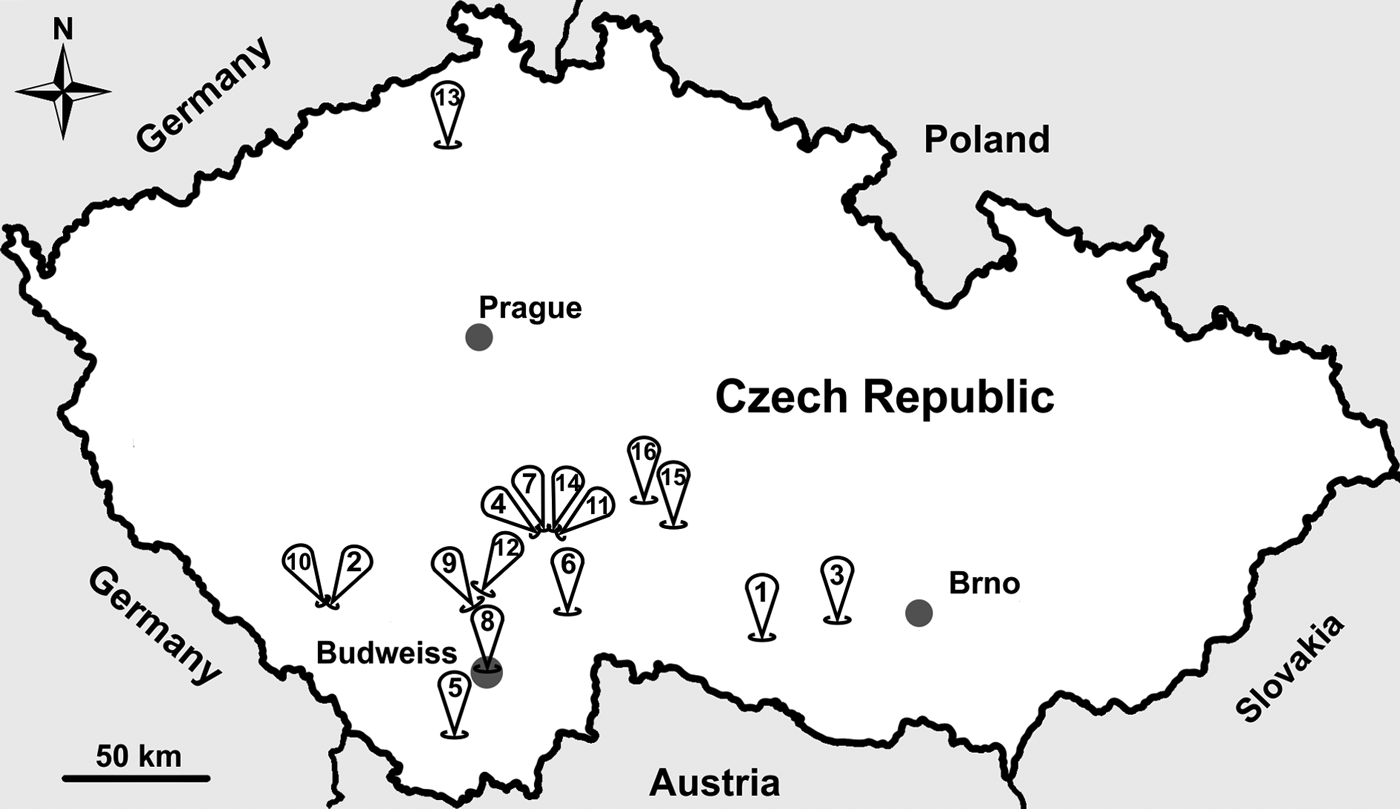
Fig. 1. Sampling locations across the study area in the Czech Republic. Sample site numbers indicate the following: (1) Dačice, (2) Výškovice, (3) Náměšť nad Oslavou, (4) Sedlečko u Tábora, (5) Dolní Třebonín, (6) Pelejovice, (7) Radimovice, (8) Budweiss, (9) Bavorovice, (10) Masákova Lhota, (11) Všechov u Tábora, (12) Opatovice, (13) Lovečkovice, (14) Soběslav, (15) Dubovice and (16) Zmišovice.
Molecular characterization
DNA was extracted from 200 mg of feces by bead disruption for 60 s at 5.5 m s−1 using 0.5 mm glass beads in a Fast Prep 24 Instrument (MP Biomedicals, Santa Ana, CA, USA) followed by isolation and purification using a commercially available kit in accordance with the manufacturer's instructions (PSP spin stool DNA Kit, Invitek, Stratec, Berlin, Germany). Purified DNA was stored at −20 °C prior to amplification by PCR.
A nested PCR approach was used to amplify a partial region of the small ribosomal subunit rRNA (SSU; ~830 bp; Xiao et al., Reference Xiao, Escalante, Yang, Sulaiman, Escalante, Montali, Fayer and Lal1999; Jiang et al., Reference Jiang, Alderisio and Xiao2005), actin (~1066 bp; Sulaiman et al., Reference Sulaiman, Lal and Xiao2002), Cryptosporidium oocyst wall protein (COWP) (~550 bp; Spano et al., Reference Spano, Putignani, McLauchlin, Casemore and Crisanti1997) and 70 kilodalton heat shock protein genes (HSP70; ~1950 bp; Sulaiman et al., Reference Sulaiman, Morgan, Thompson, Lal and Xiao2000).
The primary PCR mixtures contained 2 µL of template DNA, 2.5 U of Taq DNA Polymerase (Dream Taq Green DNA Polymerase, Thermofisher Scientific, Waltham, MA, USA), 0.5× PCR buffer (SSU) or 1× PCR buffer (actin, COWP and HSP70; Thermofisher Scientific), 6 mm MgCl2 (SSU) or 3 mm MgCl2 (actin, COWP and HSP70), 200 µ m each deoxynucleoside triphosphate, 100 mm each primer and 2 µL non-acetylated bovine serum albumin (BSA; 10 mg ml−1; New England Biolabs, Beverly, MA, USA) in 50 µL reaction volume. The secondary PCR mixtures were similar to those described above for the primary PCR, with the exception that 2 µL of the primary PCR product was used as the template, the MgCl2 concentration was 3 mm and no BSA was used. DNA of C. parvum and molecular grade water were used as positive and negative controls, respectively. Secondary PCR products were detected by 2% agarose gel electrophoresis, visualized by ethidium bromide staining and extracted using GenElute™ Gel Extraction Kit (Sigma-Aldrich, St. Louis, MO, USA). Purified secondary products were sequenced in both directions with an ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) using the secondary PCR primers and the BigDye1 Terminator v3.1 cycle sequencing kit (Applied Biosystems) in 10 µL reactions.
Phylogenetic analysis
The nucleotide sequences of each gene obtained in this study were edited using the ChromasPro 2.4.1. (Technelysium, Pty, Ltd, South Brisbane, Australia) and aligned with each other and with reference sequences from GenBank using MAFFT version 7 online server using the Q-INS-I algorithm (http://mafft.cbrc.jp/alignment/software/). Alignment adjustments were made manually to remove artificial gaps using BioEdit 7.0.5.3 (Hall, Reference Hall1999). Phylogenetic analyses were performed and the best DNA/protein phylogeny models were selected using the MEGA7 software (Guindon and Gascuel, Reference Guindon and Gascuel2003; Tamura et al., Reference Tamura, Stecher, Peterson, Filipski and Kumar2013) and Geneious v7.1.7 (http://www.geneious.com). Phylogenetic trees were inferred by maximum likelihood (ML) method, with the substitution model that best fits the alignment selected using the Bayesian information criterion. ML analysis of SSU, actin, COWP and HSP70 alignments was done in the MEGA7 software and concatenated SSU–actin–COWP alignment was done in RAxML v7.2.8 implemented in Geneious. The General Time Reversible model was selected for SSU, actin, HSP70 and concatenated SSU–actin–COWP alignment and the Tamura 3-parameter model was used of COWP alignment. All models were used under an assumption that rate variation among sites was γ distributed with invariant sites.
Bootstrap support for branching was based on 1000 replications. Phylograms were edited for style using CorelDrawX7. Sequences have been deposited in GenBank under the accession numbers (Acc. nos.) MH145308–MH145335.
Origin of specimens for transmission studies
Isolates of C. alticolis sp. n. and C. microti sp. n. were obtained from wild-caught common voles trapped at Dačice and Radimovice, respectively, in the Czech Republic. Oocysts from each species were used to infect a 6-month-old common vole (vole 0). Oocysts from vole 0 were purified using caesium chloride gradient centrifugation (Arrowood and Donaldson, Reference Arrowood and Donaldson1996) and used for analysis of oocyst morphometry and to infect other animals (see below).
Transmission studies
We experimentally determined the infectivity and pathogenicity of C. alticolis sp. n. and C. microti sp. n. for 6-month-old common voles, meadow voles (Microtus pennsylvanicus) and yellow-necked mice (Apodemus flavicollis); 2-month-old SCID (severe combined immunodeficiency), BALB/c and C57BL/6J mice (Mus musculus) and brown rats (Rattus norvegicus); and 3-day-old chickens (Gallus gallus f. domestica). Common voles and yellow-necked mice used for infectivity studies were obtained from captive colonies maintained at the Institute of Parasitology, Biology Centre of the Academy of Sciences of the Czech Republic, České Budějovice, Czech Republic. Laboratory (i.e. house mouse) mice and rats were purchased from Charles River Laboratories, Sulzfeld, Germany. Chickens originated from International Testing of Poultry, Ústrašice, Tábor, Czech Republic. Meadow voles were obtained from a captive colony maintained at Smith College, Northampton, Massachusetts, USA and used in transmission studies at North Dakota State University, USA. All other experiments were performed at the Biology Centre of the Academy of Sciences of the Czech Republic. In determining infectivity and pathogenicity, we used five individuals from each species/group. A week prior to inoculation, fecal samples from all individuals were screened daily for the presence of Cryptosporidium oocysts and specific DNA of Cryptosporidium spp. using parasitological and molecular tools (SSU) as described above. Individuals were housed separately in plastic cages with sterilized bedding and supplied with a sterilized diet and water ad libitum. Each animal was inoculated orally by gavage with 100 000 purified oocysts suspended in 200 µL of distilled water. Fecal samples from each individual were screened daily for the presence of Cryptosporidium oocysts using ACMV staining and specific DNA using nested PCR targeting the SSU gene. At least three amplicons of each target gene were sequenced directly in both directions from each infected individual.
