I. INTRODUCTION
Powder diffraction has the benefit over other material characterization techniques as it yields statistical information of the studied material. During a powder-diffraction measurement, millions of crystallites are illuminated in the X-ray beam. However, powder diffraction is limited to only measuring the average scattered intensity of this ensemble. This makes it difficult to study characteristics that make minor contributions to the pattern or are convolved with other features.
Serial femtosecond crystallography is an emerging technique which bridges the gap between single-crystal diffraction and powder diffraction. This method involves using an X-ray laser to collect a series of snapshot diffraction patterns from individual crystallites in random orientations, as the crystallites are streamed across the pulsed X-ray beam (Chapman et al., Reference Chapman, Fromme, Barty, White, Kirian, Aquila, Hunter, Schulz, DePonte, Weierstall, Doak, Maia, Martin, Schlichting, Lomb, Coppola, Shoeman, Epp, Hartmann, Rolles, Rudenko, Foucar, Kimmel, Weidenspointner, Holl, Liang, Barthelmess, Caleman, Boutet, Bogan, Krzywinski, Bostedt, Bajt, Gumprecht, Rudek, Erk, Schmidt, Hömke, Reich, Pietschner, Strüder, Hauser, Gorke, Ullrich, Herrmann, Schaller, Schopper, Soltau, Kühnel, Messerschmidt, Bozek, Hau-Riege, Frank, Hampton, Sierra, Starodub, Williams, Hajdu, Timneanu, Seibert, Andreasson, Rocker, Jönsson, Svenda, Stern, Nass, Andritschke, Schröter, Krasniqi, Bott, Schmidt, Wang, Grotjohann, Holton, Barends, Neutze, Marchesini, Fromme, Schorb, Rupp, Adolph, Gorkhover, Andersson, Hirsemann, Potdevin, Graafsma, Nilsson and Spence2011). The orientation can be determined by indexing Bragg spots, and their intensities averaged to allow the structure determination (Kirian et al., Reference Kirian, White, Holton, Chapman, Fromme, Barty, Lomb, Aquila, Maia, Martin, Fromme, Wang, Hunter, Schmidt and Spence2011; White et al., Reference White, Kirian, Martin, Aquila, Nass, Barty and Chapman2012). In contrast to conventional powder diffraction, the individual diffraction patterns which contribute to the average are available for inspection, allowing data reduction to focus on desired sub-populations of the crystallite ensemble. By summing the individual diffraction patterns, a virtual powder pattern is produced, which is often used to obtain a quick understanding of the data quality before indexing is carried out.
The point of this paper is to extend the utility of the virtual powder pattern from serial crystallography data, to show how the ability to count spots from individual diffraction images allows for new analysis methods of the materials microstructure. In particular, the methods for treating the virtual powder pattern to detect trace phases and characterize microstrain in the sample are demonstrated. This data analysis approach can yield quick information of these characteristics without requiring pattern indexing.
II. MERGING SERIAL CRYSTALLOGRAPHY DATA INTO VIRTUAL POWDER-DIFFRACTION PATTERNS
The task of generating a virtual powder-diffraction pattern from a series of serial crystallography patterns consists of integrating of the two-dimensional (2D) images into a 1D average about the image center. Although in principle this is a somewhat trivial exercise, there exists more than one method for doing so, each of which highlights different features in the diffraction images.
The first step in handling any image is correcting the systematic response of the detector. This involves subtracting a dark image, and correcting any non-uniform or non-linear gain response. These corrections are extremely important, and can be quite complicated if the detector characteristics vary from one frame to the next. Furthermore, it is important to understand the precise geometry of the detector relative to the X-ray interaction region. This includes determining the incident beam direction, or equivalently, the rotational and translational offsets of the detectors (He, Reference He2009).
The steps remaining to obtain a virtual powder pattern include background subtraction, angular integration, and normalization. However, the order in which they are carried out defines different forms of the virtual powder pattern. We will work through the different strategies and show examples of each for a sample data set.
