


Introduction
The cosmopolitan jewel beetle genus Agrilus Curtis, with its 3213 valid nominal species, is the largest conventionally recognized genus of the Animal Kingdom (as of July 31 2017; Bellamy, Reference Bellamy2008; Zoological Record®, Thomson Reuters). All Agrilus are strictly phytophagous and their astonishing diversity (figs 2a–i and 3a–i) might perhaps be linked to numerous shifts among their host plants, which is an important diversification factor in many phytophagous insects (Drès & Mallet, Reference Drès and Mallet2002; Nosil & Mooers, Reference Nosil and Mooers2005; Egan et al., Reference Egan, Nosil and Funk2008). While Agrilus adults feed on leaves, their larvae (fig. 1c) develop on living subcortical tissues of trees and shrubs to an extent sufficient to kill a host, especially when it has been already weakened by abiotic factors (Wargo, Reference Wargo1977; Dunn et al., Reference Dunn, Kimmerer and Nordin1986; Jendek & Poláková, Reference Jendek and Poláková2014). This economically significant effect is achieved by inducing subcortical necrosis that disrupts water and nutrient transport (Vansteenkiste et al., Reference Vansteenkiste, Tirry, Van Acker and Stevens2004). Moreover, Agrilus species have a proven invasive potential facilitated by their long-living immature stages easily transported with wood, while the adults are capable of long active flights. As a consequence, some Agrilus species became important invasive alien pests (Haack et al., Reference Haack, Benjamin and Haack1983; Akers et al., Reference Akers, Herms and Nielsen1986; Gordon et al., Reference Gordon, Woodford and Birch1997; Everett, Reference Everett2000) after being accidentally introduced to an area free of natural enemies (Dunn et al., Reference Dunn, Kimmerer and Nordin1986; Jones et al., Reference Jones, Reed, Mroz, Liechty and Cattelino1993; Gibbs & Greig, Reference Gibbs and Greig1997; Aukema et al., Reference Aukema, Leung, Kovacs, Chivers, Britton, Englin, Frankel, Haight, Holmes, Liebhold, McCullough and Von Holle2011). Consequently, Agrilus beetles constitute a high-risk group of organisms for quarantine entomologists, particularly in North America, and on the level similar to those of longhorned beetles (Cerambycidae) and bark and ambrosia beetles (Curculionidae: Scolytinae and Platypodinae).

Fig. 1. Agrilus planipennis (=Emerald Ash Borer), the most costly insect forestry pest in human history. (a) and (b): examples of damage: (a) North America (Canada, Ottawa); (b) Europe (Russia, Moscow Region); larva (c) and adults (d) and (e).

Fig. 2. Agrilus, diversity of Northern Hemisphere species. (a) A. acutipennis (Canada, Ontario); (b) A. anxius (Canada, Ontario); (c) A. cyaneoniger (Russia, Primorsky Krai); (d) A. dureli (China, Beijing); (e) A. egeniformis (Canada, Ontario); (f) A. fleischeri (China, Jilin); (g) A. granulatus (Canada, Ontario); (h) A. guerini (Slovakia); (i) A. hyperici (Slovakia).

Fig. 3. Agrilus, diversity of Northern Hemisphere species. (a) A. obsoletoguttatus (Canada, Ontario); (b): A. pensus (Canada, Ontario); (c) A. politus (Canada, Ontario); (d) A. pseudocoryli (Canada, Ontario); (e) A. ribbei (Russia, Primorsky Krai); (f) A. ruficollis (Canada, Ontario); (g) A. smaragdinus (China, Jilin); (h) A. viridis (Slovakia); (i) A. vittaticollis (Canada, Ontario).