All experiments were terminated 30 days post-infection (DPI). Course of infection indicators, including fecal consistency, fecal colour and infection intensity, was examined.
Histopathological and scanning electron microscopy examinations
The gastrointestinal tract of one animal from each group was examined following necropsy at 6 DPI (this time was selected based on preliminary results; data not shown). The entire small and large intestine was divided into 1 cm sections and samples were processed for histology, scanning electron microscopy (SEM) and PCR/sequencing. Specimens for histology were fixed in 4% buffered formalin and processed by the usual paraffin method. Histological sections (5 µm) were stained with haematoxylin and eosin and periodic acid–Schiff stains. The specimens for SEM were fixed overnight at 4 °C in 2.5% glutaraldehyde in 0.1 M phosphate buffer, washed three times for 15 min in the same buffer, post-fixed in 2% osmium tetroxide in 0.1 M phosphate buffer for 2 h at room temperature and finally washed three times for 15 min in the same buffer. After dehydration in a graded acetone series, specimens were dried using the critical point technique, coated with gold and examined using a JEOL JSM-7401F-FE SEM.
Oocyst morphometry
Oocysts of C. alticolis sp. n. and C. microti sp. n. were examined using differential interference contrast (DIC) microscopy, ACMV staining and fluorescence microscopy (Olympus IX70, Tokyo, Japan) following labelling with genus-specific FITC-conjugated antibodies (Cryptosporidium IF Test, Crypto Cell, Medac, Wedel, Germany). Morphometry of oocysts was determined using digital analysis of images (M.I.C. Quick Photo Pro v.3.1 software; Promicra, s.r.o., Praha, Czech Republic) collected using an Olympus Digital Colour Camera DP73. Length and width of 50 oocysts of each isolate were measured under DIC at 1000× magnification and the ratio of the length/width of each oocyst was calculated. The mean and standard deviation (s.d.) of length, width and ratio of the length/width of oocysts of each isolate were calculated.
Animal care
Animal caretakers wore disposable coveralls, shoe covers and gloves whenever entering the rooms where animals were housed. All wood-chip bedding, feces and disposable protective clothing were sealed in plastic bags, removed from the buildings and incinerated at the end of the study.
Statistical analysis
Prevalence was calculated by dividing the number of positive individuals by the total number of individuals sampled. Differences in Cryptosporidium prevalence were determined by χ 2 analysis using a 5% significance level. The hypothesis tested in the analysis of oocyst morphometry was that two-dimensional mean vectors of measurement are the same in the two populations being compared. Hotelling's T2 test was used to test the null hypothesis. Analyses were performed using program Epi Info (TM) 7.1.1.14 (Centers for Disease Control and Prevention, GA, USA) and R 3.5.0. (https://www.r-project.org/).
Results
Prevalence and infection intensity of Cryptosporidium
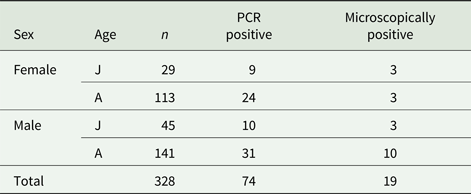
Out of 328 fecal samples from wild-caught common voles, 19 (5.8%) were microscopically positive for the presence of oocysts of Cryptosporidium sp. and 74 (22.6%) were positive for the presence of specific DNA of Cryptosporidium spp. (Table 1). All microscopically positive samples were also positive using PCR. Positive voles were trapped at 11 out of 16 localities (Table 2). There was no difference (χ 2 = 0.0153; d.f. = 1; P = 0.9016) in the prevalence of Cryptosporidium spp. in males (22.0%; 41/186) and females (23.2%; 33/142). Similarly, the prevalence did not differ (χ 2 = 0.3254; d.f. = 1; P = 0.5684) between juvenile (25.7%; 19/74) and adult voles (21.7%; 55/254; Table 2). Infection intensity, which ranged from 4000 to 42 000 OPG, did not differ (P = 0.1773) between males (2000–36 000 with mean 15 000 OPG) and females (4000–42 000 with mean 20 000 OPG). None of the trapped voles had diarrhoea.
Table 1. Number of wild-caught common voles positive for Cryptosporidium by PCR and microscopy, by sex and age
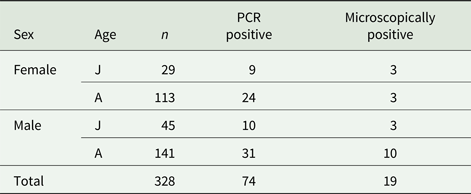
J, juvenile; A, adult.
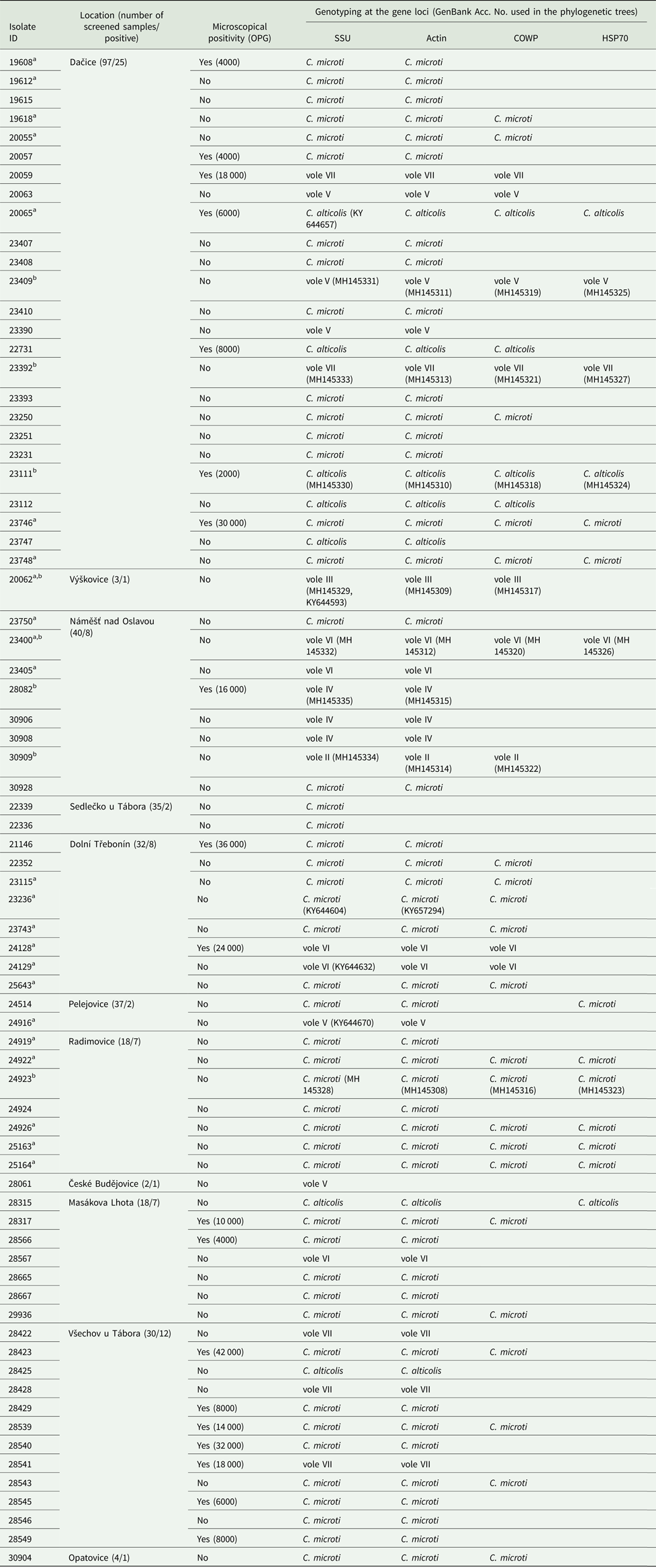
Table 2. Cryptosporidium spp. in wild common voles (Microtus arvalis)
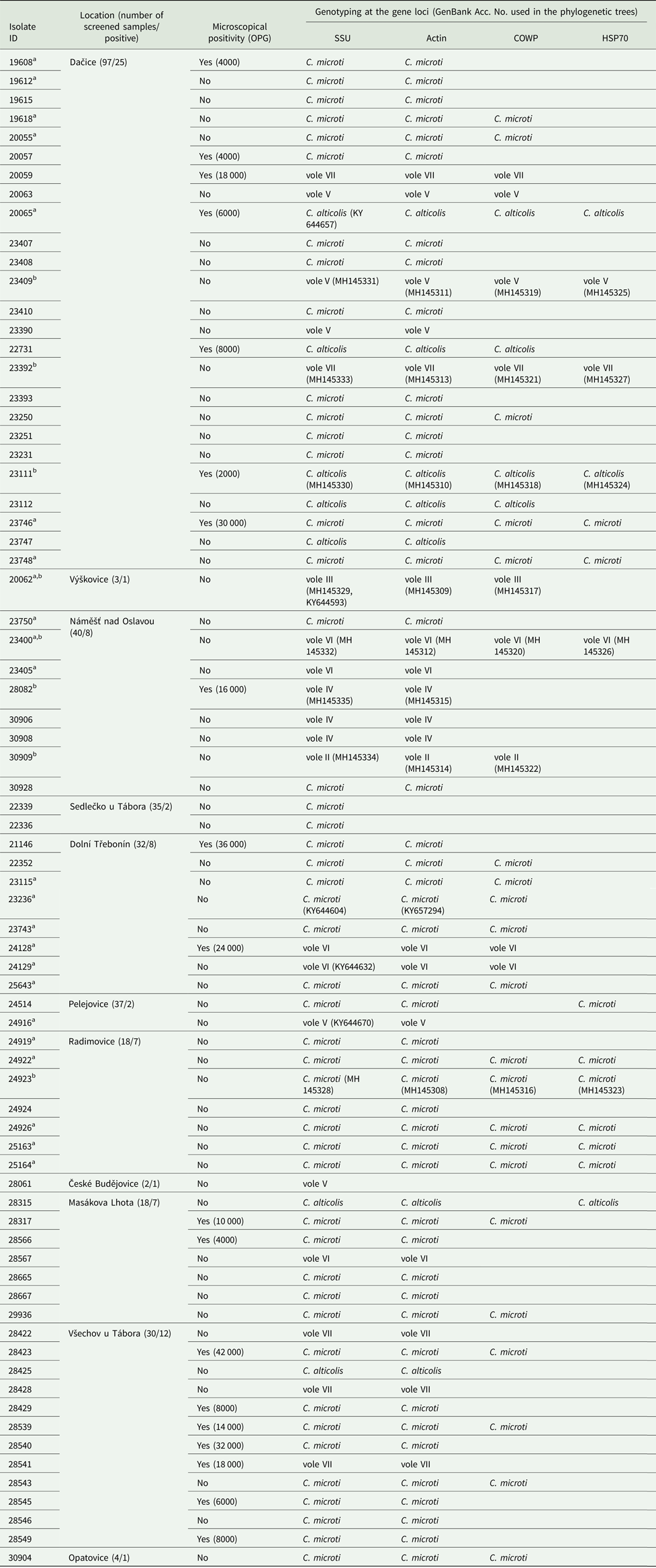
Isolates were characterized by microscopy, including infection intensity expressed as number of oocyst per gram of feces (OPG), and PCR analysis of the small ribosomal subunit rRNA (SSU), actin, Cryptosporidium oocyst wall protein (COWP) and 70 kDa heat shock protein (HSP70) genes. Only localities where Cryptosporidium-positive animals were trapped are shown.