A. Pixel-map virtual powder pattern
What we will refer to as the pixel-map virtual powder pattern is perhaps the most straightforward pattern generation procedure, and amounts to binning the intensity in each pixel directly into a powder-diffraction pattern. This operation can be expressed as (He, Reference He2009)
 $$I\lpar s\rpar =\mathop \sum \limits_{\,f\,=\,1}^{N_f } \mathop \sum \limits_{i\,=\,1}^{N_{\rm p} } \displaystyle{{\,p_{if} } \over {N_{\rm p} \lpar s \rpar N_f }}\comma \; \quad \quad s \lt s_i \lt s + \delta s$$
$$I\lpar s\rpar =\mathop \sum \limits_{\,f\,=\,1}^{N_f } \mathop \sum \limits_{i\,=\,1}^{N_{\rm p} } \displaystyle{{\,p_{if} } \over {N_{\rm p} \lpar s \rpar N_f }}\comma \; \quad \quad s \lt s_i \lt s + \delta s$$
where the inner summation bins the intensity of each pixel, p if , and is normalized by the number of pixels in the image that fall into this bin, N p(s). The scattering vector of the pixel, defined here as s i = 2sinθ i /λ, where θ is half the scattering angle to the pixel center and λ is the wavelength of incident light. The outer summation then averages this binned pattern over the serial data set containing N f images. This direct summation will only give smooth patterns when the powder bin size is larger than the pixel size (He, Reference He2009). When this is not the case, a more sophisticated algorithm which involves binning in sub-pixels and interpolation can be employed. As detailed in the supplementary materials of Sellberg et al., the pixel intensity should also be corrected for beam polarization and the pixel solid angle to obtain intensities in terms of differential scattering cross-section.
This kind of pattern can be particularly useful to study the structure of non-crystalline matter. It has allowed for the study of super-critically cooled water drops (Sellberg et al., Reference Sellberg, Huang, McQueen, Loh, Laksmono, Schlesinger, Sierra, Nordlund, Hampton, Starodub, DePonte, Beye, Chen, Martin, Barty, Wikfeldt, Weiss, Caronna, Feldkamp, Skinner, Seibert, Messerschmidt, Williams, Boutet, Pettersson, Bogan and Nilsson2014), and the ultra-fast dynamics of light-sensitive proteins in solution (Arnlund et al., Reference Arnlund, Johansson, Wickstrand, Barty, Williams, Malmerberg, Davidsson, Milathianaki, DePonte, Shoeman, Wang, James, Katona, Westenhoff, White, Aquila, Bari, Berntsen, Bogan, van Driel, Doak, Kjær, Frank, Fromme, Grotjohann, Henning, Hunter, Kirian, Kosheleva, Kupitz, Liang, Martin, Nielsen, Messerschmidt, Seibert, Sjöhamn, Stellato, Weierstall, Zatsepin, Spence, Fromme, Schlichting, Boutet, Groenhof, Chapman and Neutze2014). For the case of crystalline matter in solution, it is best to perform some background subtraction before binning the intensity; otherwise the resulting virtual powder pattern will be dominated by the background signal.
After averaging a sufficient number of images, the resulting virtual powder pattern from this method mimics a more standard powder-diffraction measurement. That is to say, that for crystalline matter, the intensity neglecting the Lorentz factor is given by
 $$\eqalign{I\lpar s\rpar =& \lt I_0 \gt \lt N_{\rm c}\gt \cr& P\lpar \theta \rpar A\left(\theta \right)D\left(s \right) \left\vert {F\lpar s\rpar } \right\vert^2 \sum\limits_{hkl} m_{hkl} S_{hkl} \lpar s - s_{hkl} \rpar}$$
$$\eqalign{I\lpar s\rpar =& \lt I_0 \gt \lt N_{\rm c}\gt \cr& P\lpar \theta \rpar A\left(\theta \right)D\left(s \right) \left\vert {F\lpar s\rpar } \right\vert^2 \sum\limits_{hkl} m_{hkl} S_{hkl} \lpar s - s_{hkl} \rpar}$$
This expression includes the peak shape function, s
hkl
(s
−
s
hkl
), for each of the Bragg reflection which is composed of a convolution of crystallite size, strain and instrumental peak shapes (Scardi and Leoni, Reference Scardi and Leoni2001). The peaks are then modulated by the continuous powder structure factor,
 $\left\vert {F\lpar s\rpar } \right\vert ^2 $
, the Debye–Waller factor, D(s), an absorption factor, A(θ), and a polarization factor, P(θ). Finally, the peaks are scaled by the spot multiplicity, m
hkl
(which might also contain some effects of texture, or preferential alignment), the average incident intensity, <I
0>, and the average number of crystals in an image, <N
c>.
$\left\vert {F\lpar s\rpar } \right\vert ^2 $
, the Debye–Waller factor, D(s), an absorption factor, A(θ), and a polarization factor, P(θ). Finally, the peaks are scaled by the spot multiplicity, m
hkl
(which might also contain some effects of texture, or preferential alignment), the average incident intensity, <I
0>, and the average number of crystals in an image, <N
c>.
A pattern generated from these data has a few advantages over the normal powder-diffraction data. The background can be dealt with on an image-by-image basis, which can improve the signal contrast in the averaged pattern. Secondly, in the dilute case, each image may contain only a few crystallites, which means that these images can be preselected to focus on different points of interest (i.e. sorting out frames containing different phases, or selecting only images with spots of a certain size).