Indeed, human-mediated range expansions are well documented in Agrilus with at least 11 Eurasian species having been introduced to North America: Agrilus cuprescens (Ménétriés), Agrilus cyanescens (Ratzeburg), Agrilus derasofasciatus Lacordaire, Agrilus hyperici (Creutzer), Agrilus pilosovittatus Saunders, Agrilus planipennis Fairmaire, Agrilus ribesi Schaefer, Agrilussinuatus (Olivier), Agrilus smaragdifrons Ganglbauer, Agrilus subrobustus Saunders, and Agrilus sulcicollis Lacordaire (Jendek & Grebennikov, Reference Jendek and Grebennikov2009; Jendek, Reference Jendek2016; Hoebeke et al., Reference Hoebeke, Jendek, Zablotny, Rieder, Yoo, Grebennikov and Ren2017). The European species, Agrilus angustulus (Illiger), might have been introduced to the Russian Far East, while the North American species, Agrilus bilineatus (Weber), is the first among the Nearctic species to reach the Palaearctic Region (based on the single record in Turkey, Jendek Reference Jendek2016). Among these adventive Agrilus taxa, one has become most popular and feared. This is the infamous A. planipennis, the Emerald Ash Borer (figs 1a–e), which is a prime example of a large-scale and continent-wide biological and economic disaster caused by an introduction of a ‘bad’ jewel beetle to two new continents (North America and Europe; fig. 1a, b, respectively). Damage caused by this notorious pest was partly ascribed to the high susceptibility of at least the North American Ash trees to beetle's attacks, and partly due to the absence of natural enemies (Haack et al., Reference Haack, Jendek, Liu, Marchant, Petrice, Poland and Ye2002; Liu et al., Reference Liu, Bauer, Gao, Zhao, Petrice and Haack2003). The negative economic consequences caused by this beetle in North America have been indeed enormous and variously estimated to exceed 10 billion USD (Kovacs et al., Reference Kovacs, Mercader, Haight, Siegert, McCullough and Liebhold2011; Herms & McCullough, Reference Herms and McCullough2014).
Rapid detection and taxonomic identification of an invasive species is the foremost step that leads to appropriate control measures to follow. The lack of Agrilus taxonomic expertise and absence of adequate diagnostic tools result in a significant delay between the establishment of an invasive species and its first positive identification (Jendek & Grebennikov, Reference Jendek and Grebennikov2011). For example, it took more than 5 years to detect and identify A. planipennis in North America (Haack et al., Reference Haack, Jendek, Liu, Marchant, Petrice, Poland and Ye2002). Still worst, the occurrence of the introduced A. ribesi had been overlooked in the USA and Canada for almost a century, while the repeatedly observed damage was erroneously assigned to a native species (Jendek et al., Reference Jendek, Grebennikov and Bocak2015). Since the number of non-native pests raises continuously (Aukema et al., Reference Aukema, McCullough, Von Holle, Liebhold, Britton and Frankel2010), it becomes of prime economic importance to develop DNA-based pest diagnostic tools to facilitate their rapid and reliable identification.
The sheer number of species within the enormously diverse genus Agrilus generates its unique challenge, which in turn inhibits an efficient use of generic taxonomy. The current internal classification of the genus is highly inconsistent and incomplete. All 35 nominal Agrilus subgenera proposed so far (e.g., Alexeev, Reference Alexeev1998; Bellamy, Reference Bellamy2008) were introduced on a regional (mainly European) and non-phylogenetic basis by using the vague criterion of general similarity among adult beetles. In the absence of a comprehensive worldwide taxonomic treatment of the genus, the vast majority of nominal Agrilus species remain unassigned to any of these ‘subgenera’. As a result, the introduction of formal subgenera arguably caused more inconsistencies and taxonomic confusion, compared with not being introduced at all (Jendek & Grebennikov, Reference Jendek and Grebennikov2011). The undeveloped and rigid (in terms of zoological nomenclature) sub-generic approach has been recently substituted by the use of informal species-groups serving as temporary taxonomic labels preventing the proliferation of unnecessary genus-group names (Jendek & Grebennikov, Reference Jendek and Grebennikov2011).