a Sequences of SSU and actin previously obtained in the study of Stenger et al. (Reference Stenger, Horčičková, Clark, Kváč, Čondlová, Khan, Widmer, Xiao, Giddings, Pennil, Stanko, Sak and McEvoy2018).
b Sequence of isolates used in phylogeny trees.
Out of 74 voles positive for Cryptosporidium, 74, 71, 33 and 14 were genotyped by sequence analysis of SSU, actin, COWP and HSP70 genes, respectively (Table 2, Fig. 2 and online Supplementary Figs S1–S4). The remaining positive samples yielded sequences of insufficient quality to include in analyses (three actin sequences) or failed to amplify at COWP (n = 41) and HSP70 (n = 60) loci.

Fig. 2. A maximum likelihood (ML) tree based on concatenated small subunit rRNA (SSU), actin and Cryptosporidium oocyst wall protein (COWP) gene sequences. A representative of each SSU, actin and COWP species/genotype from wild-caught common voles from this study is highlighted in bold. GenBank accession numbers are shown in parenthesis after the isolate identifier. Numbers at the nodes represent the bootstrap values gaining more than 50% support. Branch length scale bar indicates the number of substitutions per site.
Sequence analysis revealed the presence of eight genotypes of Cryptosporidium, of which two are described here as new species (Table 2). ML trees inferred from sequences of SSU, actin, COWP and HSP70 genes individually or SSU, actin and COWP in concatenation formed three major phylogenetic groups (Fig. 2 and online Supplementary Figs S1–S4). Group 1 included C. microti sp. n. and Cryptosporidium vole genotypes II, III, VI and VII. Cryptosporidium microti (n = 47) was identical to Cryptosporidium sp. isolate 19608-Miar-EU previously recovered from a wild-caught common vole in the Czech Republic [Acc. No. KY657290] and was closely related to Cryptosporidium muskrat genotype II [Acc. No. AY737571], Cryptosporidium sp. isolate 1857-Mipe-NA from a wild-caught meadow vole [Acc. No. KY644574] and Cryptosporidium sp. isolate 1544-Pero-NA from a wild-caught Peromyscus mouse [Acc. No. KY644565] in the USA, sharing 99.2%, 98.8% and 98.6% sequence identity, respectively.
Cryptosporidium vole genotype III (n = 1) was identical to Cryptosporidium sp. isolate 20062-Miar-EU from a wild-caught common vole in the Czech Republic (Acc. No. KY644593) and clustered with Cryptosporidium sp. isolate 10482-Mygl-EU from a wild-caught bank vole (Acc. No. KY644595) and Cryptosporidium sp. isolate 2035-Myga-NA from a wild-caught Southern red-backed vole (Acc. No. KY644592) in Slovakia and the USA, respectively, sharing 99.8 and 99.5% sequence identity.
Cryptosporidium vole genotype VI (n = 5) was identical to Cryptosporidium sp. isolate 24129-Miar-EU from a wild-caught common vole in the Czech Republic (Acc. No. KY644632) and clustered with Cryptosporidium vole genotype II (n = 1) from the present study (Acc. No. MH145334), sharing 99.1% sequence identity. Cryptosporidium vole genotype VII (n = 5), a genotype that was first identified in this study, clustered with the Cryptosporidium vole genotype (Acc. No. EF641020) and Cryptosporidium sp. isolate 1947-Mipe-NA (Acc. No. KY644626), both from wild-caught meadow voles in the USA, sharing 98.9 and 98.5% sequence identity, respectively. C. alticolis sp. n. (n = 7), the only member of group 2, was identical to Cryptosporidium sp. isolate 20065-Miar-EU from a wild-caught common vole in the Czech Republic (Acc. No. KY644657), and clustered with Cryptosporidium sp. isolate 2333-Pero-NA from a wild-caught meadow vole in the USA (Acc. No. KY644655) and Cryptosporidium sp. isolate Mrb001 from a grey red-backed vole in Japan (Acc. No. AB477098), sharing 97.3 and 97.5% sequence identity, respectively.
Group 3 comprised Cryptosporidium vole genotype IV (n = 3) and vole genotype V (n = 5). Cryptosporidium genotype vole V was identical to Cryptosporidium sp. isolate 24916-Miar-EU from a wild-caught common vole in the Czech Republic (Acc. No. KY644670) and formed a sister group with muskrat genotype I (Acc. No. EF641013) and Cryptosporidium sp. isolate 1962-Mipe-NA from a wild-caught meadow vole (Acc. No. KY644685), both in the USA, sharing 98.1 and 98.0% sequence identity, respectively. Cryptosporidium vole genotype IV, which was reported for the first time in this study, clustered outside of this group.
Based on evidence that they are genetically and biologically distinct from known Cryptosporidium species, we describe C. alticolis sp. n. and C. microti sp. n. as new species of the genus Cryptosporidium. Descriptions of C. alticolis sp. n. and C. microti sp. n. follow.
Cryptosporidium alticolis sp. n.
Prevalence and infection intensity. Seven voles (2.1%) from three localities had DNA of C. alticolis sp. n. detectable by PCR, of which three had oocysts that were detectable by microscopy with an infection intensity of 2000–8000 OPG (Table 2).
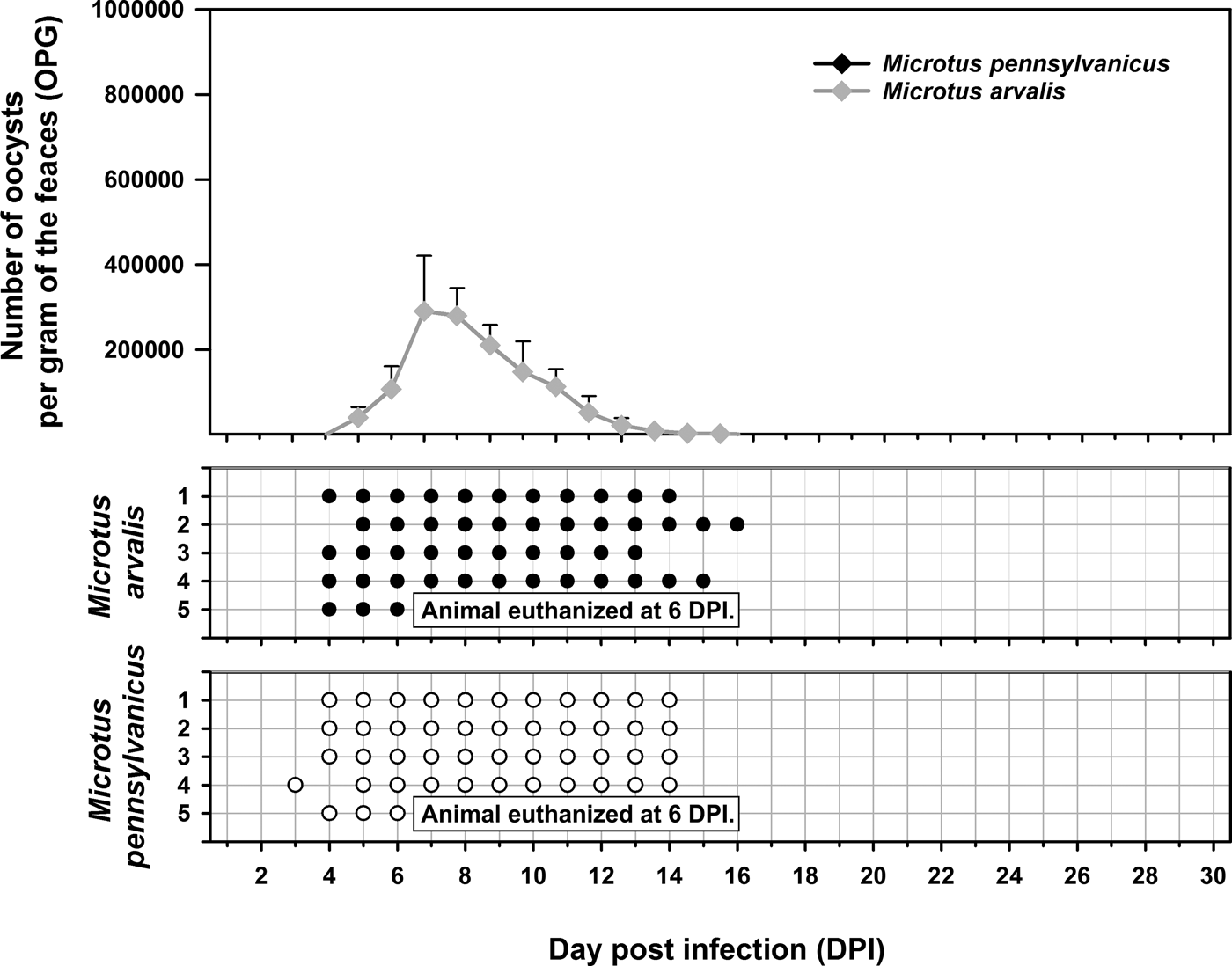
Experimental transmission. Oocysts of C. alticolis sp. n. from naturally infected common voles were infectious for common and meadow voles, but not for yellow-necked mice, SCID mice, BALB/c mice, C57BL/6J mice, brown rats or chickens. The prepatent period of C. alticolis sp. n. in common and meadow voles was 3–4 DPI (Fig. 3). Whereas common voles shed oocysts of C. alticolis sp. n. continuously during the patent period (12–15 DPI), meadow voles shed oocysts sporadically up to 12 DPI (Fig. 3). The infection intensity of C. alticolis sp. n. in common voles (2000–1000 000 OPG) was higher than in meadow voles (2000–50 000 OPG). No macroscopical changes were observed in the gastrointestinal tract of common or meadow voles infected with C. alticolis sp. n. and the surface epithelium remained intact. DNA of C. alticolis sp. n. was detected throughout the small and large intestine of common and meadow voles; however, endogenous developmental stages were detected only in the jejunum and ileum by histology and electron microscopy (Figs 4 and 5). Cryptosporidium alticolis sp. n. was not detected in the stomach and other organs (liver, pancreas, kidneys, lungs and spleen). None of the experimentally infected common or meadow voles were diarrhoeic. The lamina propria in the jejunum and ileum was slightly oedematous with occasional dilatation of lymphatic vessels (data not shown). Sequences of SSU, actin, COWP and HSP70 genes from experimentally infected hosts shared 100% identity with the isolate used in the inoculum.