B. Integrated-spot virtual powder pattern
The second kind of virtual powder pattern that can be generated from serial crystallography data is rooted in the methodology of single-crystal diffraction. This involves integrating the intensity in each spot, and then binning the intensities into a virtual powder pattern. This binning method can be expressed as
 $$I\lpar s\rpar =\mathop \sum \limits_{\,f\,=\,1}^{N_f } \mathop \sum \limits_{\,j\,=\,1}^{N_s } \displaystyle{{I_{\,jf} } \over {N_{\rm p} \lpar s \rpar N_f }}\comma \; \quad \quad s\lt s_{\,jf}\lt s+\delta s$$
$$I\lpar s\rpar =\mathop \sum \limits_{\,f\,=\,1}^{N_f } \mathop \sum \limits_{\,j\,=\,1}^{N_s } \displaystyle{{I_{\,jf} } \over {N_{\rm p} \lpar s \rpar N_f }}\comma \; \quad \quad s\lt s_{\,jf}\lt s+\delta s$$
where now the integrated intensity of each spot, I jf , is binned according to the scattering vector magnitude of its center, s jf . It is also worth noting that the intensity of each image is normalized by the number of pixels in the image with a given scattering vector, N p(s). This corrects for the increased sampling of a 2D detector at higher scattering angles, as well as, any regions which might be masked or missing. During the preceding spot integration, background subtraction, as well as, the previously mentioned pixel corrections should be taken into account.
The earliest example of time-resolved serial crystallography used this powder pattern generation method to show a time-dependent light-induced structural change in the membrane protein Photosystem I–ferredoxin cocrystal (Aquila et al., Reference Aquila, Hunter, Doak, Kirian, Fromme, White, Andreasson, Arnlund, Bajt, Barends, Barthelmess, Bogan, Bostedt, Bottin, Bozek, Caleman, Coppola, Davidsson, DePonte, Elser, Epp, Erk, Fleckenstein, Foucar, Frank, Fromme, Graafsma, Grotjohann, Gumprecht, Hajdu, Hampton, Hartmann, Hartmann, Hau-Riege, Hauser, Hirsemann, Holl, Holton, Homke, Johansson, Kimmel, Kassemeyer, Krasniqi, Kuhnel, Liang, Lomb, Malmerberg, Marchesini, Martin, Maia, Messerschmidt, Nass, Reich, Neutze, Rolles, Rudek, Rudenko, Schlichting, Schmidt, Schmidt, Schulz, Seibert, Shoeman, Sierra, Soltau, Starodub, Stellato, Stern, Suder, Timneanu, Ullrich, Wang, Williams, Weidenspointner, Weierstall, Wunderer, Barty, Spence and Chapman2012), and it has also been useful to study the observable damage in a protein crystal from exposure to an extremely bright femtosecond X-ray pulse (Lomb et al., Reference Lomb, Barends, Kassemeyer, Aquila, Epp, Erk, Foucar, Hartmann, Rudek, Rolles, Rudenko, Shoeman, Andreasson, Bajt, Barthelmess, Barty, Bogan, Bostedt, Bozek, Caleman, Coffee, Coppola, Deponte, Doak, Ekeberg, Fleckenstein, Fromme, Gebhardt, Graafsma, Gumprecht, Hampton, Hartmann, Hauser, Hirsemann, Holl, Holton, Hunter, Kabsch, Kimmel, Kirian, Liang, Maia, Meinhart, Marchesini, Martin, Nass, Reich, Schulz, Seibert, Sierra, Soltau, Spence, Steinbrener, Stellato, Stern, Timneanu, Wang, Weidenspointner, Weierstall, White, Wunderer, Chapman, Ullrich, Strüder and Schlichting2011; Barty et al., Reference Barty, Caleman, Aquila, Timneanu, Lomb, White, Andreasson, Arnlund, Bajt, Barends, Barthelmess, Bogan, Bostedt, Bozek, Coffee, Coppola, Davidsson, DePonte, Doak, Ekeberg, Elser, Epp, Erk, Fleckenstein, Foucar, Fromme, Graafsma, Gumprecht, Hajdu, Hampton, Hartmann, Hartmann, Hauser, Hirsemann, Holl, Hunter, Johansson, Kassemeyer, Kimmel, Kirian, Liang, Maia, Malmerberg, Marchesini, Martin, Nass, Neutze, Reich, Rolles, Rudek, Rudenko, Scott, Schlichting, Schulz, Seibert, Shoeman, Sierra, Soltau, Spence, Stellato, Stern, Strueder, Ullrich, Wang, Weidenspointner, Weierstall, Wunderer and Chapman2012).