The species-level identification of Agrilus beetles outside of a few well-studied regions, such as Europe and temperate North America, is often a significant challenge and in many cases is nearly, or outright, impossible. This is due to the immense number of Agrilus species, as well as to the much-constrained adult morphological variability resulting in a high number of externally similar species (Jendek & Grebennikov, Reference Jendek and Grebennikov2011). As a result, reliable Agrilus identification requires expert skills and an extensive reference collection. An experienced entomologist not particularly specialized in jewel beetles is normally able to identify adults of locally native Agrilus species, but will almost always fail to do so for any larva or for non-native adults, particularly from a geographically distant region. Furthermore, the presence of a well-curated and extensive voucher collection of Agrilus will always be required for such identifications. Overall, putting a correct species name on an Agrilus beetle, adult or immature, is a task beyond the current diagnostic capacity of many entomological professionals, research organizations or even that of some developed countries (Jendek & Grebennikov, Reference Jendek and Grebennikov2011).
Recently mitochondrial DNA-based species identification methods have become increasingly important as a practical alternative to the classical morphology-based identification (e.g., Hebert et al., Reference Hebert, Ratnasingham and deWaard2003; Riedel et al., Reference Riedel, Sagata, Surbakti, Rene and Balke2013a, Reference Riedel, Sagata, Suhardjono, Tänzler and Balkeb; Ashfaq & Hebert, Reference Ashfaq and Hebert2016). Although there is a possibility that nuclear-encoded mitochondrial pseudogenes can be amplified instead of the mitochondrial target fragment or that several sequence types might be present in cells of a single individual, the method can be used when high numbers of individuals and representatives of different populations are sequenced to identify the dominant protein coding haplotype (Kang et al., Reference Kang, Kim, Park, Kim and Kim2016). The sequencing of short mtDNA fragments with conservative primers has been repeatedly demonstrated to be a rapid, reliable and cost-efficient alternative, even though a subsequent critical evaluation of the results by using classical morphology by a taxonomic expert remains desirable. Besides being rapid and expertise-free, DNA-based methods enable the identification of immature stages, which may be the only available representatives of a species when the economically significant damage is identified for the first time (Ahrens et al., Reference Ahrens, Monaghan and Vogler2007). In ecological and biodiversity research, putative biological species can be preliminarily delimited and identified using various DNA-based methods. The simplest of such methods is the one based on pairwise genetic distances. The species-delimitation results, however, might differ substantially when various thresholds are applied (Hebert et al., Reference Hebert, Ratnasingham and deWaard2003). Since the method was proposed, 3% of the uncorrected pairwise distance threshold has been repeatedly rejected as a ‘universal’ value, mainly because extreme deviations from this rate have been detected in either direction. Furthermore, the cox1–5′ fragment (=DNA barcode) of morphologically or ecologically distinct species might be identical or, conversely, genetically highly divergent populations might be detected within the same ‘good’ morphological species (Baselga et al., Reference Baselga, Gomez-Rodriguez, Novoa and Vogler2013; Zahiri et al., Reference Zahiri, Lafontaine, Schmidt, de Waard, Zakharov and Hebert2014; Li et al., Reference Li, Gunter, Pang and Bocak2015; Kusy et al., Reference Kusy, Sklenarova and Bocak2018). Since such information for Agrilus beetles has been limited, in this study we attempt to present a large database of mtDNA fragments to evaluate the levels of genetic variability within this genus.