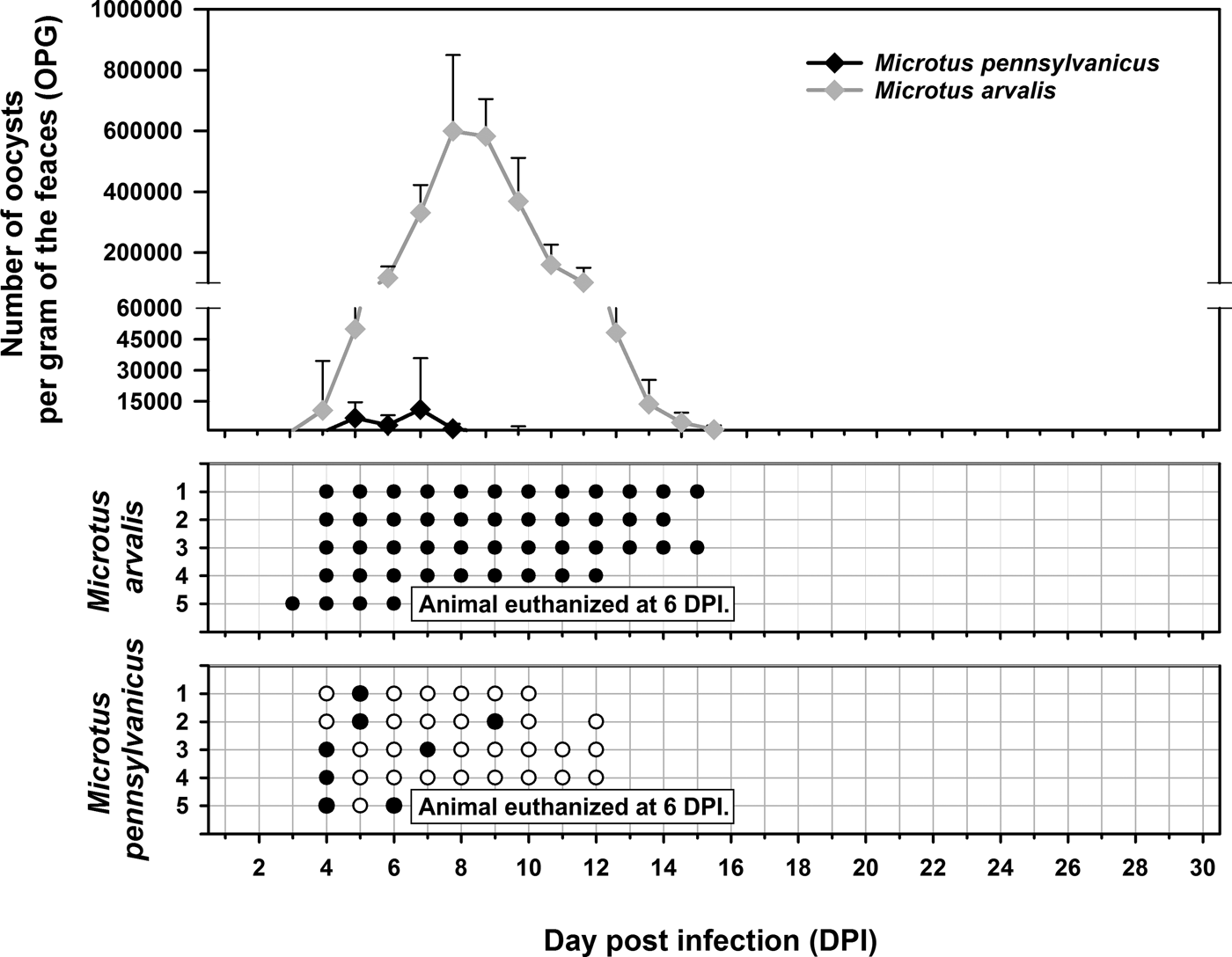
Fig. 3. Course of infection of Cryptosporidium alticolis sp. n. in experimentally infected common voles (Microtus arvalis) and in meadow voles (Microtus pennsylvanicus) based on coprological and molecular examination of feces. Any circles indicate detection of specific DNA, black circle indicates microscopic detection of oocysts.
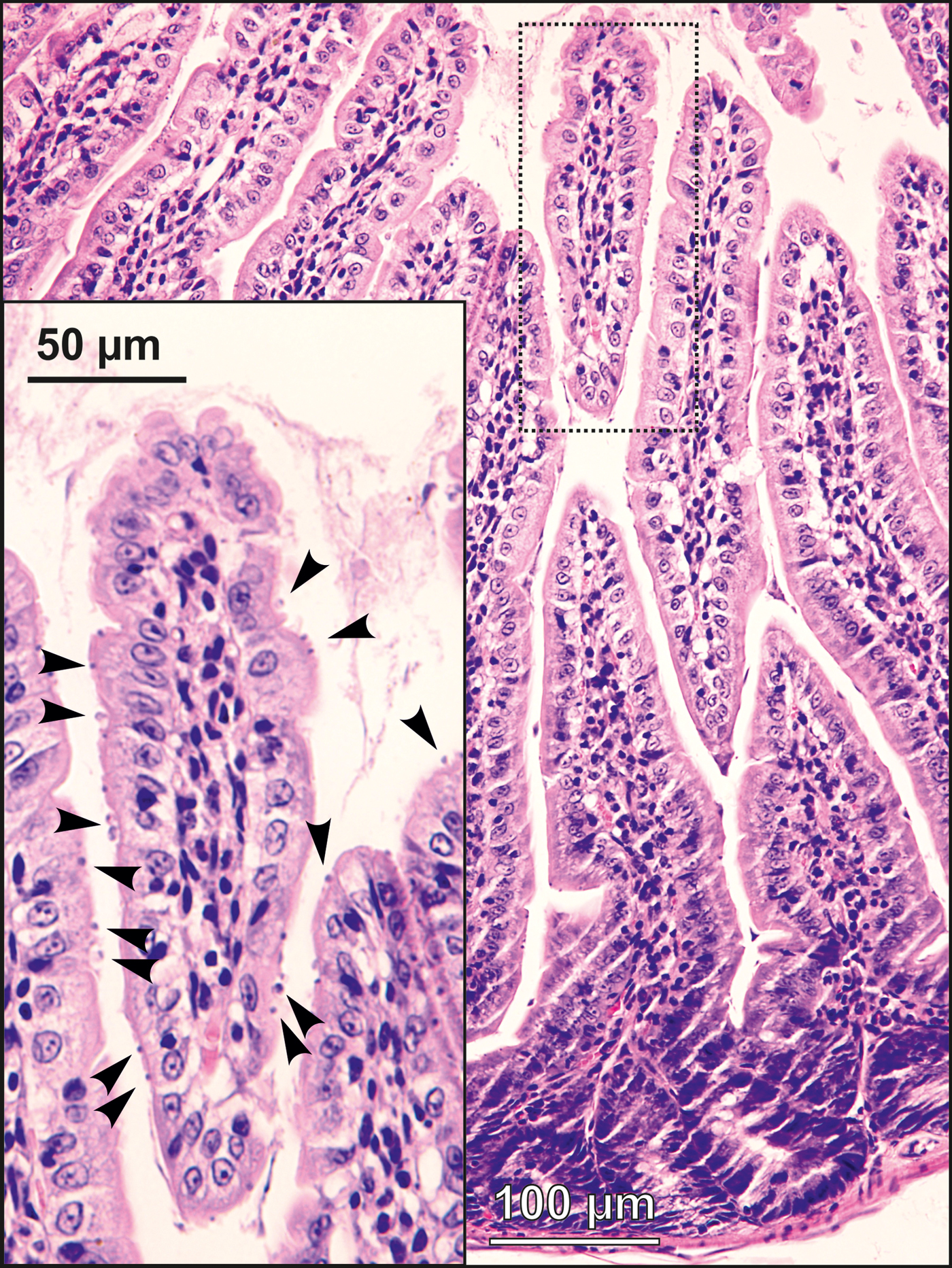
Fig. 4. Developmental stages (arrowheads) of Cryptosporidium alticolis sp. n. in mucosal glandular epithelium from the duodenum of an experimentally infected common vole (Microtus arvalis). Bar included in each picture.

Fig. 5. Scanning electron photomicrograph of the jejunal epithelium of an experimentally infected common vole (Microtus arvalis). Attached developmental stage of Cryptosporidium alticolis sp. n. (arrowhead; detail in the upper right corner).