Performing spot integration before binning, results in peak shapes which are different than those resulting from the pixel-map method. This powder intensity is then expressed as
 $$\eqalign{I\lpar s\rpar =&\lt I_{\rm 0}\gt \lt N_{\rm c}\gt \cr& P\lpar \theta \rpar A\lpar \theta \rpar \; D\lpar s\rpar \; \mathop \sum \limits_{hkl} m_{hkl} \left\vert {F\lpar s_{hkl} \rpar } \right\vert ^2 P_{hkl} \lpar s - s_{hkl} \rpar} \;$$
$$\eqalign{I\lpar s\rpar =&\lt I_{\rm 0}\gt \lt N_{\rm c}\gt \cr& P\lpar \theta \rpar A\lpar \theta \rpar \; D\lpar s\rpar \; \mathop \sum \limits_{hkl} m_{hkl} \left\vert {F\lpar s_{hkl} \rpar } \right\vert ^2 P_{hkl} \lpar s - s_{hkl} \rpar} \;$$
Compared to Eq. (2), this relationship contains the structure factor at the Bragg condition,
 $\left\vert {F\lpar s_{hkl} \rpar } \right\vert ^2 $
, and the peak shape, given by the distribution of observed spot centers for a given hkl reflection, P
hlk
(s − s
hkl
). This distribution can be determined by a changing average lattice constant between crystals, fluctuations in the incident X-ray beam Poynting vector, and the apparent shift of the spot center because of the partial intersection of the 3D reciprocal lattice spot by the Ewald sphere (partiality). It is expected that an internal crystallite strain gradient, crystallite size and instrumental broadening will have a reduced effect in this pattern, and only contribute through partiality. Then by comparing peak shapes from this pattern to the pixel-map pattern, the microstrain broadening because of changing average unit-cell constant can be separated from that due to intra-crystallite strain gradients or defects. Furthermore, binning the intensity by the spot centroid should result in a narrower profile, which can lead to better peak separation. Examples of how these profiles are different from the pixel-map pattern will be demonstrated later.
$\left\vert {F\lpar s_{hkl} \rpar } \right\vert ^2 $
, and the peak shape, given by the distribution of observed spot centers for a given hkl reflection, P
hlk
(s − s
hkl
). This distribution can be determined by a changing average lattice constant between crystals, fluctuations in the incident X-ray beam Poynting vector, and the apparent shift of the spot center because of the partial intersection of the 3D reciprocal lattice spot by the Ewald sphere (partiality). It is expected that an internal crystallite strain gradient, crystallite size and instrumental broadening will have a reduced effect in this pattern, and only contribute through partiality. Then by comparing peak shapes from this pattern to the pixel-map pattern, the microstrain broadening because of changing average unit-cell constant can be separated from that due to intra-crystallite strain gradients or defects. Furthermore, binning the intensity by the spot centroid should result in a narrower profile, which can lead to better peak separation. Examples of how these profiles are different from the pixel-map pattern will be demonstrated later.
C. Normalized integrated spot virtual powder pattern
As mentioned previously, serial crystallography allows one to determine not only the average intensity, but also the number of spots contributing to a reflection. The integrated spot pattern discussed in the previous section can then be normalized by this value, as described by the expression
 $$I\lpar s\rpar =\mathop \sum \limits_{\,f\,=\,1}^{N_f } \mathop \sum \limits_{\,j\,=\,1}^{N_s } \displaystyle{{I_{\,jf} } \over {N_s \lpar s \rpar N_f }}\comma \; \quad \quad s\lt s_{\,jf}\lt s+\delta s$$
$$I\lpar s\rpar =\mathop \sum \limits_{\,f\,=\,1}^{N_f } \mathop \sum \limits_{\,j\,=\,1}^{N_s } \displaystyle{{I_{\,jf} } \over {N_s \lpar s \rpar N_f }}\comma \; \quad \quad s\lt s_{\,jf}\lt s+\delta s$$
Now, the number of spots observed at a given scattering angle, N s (s), is the normalizing term, yielding an average intensity per bin. This intrinsically accounts for any detector sampling angular dependence, as well as, the multiplicity and any preferred orientation of the observed reflection. The obtained average intensity is then given by the expression
 $$\eqalign{I\lpar s\rpar =& \lt I_{\rm 0}\gt \cr & P\lpar \theta \rpar A\lpar \theta \rpar \; D\lpar s\rpar \; \mathop \sum \limits_{hkl} \left\vert {F\lpar s_{hkl} \rpar} \right\vert^2 P^{\prime}_{hkl} \left(s - s_{hkl} \right)}$$
$$\eqalign{I\lpar s\rpar =& \lt I_{\rm 0}\gt \cr & P\lpar \theta \rpar A\lpar \theta \rpar \; D\lpar s\rpar \; \mathop \sum \limits_{hkl} \left\vert {F\lpar s_{hkl} \rpar} \right\vert^2 P^{\prime}_{hkl} \left(s - s_{hkl} \right)}$$
which after correcting for polarization, absorption, and the Debye–Waller effect, is directly proportional to the structure factor at the Bragg condition. The peak shape functions,
 $P^{\prime}_{hkl} \left({s - s_{hkl} } \right)$
, will also be normalized by the relative number of spots with a given scattering vector, and should then ideally resemble top-hat functions with a width related to variance in the spot position – assuming the structure factor is on average independent of the spot position. This is a new data type for powder diffraction which will be shown to allow for the trace phase detection, but also has potential to be useful for structure determination in crystallite systems which contain texture.
$P^{\prime}_{hkl} \left({s - s_{hkl} } \right)$
, will also be normalized by the relative number of spots with a given scattering vector, and should then ideally resemble top-hat functions with a width related to variance in the spot position – assuming the structure factor is on average independent of the spot position. This is a new data type for powder diffraction which will be shown to allow for the trace phase detection, but also has potential to be useful for structure determination in crystallite systems which contain texture.
III. CASE STUDY: THE MICROSTRUCTURE OF Pr0.5Ca0.5MnO3 (PCMO) POWDER
To illustrate the proposed data treatment methods, data from a serial femtosecond crystallography (SFX) experiment was taken to study the structure of PCMO and analyzed. The experiment was performed at the Coherent X-ray Imaging (CXI) instrument (Boutet and Williams, Reference Boutet and Williams2010) of the Linac Coherent Light Source (LCLS). The experimental geometry followed that of other SFX experiments (Boutet et al., Reference Boutet, Lomb, Williams, Barends, Aquila, Doak, Weierstall, DePonte, Steinbrener, Shoeman, Messerschmidt, Barty, White, Kassemeyer, Kirian, Seibert, Montanez, Kenney, Herbst, Hart, Pines, Haller, Gruner, Philipp, Tate, Hromalik, Koerner, van Bakel, Morse, Ghonsalves, Arnlund, Bogan, Caleman, Fromme, Hampton, Hunter, Johansson, Katona, Kupitz, Liang, Nass, Martin, Redecke, Stellato, Timneanu, Wang, Zatsepin, Schafer, Defever, Neutze, Fromme, Spence, Chapman and Schlichting2012; Redecke et al., Reference Redecke, Nass, Deponte, White, Rehders, Barty, Stellato, Liang, Barends, Boutet, Williams, Messerschmidt, Seibert, Aquila, Arnlund, Bajt, Barth, Bogan, Caleman, Chao, Doak, Fleckenstein, Frank, Fromme, Galli, Grotjohann, Hunter, Johansson, Kassemeyer, Katona, Kirian, Koopmann, Kupitz, Lomb, Martin, Mogk, Neutze, Shoeman, Steinbrener, Timneanu, Wang, Weierstall, Zatsepin, Spence, Fromme, Schlichting, Duszenko, Betzel and Chapman2013), as a jet of liquid containing crystallites was probed by a femtosecond X-ray pulse at a pulse repetition rate of 120 Hz. The 2D diffraction pattern from each pulse was measured in transmission, and the detector was read out between pulses. For this experiment X-ray pulses having a 10 keV photon energy, 0.1–0.3 mJ pulse energy, and 10 fs pulse length were focused to a nominal 2 µm spot size on the jet.
A sample of polycrystalline PCMO powder was prepared from precursor powders of Pr6O11, CaCO3, and Mn2O3 by repeated calcination at 1050 °C for 12 h followed by ball milling. The cycle of calcination and ball milling was carried out three times, until powder-diffraction measurements did not find evidence of the precursor phases. The powder was then dispersed in pure ethanol and filtered using syringe PEEKTM filters from the Upchurch Scientific to remove any particles larger than 500 nm. This solution was concentrated by centrifugation to 1011 particles ml−1 as measured by a NanosightTM microscope. The solution was delivered to the X-ray interaction region using a gas dynamic virtual nozzle (DePonte et al., Reference DePonte, Weierstall, Schmidt, Warner, Starodub, Spence and Doak2008) to create a liquid jet that was nominally 5 µm in diameter. The nozzle was encased in a cooling block, to allow for the liquid to be cooled near the freezing point of ethanol, 200 K. However, all data presented here were taken at room temperature.
The collected diffraction patterns were sorted into those containing spots, and those without spots, corrected for detector dark current and persistent background signal using the program Cheetah (Barty et al., Reference Barty, Kirian, Maia, Hantke, Yoon, White and Chapman2014). An example of diffraction pattern after applying these corrections is depicted in Figure 1. The patterns proved to have too few spots to allow for Fourier-based indexing, and serial crystallography structure solution. Nonetheless, one can learn about the PCMO structure by studying the virtual powder-diffraction patterns.

Figure 1. (Colour online) An example raw diffraction image collected during the LCLS experiment is shown, depicting the low number of spots observed in an image. The spot that is indicated with a dashed square is shown in the inset and has a scattering vector magnitude of 0.382 A−1, which does not line up with the PCMO phase.