Taxonomic summary
ZooBank number for species: urn:lsid:zoobank.org:act:D12C78AA-222E-4E07-A7CE-51AA6A747BC6
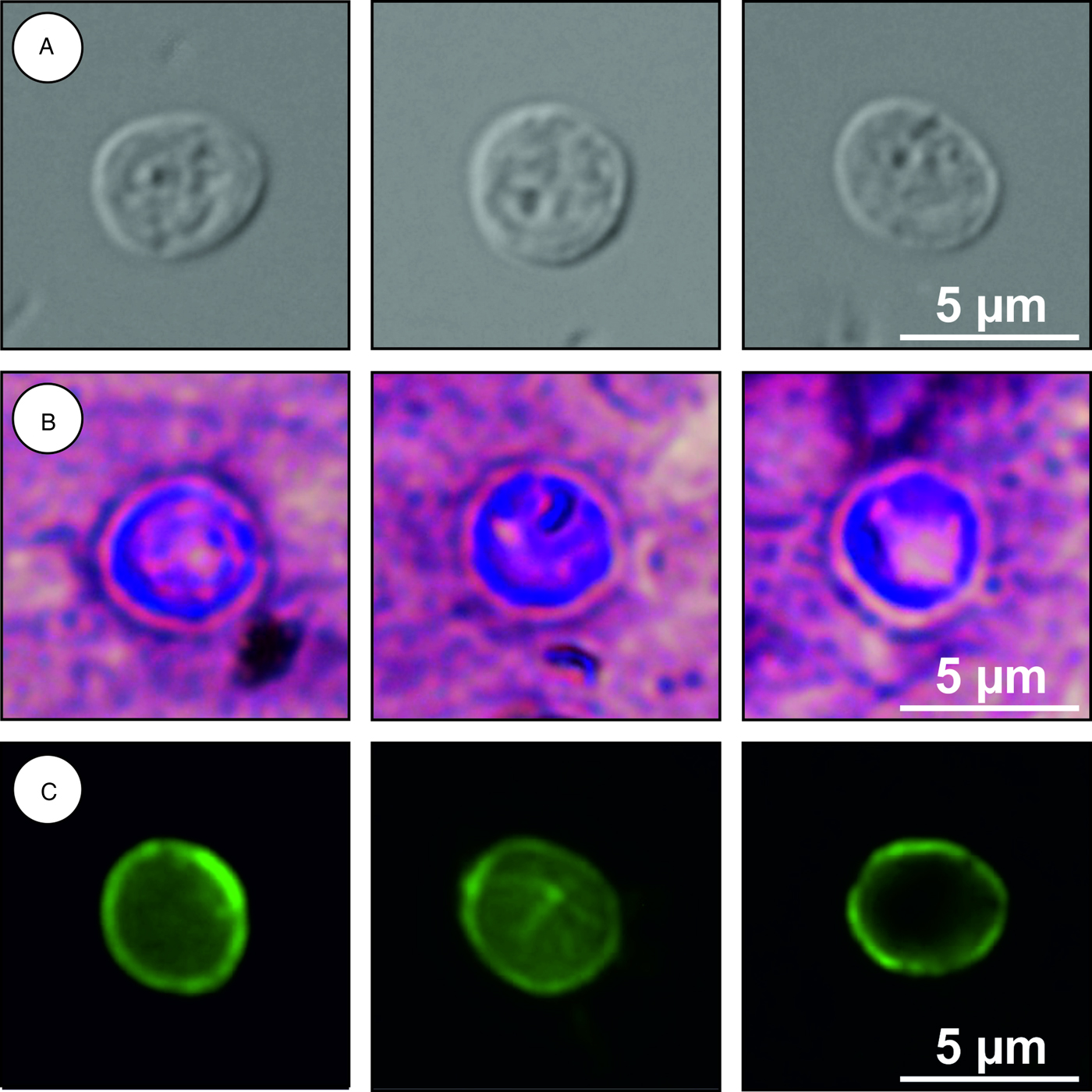
Description: Oocysts are shed fully sporulated with four sporozoites and an oocyst residuum. Sporulated oocysts (n = 50) measure 4.9–5.7 µm (mean ± s.d. = 5.4 ± 0.2 µm) × 4.6–5.2 µm (mean ± s.d. = 4.9 ± 0.2 µm) with a length/width ratio of 1.00–1.20 (mean ± s.d. = 1.10 ± 0.05) (Fig. 6). Morphology and morphometry of other developmental stages are unknown.
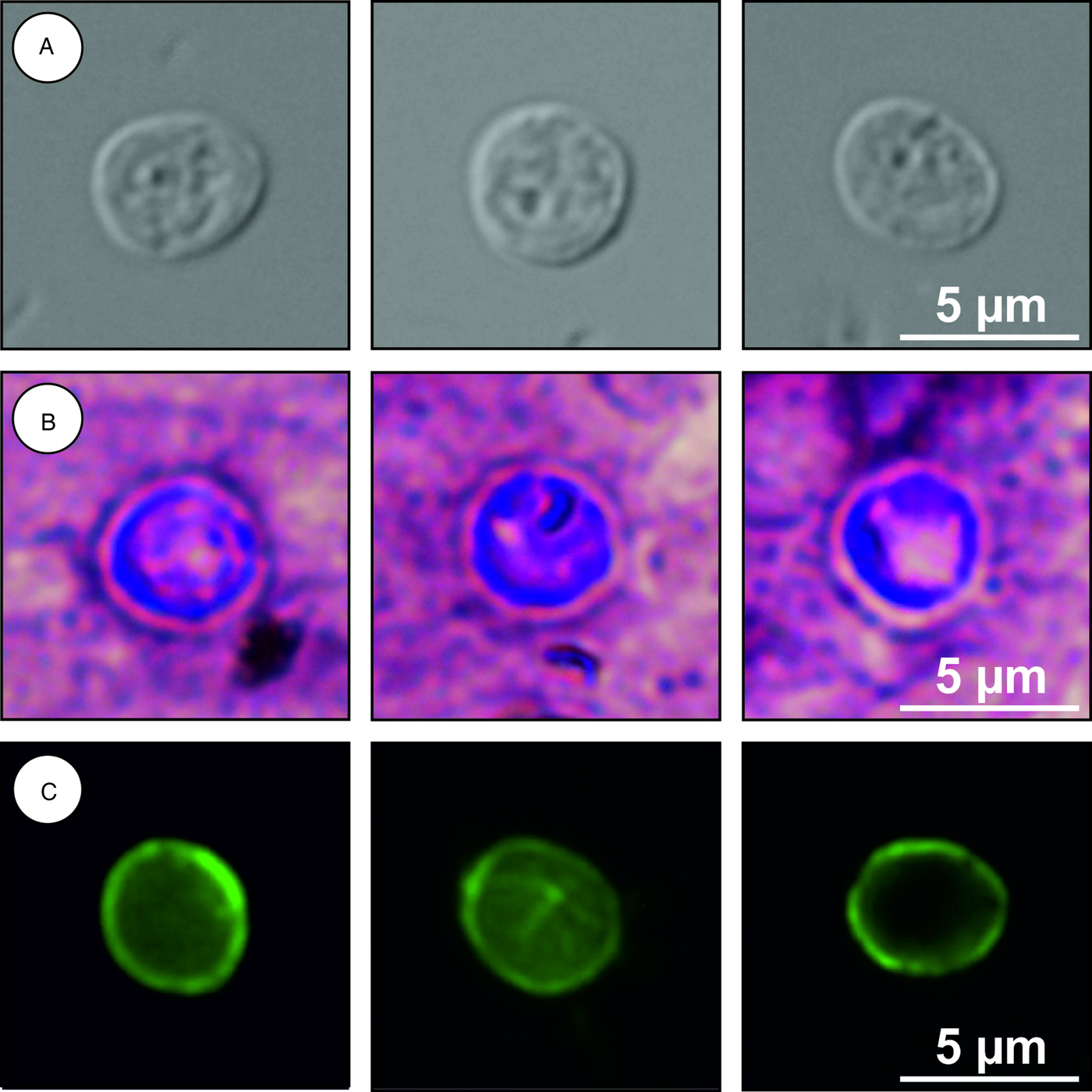
Fig. 6. Cryptosporidium alticolis sp. n. oocysts visualized in various preparations: (A) differential interference contrast microscopy and stained by (B) aniline–carbol–methyl violet and (C) anti-Cryptosporidium FITC-conjugated antibody. Bar included in each picture.
Type host: common vole (M. arvalis)
Type locality: Dačice (Czech Republic)
Other localities: Masákova Lhota and Všechov (Czech Republic)
Site of infection: jejunum and ileum (Figs 4 and 5)
Distribution: Czech Republic
Type material/hapanotype: Tissue samples in 10% formaldehyde and histological sections of infected jejunum (nos. 174/2016, 175/2016, 176/2016 and 177/2016) and ileum (nos. 178/2016 and 179/2016); genomic DNA isolated from fecal samples of naturally (isolation no. 23111) and experimentally (isolation no. 27124) infected M. arvalis; genomic DNA isolated from jejunal and ileal tissue of experimentally infected M. arvalis (isolation nos. 27035 and 27037, respectively); digital photomicrographs (nos. DIC 1-13/23111, MV 1-11/23111, IF 1-9/23111, HI 1-3/27124 and SEM 1-3/27124) and fecal smear slides with oocysts stained by ACMV staining from experimentally infected M. arvalis (nos. 27124/3, 27124/4, 27124/5 and 27124/6). Specimens deposited at the Institute of Parasitology, Biology Centre of the Czech Academy of Sciences, Czech Republic.
Reference sequences: Partial sequences of SSU, actin, COWP and HSP70 genes were deposited at GenBank under Acc. Nos. MH145330, MH145310, MH145318 and MH145324, respectively.
Etymology: The species name alticolis is derived from the Latin noun ‘alticola’ (meaning a vole).
Differential diagnosis: Oocysts of C. alticolis are larger than those of C. microti (P = 0.001), have similar ACMV staining to other species of Cryptosporidium and cross-react with antibodies developed primarily for C. parvum (Fig. 6). It can be differentiated genetically from other Cryptosporidium spp. based on sequences of SSU, actin, COWP and HSP70 genes. Endogenous development of C. alticolis sp. n. takes place in the small intestine, whereas C. microti develops in the large intestine.
Cryptosporidium microti sp. n.
Prevalence. Forty-seven wild-caught common voles (14.3%) from nine localities were positive for C. microti sp. n. by PCR, of which 12 had oocysts detectable by microscopy. The infection intensity ranged from 4000 to 42 000 OPG.