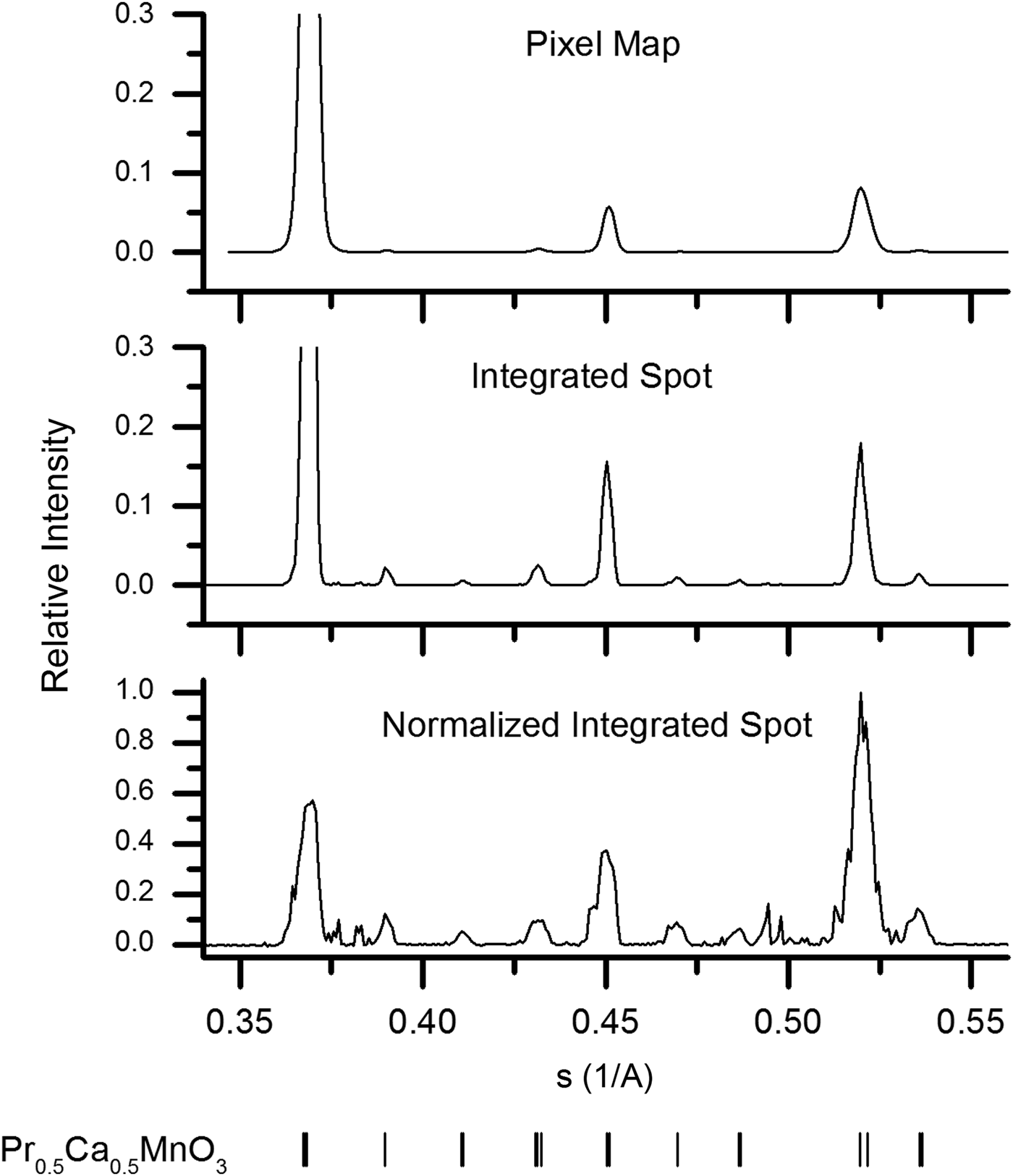
Calculated powder patterns of each of the three types previously discussed are presented in Figure 2. The same set of 22 000 images were used to generate each powder-diffraction pattern. A set of programs have been developed for calculating these powder patterns from serial crystallography data based on the shared library component of CrystFEL (White et al., Reference White, Kirian, Martin, Aquila, Nass, Barty and Chapman2012). Each program begins with the found spot positions and defines a window around each for integration and local background estimation. The average background is then subtracted from the observed pixel intensities before mapping the spot window into the powder domain.
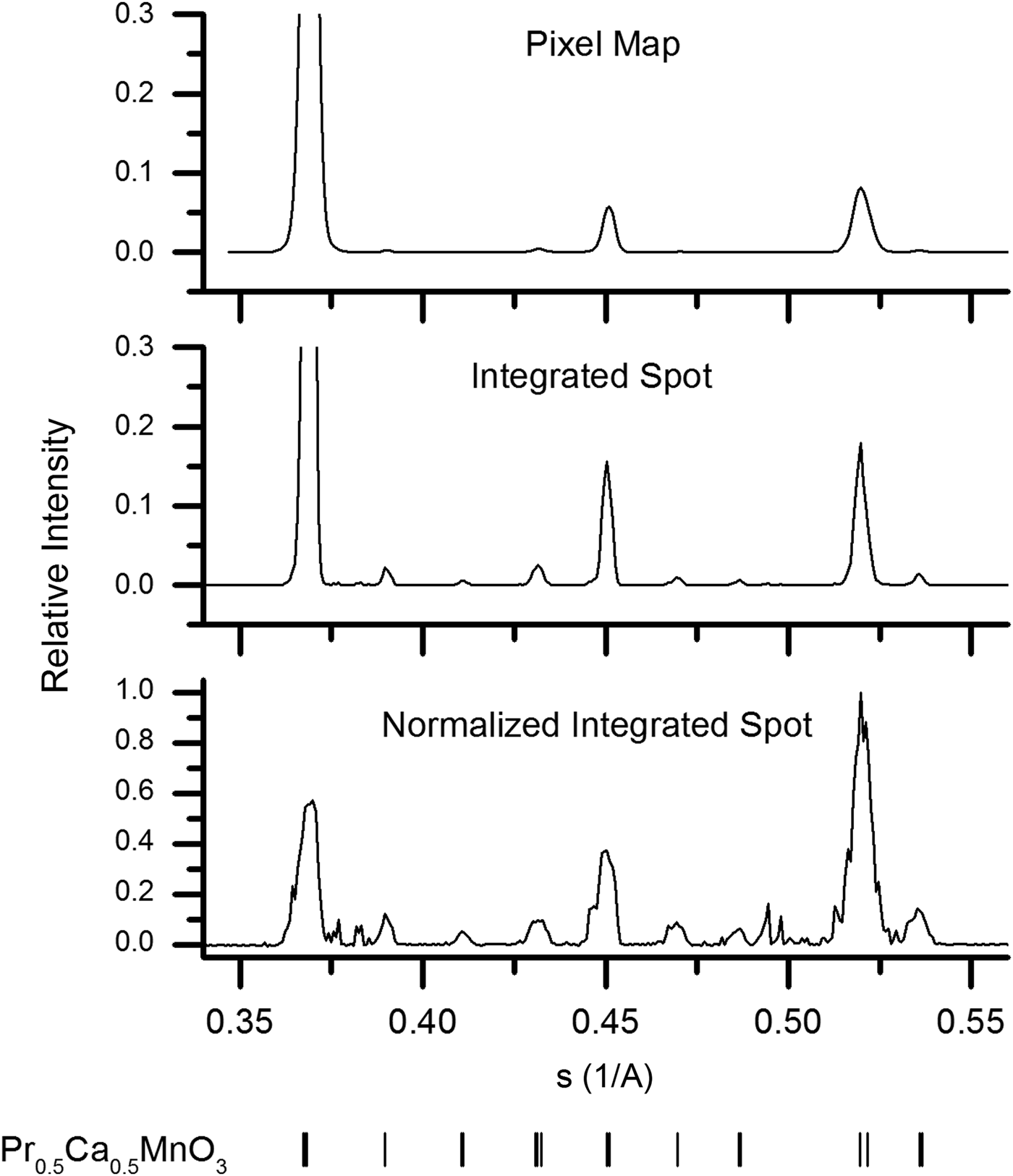
Figure 2. Virtual powder patterns calculated from a collection of 22 000 diffraction images using the discussed methods for normalization. The peak positions in each case can be compared to the peak positions of room temperature PCMO shown at the bottom of the figure.
Insight into the microstructure of the PCMO sample comes from comparing each of the three generated powder patterns in Figure 2. For instance, the peak broadening in the pixel map pattern is larger than that in the integrated spot pattern. This is quantitatively shown in the Williamson–Hall plots (Williamson and Hall, Reference Williamson and Hall1953) of the integral breadths depicted in Figure 3. The slope of these linear fits is generally taken as a measure of microstrain, while the intercept is related to the average crystallite size (Scardi et al., Reference Scardi, Leoni and Delhez2004). However, in this case it is evident that the linear fit is a poor approximation to the data, and the values obtained merely indicate the magnitude of the broadening. The slope of the linear fit for the pixel-map data is 0.0043, whereas that of the integrated-spot case is 0.0022, implying a significant difference in the dependence of the broadening on the scattering vector magnitude. As already mentioned, this may be explained by the hypothesis that pixel-map pattern peaks are a convolution of size and strain effects, while the integrated-spot pattern profiles are primarily given by the different average unit-cell parameters between crystallites. Furthermore, the changing difference of the peak broadening for spot size from peak-to-peak indicates the anisotropic nature of this internal microstrain. It is interesting to note that the intercepts of the fitted lines to the two sets of data are both nearly 0.0020 A−1. The similarity of this value from the two methods is not entirely consistent with the argument that the crystallite size contribution is not present in the integrated-spot pattern profile. However, as already mentioned this could be due to the poor linear approximation of the data, and more detailed understanding of the partiality component of the integrated-spot peak shape is necessary before decisive conclusions can be drawn.