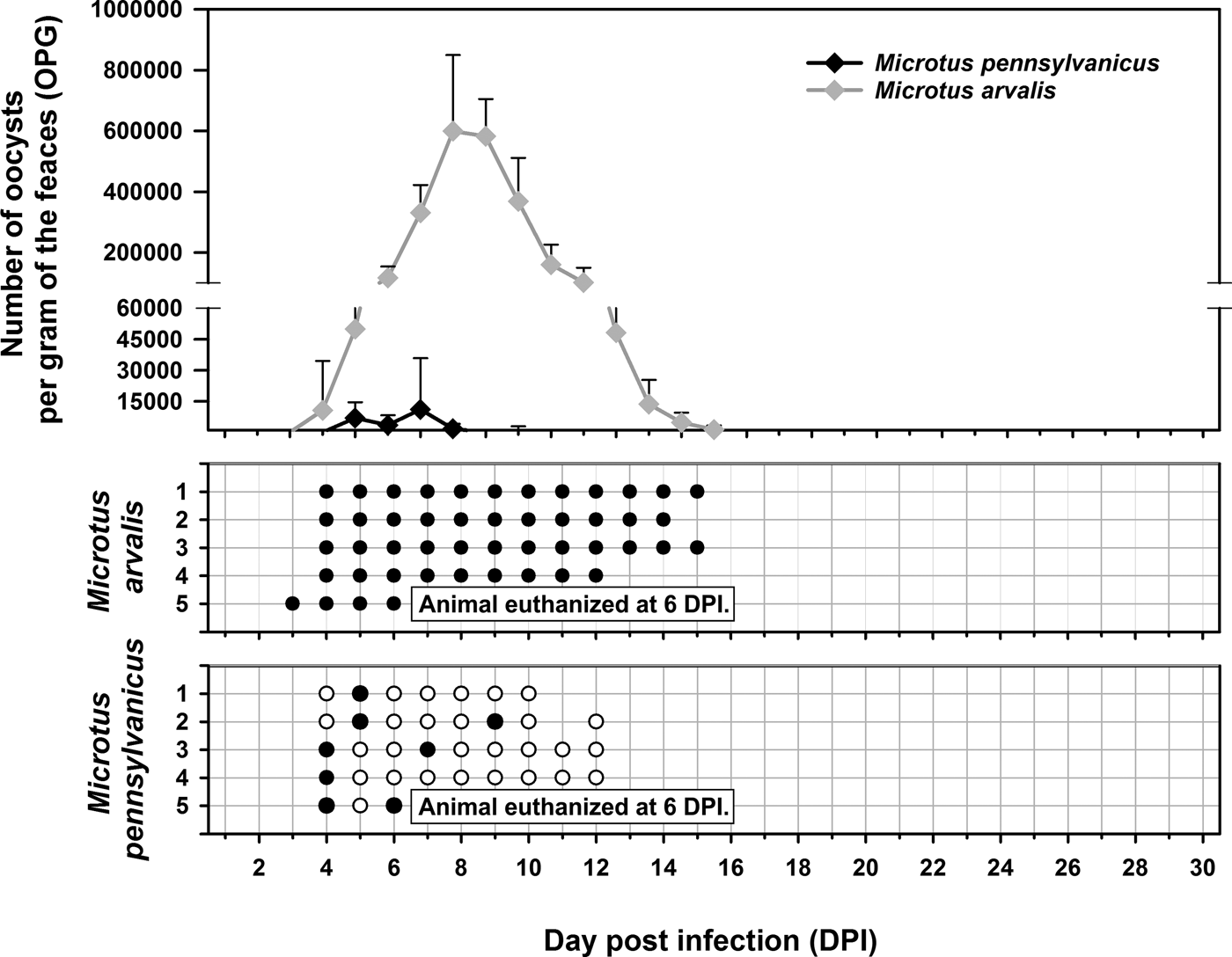
Experimental transmission. Oocysts of C. microti sp. n. from naturally infected common voles were infectious for common and meadow voles, but not for yellow-necked mice, SCID mice, BALB/c mice, C57BL/6J mice, brown rats or chickens. Common voles shed C. microti sp. n. from 4 to 16 DPI, with oocysts detectable by microscopy throughout this period. The infection intensity ranged from 2000 to 430 000 OPG with maximum shedding at 6–7 DPI (Fig. 7). In meadow voles, DNA of C. microti sp. n. was detected from 4 to 14 DPI; however, oocysts were not detectable by microscopy at any time during the patent period.
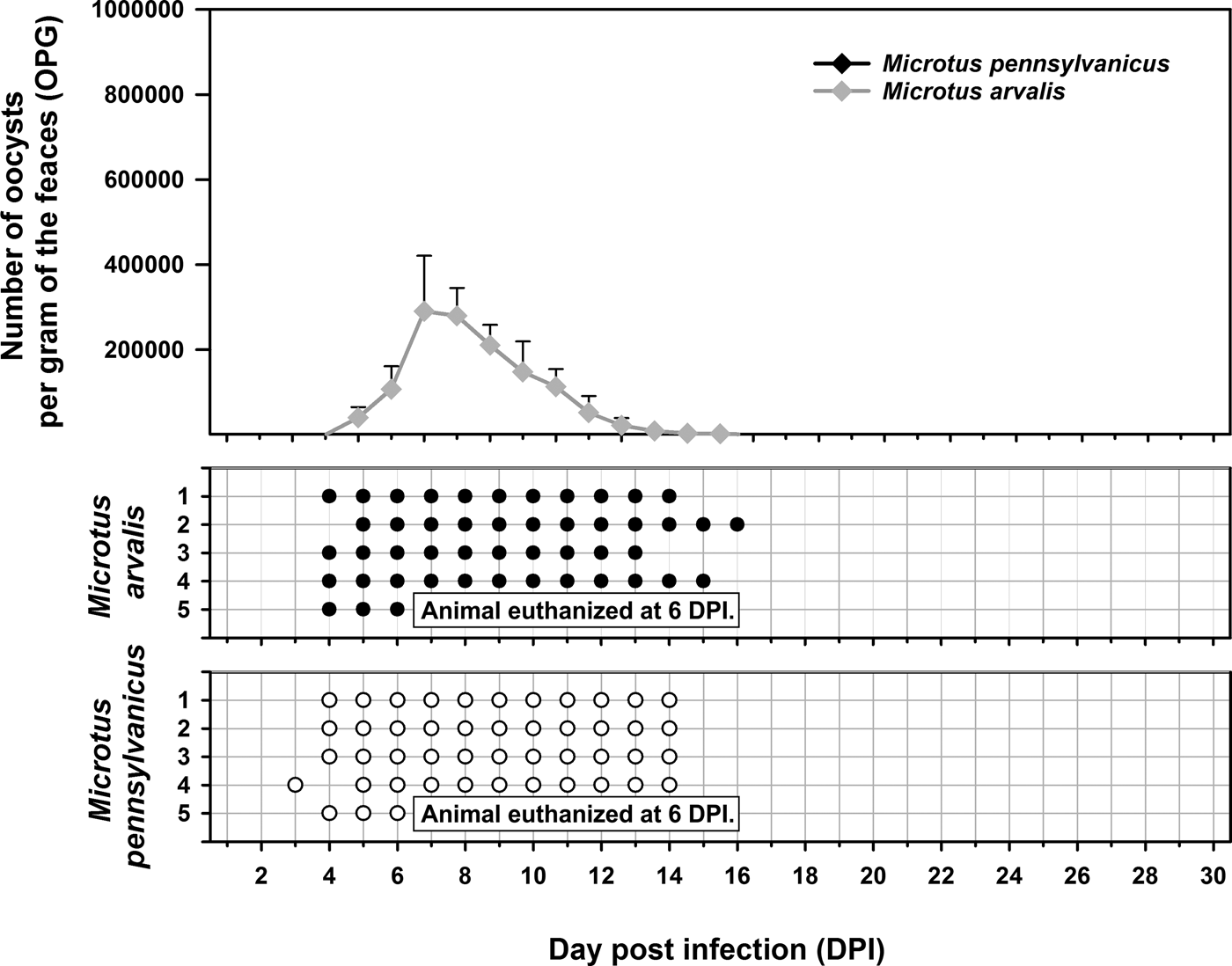
Fig. 7. Course of infection of Cryptosporidium microti sp. n. in experimentally infected common voles (Microtus arvalis) and in meadow voles (Microtus pennsylvanicus) based on coprological and molecular examination of feces. Any circles indicate detection of specific DNA, black circle indicates microscopic detection of oocysts.
Sequences of SSU, actin, COWP and HSP70 genes from experimentally infected hosts shared 100% identity with the isolate used in the inoculum. Specific DNA of C. microti sp. n. was found exclusively in the caecum and colon of common and meadow voles. Endogenous developmental stages were detected in the caecum and colon of the common vole (Figs 8 and 9), but were not detected in the meadow vole. Infections were not associated with macroscopical or pathological changes in the digestive tract of common or meadow voles and these animals showed no signs of diarrhoea.
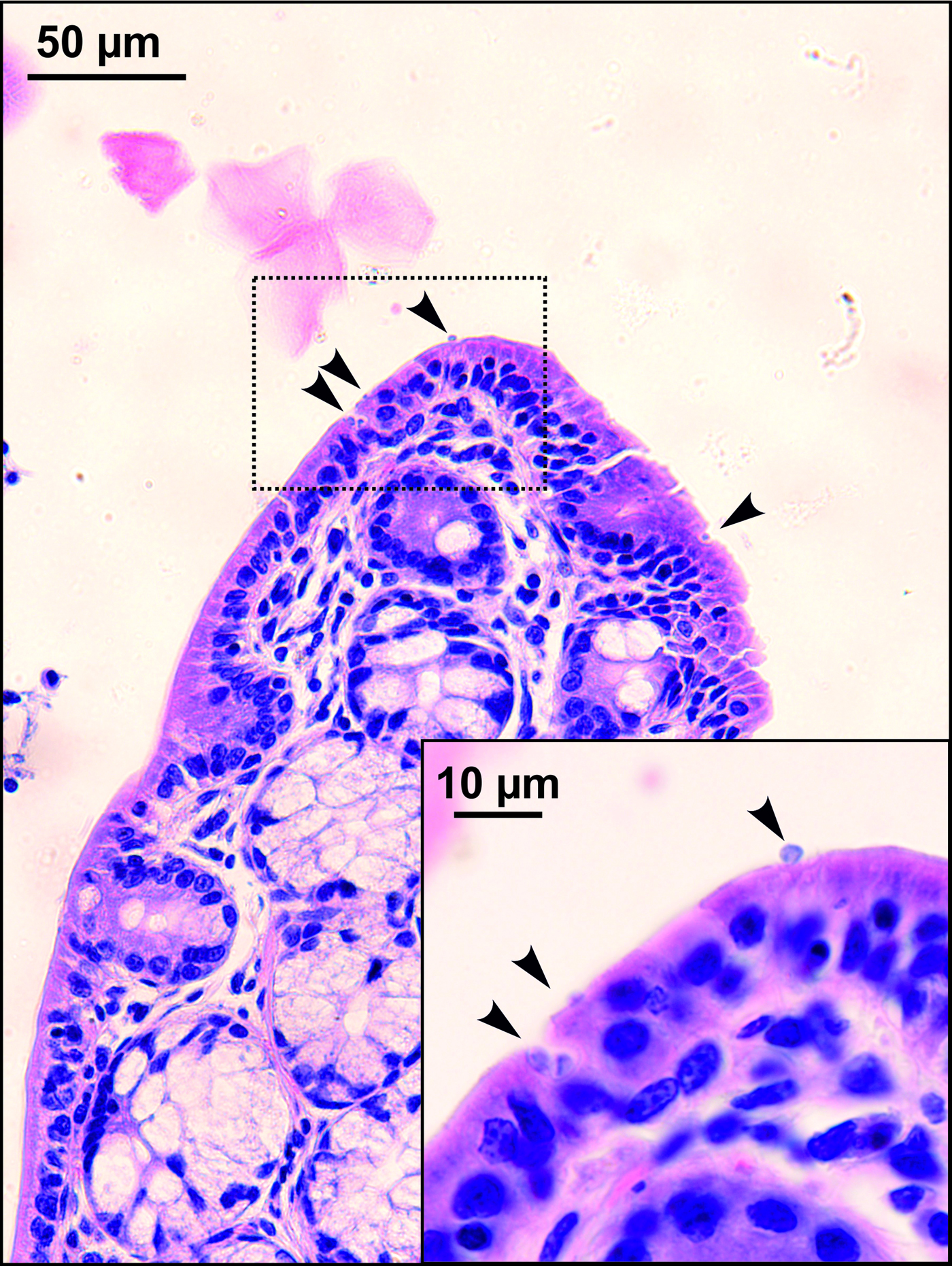
Fig. 8. Developmental stages (arrowheads) of Cryptosporidium microti sp. n. in mucosal glandular epithelium from the colon of an experimentally infected common vole (Microtus arvalis). Bar included in each picture.

Fig. 9. Scanning electron photomicrograph of the colon epithelium of a common vole (Microtus arvalis). Attached developmental stage of Cryptosporidium microti sp. n. (arrowhead; detail in the upper right corner).
Taxonomic summary
ZooBank number for species: urn:lsid:zoobank.org:act:4FD6136C-3932-4881-BE49-4714A5AB488A
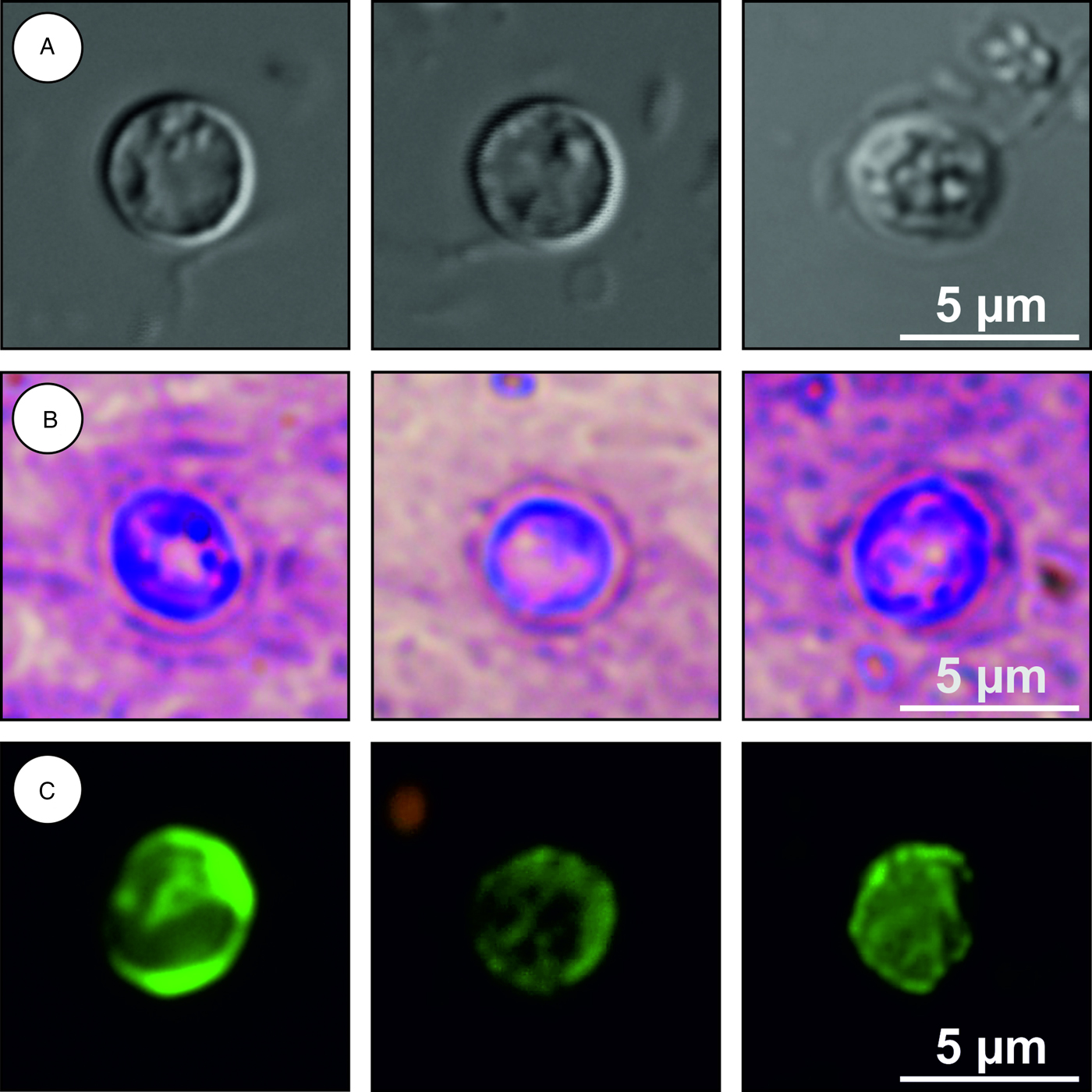
Description: Oocysts are shed fully sporulated with four sporozoites and an oocyst residuum. Sporulated oocysts (n = 50) measure 3.9–4.7 µm (mean ± s.d. = 4.3 ± 0.1 µm) × 3.8–4.4 µm (mean ± s.d. = 4.1 ± 0.1 µm) with length/width ratio of 1.00–1.06 (mean ± s.d. = 1.03 ± 0.02) (Fig. 10). Morphology and morphometry of other developmental stages are unknown.
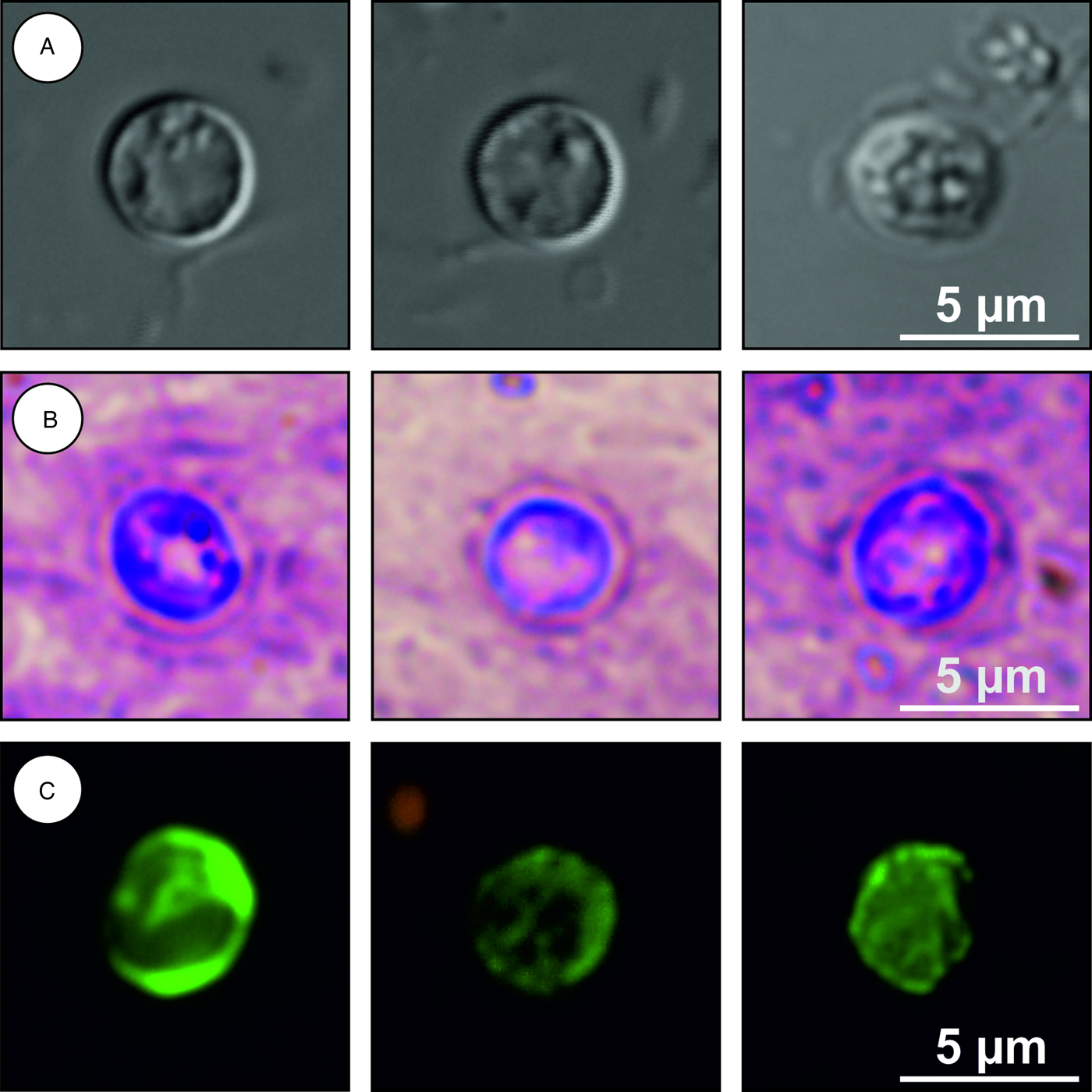
Fig. 10. Cryptosporidium microti sp. n. oocysts visualized in various preparations: (A) differential interference contrast microscopy and stained by (B) aniline–carbol–methyl violet and (C) anti-Cryptosporidium FITC-conjugated antibody. Bar included in each picture.