Figure 3. (Colour online) Two Williamson–Hall plots are overlaid, comparing the peak integral breadths from the pixel map versus integrated spot patterns. The linear fit of the pixel-map data (solid line) has an intercept of 0.0019 A−1 and a slope of 0.0043, while those of the integrated-spot data (dashed line) are 0.0020 A−1 and 0.0022, respectively.
Turning our attention to the normalized-spot pattern in Figure 2, the observed positions of the largest peaks in this pattern are consistent with those given at the bottom of the figure for room temperature PCMO (Jirák et al., Reference Jirák, Krupička, Šimša, Dlouhá and Vratislav1985). It is also evident that the PCMO peaks that were hardly visible in the other two patterns become more apparent in this case. In addition, a lot of new sharp peaks become visible around scattering vectors of 0.38 and 0.5 A−1 which were not discernable in the other two patterns. These peaks correspond with bright, well-defined observed spots in the collected images – an example of one is depicted in the inset of Figure 1. Some of the new observed peaks are found to be consistent with the precursor phases of Pr6O11 (JCPDS 42-1121) and α-Mn2O3 (Geller, Reference Geller1971), which may indicate an incomplete solid-state reaction. An approximate trace phase crystallite concentration of 1% was determined by simply counting the number of spots contributing to each peak in the normalized-spot pattern. This is at the limit of conventional powder diffraction and quantitative phase determination (Madsen et al., Reference Madsen, Scarlett, Cranswick and Lwin2001), yet clear evidence of these phases is found by the described method. Our presented method for trace phase detection thus offers higher sensitivity to small amounts of trace phases. However, refinement of the trace phases is limited when data are collected from only a few grains.
IV. CONCLUSIONS
In this paper, we have presented the underlying assumptions and consequences of different strategies to assemble a virtual powder pattern from serial crystallography data. The three presented schemes have been demonstrated for the case of serial femtosecond crystallography data collected on a powder sample of PCMO. This allows an insight into the internal crystallite strain and the presence of trace amounts of precursor phases in the microstructure.
In particular, the prospect of extremely trace phase detection by this method is promising. The minimum concentration, which this serial method can detect, is in principle only limited by the diffraction signal strength from an individual crystallite in the beam. If the spot position of a phase is unique, then a single pattern can be found in a stack of millions. In this regard, a clear advantage of serial crystallography data collection is that the patterns containing these rare spots can be recalled and further analyzed for a better understanding of these trace phases.
Although the high intensity of the X-ray free-electron laser (XFEL) radiation allowed for an abundance of signal from this sample, the sample delivery, data collection, and analysis strategies that are described here are not limited to this X-ray source. A similar serial method can be adopted during measurements at synchrotrons with a microfocused beam, such as raster scanning a fixed target (Gati et al., Reference Gati, Bourenkov, Klinge, Rehders, Stellato, Oberthür, Yefanov, Sommer, Mogk, Duszenko, Betzel, Schneider, Chapman and Redecke2014) or a compact in-air flowing device (Stellato et al., Reference Stellato, Oberthür, Liang, Bean, Gati, Yefanov, Barty, Burkhardt, Fischer, Galli, Kirian, Meyer, Panneerselvam, Yoon, Chervinskii, Speller, White, Betzel, Meents and Chapman2014). If the interrogated crystals diffract strongly enough, then a rotating anode laboratory X-ray source with a 2D detector may even be sufficient.
ACKNOWLEDGEMENTS
Portions of this research were carried out on the CXI Instrument at the Linac Coherent Light Source (LCLS), a division of SLAC National Accelerator Laboratory and an Office of Science user facility operated by Stanford University for the U.S. Department of Energy. The authors acknowledge the contributions of the technical staff at the CXI instrument that prepared the experimental chamber and helped to install the specialized injector system. The authors also thank Michael Bogan who allowed to use of his laboratory for sample characterization and Raymond Sierra for help with the sample concentration analysis. NAZ acknowledges funding from grant NSF MCB 1021557. JPK acknowledges funding and support from the University of Hamburg and Deutscher Akademischer Austausch Dienst (DAAD) Research Internships in Science and Engineering (RISE) program.