Type host: common vole (M. arvalis)
Type locality: Radimovice (Czech Republic)
Other localities: Dačice, Zňátky, Sedlečko, Dolní Třebonín, Pelejovice, Masákova Lhota, Všechov and Opatovice (Czech Republic)
Site of infection: caecum and colon (Figs 8 and 9)
Distribution: Czech Republic
Type material/hapanotype: Tissue samples in 10% formaldehyde and histological sections of infected caecum (nos. 97/2016 and 98/2016) and colon (nos. 99/2016 and 100/2016), genomic DNA isolated from fecal samples of naturally (isolation no. 24923) and experimentally (isolation no. 28063) infected M. arvalis; genomic DNA isolated from ceacal and colonical tissue of experimentally infected M. arvalis (isolation nos. 29751 and 29753, respectively); digital photomicrographs (nos. DIC 1-11/24923, MV 1-9/24923, IF 1-9/24923, HI 1-3/28063 and SEM 1-3/28063) and fecal smear slides with oocysts stained by ACMV staining from experimentally infected M. arvalis (nos. 28063/3, 28063/4, 28063/5 and 28063/6). Specimens deposited at the Institute of Parasitology, Biology Centre of the Czech Academy of Sciences, Czech Republic.
Reference sequences: Partial sequences of SSU, actin, COWP and HSP70 genes were deposited at GenBank under Acc. Nos. MH145328, MH145308, MH145316 and MH145323, respectively.
Etymology: The species name microti is derived from the Latin noun ‘microtus’ (meaning a vole).
Differential diagnosis: Oocysts of C. microti sp. n. are smaller than those of C. alticolis sp. n. (P = 0.001), have similar ACMV staining to other species of Cryptosporidium and cross-react with antibodies developed primarily for C. parvum (Fig. 10). It can be differentiated genetically from other Cryptosporidium spp. based on sequences of SSU, actin, COWP and HSP70 genes. Endogenous development of C. microti sp. n. takes place in the large intestine, whereas C. alticolis sp. n. develops in the small intestine.
Discussion
This and other genotyping studies have shown that voles host several Cryptosporidium species and genotypes that appear to be host specific and not infectious for humans, but they rarely host C. parvum (Feng et al., Reference Feng, Alderisio, Yang, Blancero, Kuhne, Nadareski, Reid and Xiao2007; Stenger et al., Reference Stenger, Horčičková, Clark, Kváč, Čondlová, Khan, Widmer, Xiao, Giddings, Pennil, Stanko, Sak and McEvoy2018; Ziegler et al., Reference Ziegler, Wade, Schaaf, Chang and Mohammed2007a, Reference Ziegler, Wade, Schaaf, Stern, Nadareski and Mohammed2007b). The finding that oocysts of C. alticolis sp. n. and C. microti sp. n. are indistinguishable from oocysts of C. parvum suggests that earlier detections of C. parvum, which were not supported by genotyping data, were misidentifications. Oocyst size is generally only useful for differentiating intestinal (smaller and rounder) and gastric (larger and more oval) species of Cryptosporidium (Ryan and Xiao, Reference Ryan, Xiao, Cacciò and Widmer2014).
Cryptosporidium microti sp. n. and Cryptosporidium vole genotypes II, III, VI and VII clustered as part of a large heterogeneous group in ML trees. This is generally consistent with the report by Stenger et al. (Reference Stenger, Horčičková, Clark, Kváč, Čondlová, Khan, Widmer, Xiao, Giddings, Pennil, Stanko, Sak and McEvoy2018) that Cryptosporidium genotypes from voles in the Europe and North America formed between three and four phylogenetic groups in ML trees.
Cryptosporidium alticolis sp. n. and C. microti sp. n. are genetically distinct from other known species of Cryptosporidium. Cryptosporidium alticolis sp. n. shares 95.2, 94.7 and 94.3% sequence identity, respectively, with C. canis, C. suis and C. parvum at the SSU locus; 87.9, 90.5 and 89.7%, respectively, at the actin locus; and 84.5, 91.2 and 90.5%, respectively, at the HSP70 locus. At the COWP locus, C. alticolis sp. n. shared 88.1 and 89.9% sequence identity, respectively, with C. canis and C. parvum. Cryptosporidium microti sp. n. shared 95.5, 98.8 and 96.4% sequence identity, respectively, with C. canis, C. suis and C. parvum at the SSU locus; 85.6, 91.6 and 90.5%, respectively, at the actin locus; and 84.2, 93.1 and 92.6%, respectively, at the HSP70 locus. At the COWP locus, C. microti sp. n. shared 86.7 and 91.5% sequence identity, respectively, with C. canis and C. parvum. In comparison, C. hominis and C. parvum share 98–99% identity and C. muris and C. andersoni share 96–99% identity at these loci.
The prevalence of Cryptosporidium in voles ranges from 1 to 100% (Laakkonen et al., Reference Laakkonen, Soveri and Henttonen1994; Perz and Le Blancq, Reference Perz and Le Blancq2001; Bajer et al., Reference Bajer, Bednarska, Pawelczyk, Behnke, Gilbert and Sinski2002, Reference Bajer, Caccio, Bednarska, Behnke, Pieniazek and Sinski2003; Zhou et al., Reference Zhou, Fayer, Trout, Ryan, Schaefer and Xiao2004). The prevalence in wild-caught common voles in the present study (23%) was greater than the 14% reported by Stenger et al. (Reference Stenger, Horčičková, Clark, Kváč, Čondlová, Khan, Widmer, Xiao, Giddings, Pennil, Stanko, Sak and McEvoy2018) using similar detection methods, and much lower than the 62–73% reported by Bajer et al. (Reference Bajer, Bednarska, Pawelczyk, Behnke, Gilbert and Sinski2002) and Bajer (Reference Bajer2008) using microscopic detection, a method that is less sensitive than PCR. The prevalence of Cryptosporidium can be affected by factors such as age, season, population density, location, weather and climate, diet and water consumption (Nichols et al., Reference Nichols, Chalmers, Hadfield, Cacciò and Widmer2014).
Cryptosporidium microti sp. n. dominated at most locations in this study. Mixed infections were not detected, but they cannot be ruled out because the methods used were not effective at detecting multi-species infections. Microscopy cannot differentiate among species with similar sized oocysts and PCR preferentially amplifies DNA from the dominant species/genotype (Santín and Zarlenga, Reference Santín and Zarlenga2009; Jeníková et al., Reference Jeníková, Němejc, Sak, Květoňová and Kváč2011; Ma et al., Reference Ma, Cai, Ma, Feng and Xiao2014; Qi et al., Reference Qi, Wang, Jing, Wang, Wang and Zhang2015).
Cryptosporidium alticolis sp. n. infects the small intestine, which is similar to most intestinal Cryptosporidium spp. of mammals (Ryan and Xiao, Reference Ryan, Xiao, Cacciò and Widmer2014). In contrast, C. microti is only the third species, after C. suis in pigs and C. oculltus in rats, reported to infect the colon (Ryan et al., Reference Ryan, Monis, Enemark, Sulaiman, Samarasinghe, Read, Buddle, Robertson, Zhou, Thompson and Xiao2004; Vítovec et al., Reference Vítovec, Hamadejová, Landová, Kváč, Květoňová and Sak2006; Kváč et al., Reference Kváč, Vlnatá, Ježková, Horčičková, Konečný, Hlásková, McEvoy and Sak2018). Similar to C. oculltus (Kváč et al., Reference Kváč, Vlnatá, Ježková, Horčičková, Konečný, Hlásková, McEvoy and Sak2018), C. microti sp. n. localizes to the mucosal surface in the large intestine. In contrast, C. suis predominates in the glandular epithelium of the submucosal colonic lymphoglandular complexes in pigs (Vítovec et al., Reference Vítovec, Hamadejová, Landová, Kváč, Květoňová and Sak2006).
Neither C. alticolis sp. n. nor C. microti sp. n. developed clinical signs in common voles or meadow voles under experimental conditions in the present study. This is consistent with the reports that wild animals rarely display signs of clinical cryptosporidiosis (Sturdee et al., Reference Sturdee, Chalmers and Bull1999; Hikosaka and Nakai, Reference Hikosaka and Nakai2005; Castro-Hermida et al., Reference Castro-Hermida, García-Presedo, González-Warleta and Mezo2011; Němejc et al., Reference Němejc, Sak, Květoňová, Hanzal, Jeníková and Kváč2012; Čondlová et al., Reference Čondlová, Horčičková, Sak, Květoňová, Hlásková, Konečný, Stanko, McEvoy and Kváč2018).
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182018001142
Financial support
This study was funded by Ministry of Education, Youth and Sports of the Czech Republic (LTAUSA17165), the US National Institute of Allergy and Infectious Diseases (1R15AI122152-01A1), Grant Agency of University of South Bohemia (098/2016/Z and 002/2016/Z) and supported by MEYS CR (LM2015062 Czech-BioImaging). We thank Annaliese Beery, Jennifer Christensen, Nancy Owen and Karen Swiecanski of Smith College for providing meadow voles and for assistance with transporting voles to North Dakota State University for the study.
Conflicts of interest
None.
Ethical standards
The research was conducted under ethical protocols approved by the Institute of Parasitology, Biology Centre and Central Commission for Animal Welfare, Czech Republic (protocol nos. 071/2010 and 114/2013) and Institutional Animal Care and Use Committee North Dakota State University, ND, USA (protocol no. A18014).