The tree-based methods of species identification (Satler et al., Reference Satler, Carstens and Hedin2013) form a phylogenetically-sound alternative to the threshold-based approach. They integrate morphological and molecular methods in insect systematics of predominantly highly diversified clades, with the aim of producing robust species-level phylogenetic trees (e.g., Pons et al., Reference Pons, Barraclough, Gomez-Zurita, Cardoso, Duran, Hazell, Kamoun, Sumlin and Vogler2006; Riedel et al., Reference Riedel, Sagata, Suhardjono, Tänzler and Balke2013b; Bocek & Bocak, Reference Bocek and Bocak2016). Unlike the DNA-barcode threshold approach, these methods were not primarily designated for the routine identification of an unknown sample against the reference database. Instead, they enable the positioning of an unknown sample with a high degree of confidence in the phylogenetic framework and among its closest relatives (Vogler & Monaghan, Reference Vogler and Monaghan2007; Monaghan et al., Reference Monaghan, Wild, Elliot, Fujisawa, Balke, Inward, Lees, Ranaivosolo, Eggleton, Barraclough and Vogler2009). So performed, the tree-based methods are superior in their diagnostic capacity to the distance-based matching approach, partly because they can handle unknown unknowns, that are representatives of yet undescribed species. Instead of a numerical and somewhat arbitrary genetic distance threshold, the tree-based methods use the shape of the phylogenetic tree, which might be either a dated tree (obtained with the General Mixed Coalescent model, GMYC; Pons et al., Reference Pons, Barraclough, Gomez-Zurita, Cardoso, Duran, Hazell, Kamoun, Sumlin and Vogler2006; Fujisawa & Barraclough, Reference Fujisawa and Barraclough2013) or a phylogram (obtained with the Bayesian Poisson Tree process, Zhang et al., Reference Zhang, Kapli, Pavlidis and Stamatakis2013). The results of all tree-based analyses are not necessarily identical when different methods are used (Carstens et al., Reference Carstens, Pelletier, Reid and Satler2013), and various levels of success have been reported in unrelated animal lineages (Baselga et al., Reference Baselga, Gomez-Rodriguez, Novoa and Vogler2013; Li et al., Reference Li, Gunter, Pang and Bocak2015; Bocek & Bocak, Reference Bocek and Bocak2016). Nevertheless, the performance of the tree-based methods is at least comparable with the identification by a trained non-specialist, while the even greater reliability of the tree-based identification can be expected with a large reference dataset. Another important advantage of a tree-based approach is the possibility of identifying the geographic origin of a non-native sample, either within the intraspecific variability (if samples from different parts of the range are available) or, when a lineage has a phylogeographic structure, the closest relatives can be expected in the same region. In the present work, therefore, we will attempt to investigate the applicability of the tree-based identification for Agrilus pests by recovering geographically-delimited clades and morphologically pre-defined groups of closely related species.
In summary, the principal aim of the present study is to report and make publicly available the extensive database of three mitochondrial DNA fragments and new barcode sequences for a large number of Agrilus species analyzed so far. The recovered phylogenetic trees provide information on relationships among principal Agrilus lineages and the newly generated data can be used for DNA-based identification of unknown Agrilus samples originating from anywhere within the temperate zone of the Northern Hemisphere and irrespective of the beetle's life stage. Furthermore, we test the performance of various DNA-based species delimitation methods against the dataset of morphologically identified Agrilus species. Specifically, we test if the results are similar when different species delimitation methods are applied and, additionally, which particular species is conflictingly delimited using morphology vs DNA data. If a single species is represented by multiple distantly related lineages, the current species concept is unacceptable in the biological classification (Vences et al., Reference Vences, Guayasamin, Miralles and De la Riva2013) and the limits of such a species need further study. Additionally, we investigate the extent in which the closely related species might occur in distant zoogeographical regions, that is in Europe, in East Asia and in Northern America. Overall this work should serve as a starting point for building the densely sampled public online Agrilus database in support of the DNA-based identification of economically significant pests from the temperate zone of the Northern Hemisphere.
Material and methods
Taxon sampling
In the course of this study a total of 475 Agrilus individuals of over 100 species were collected by various collectors in 2000–2015 in Eastern Europe and in Central Europe north of the Alps (111 individuals), the Mediterranean Region (57 individuals), Russian Far East (111 individuals), China, Korea and Japan (23 individuals), the Oriental Region (11 individuals) and North America (154 individuals). All these specimens were identified to species level by one of us (EJ). Voucher numbers, NCBI GenBank accession numbers and detailed locality and host plant data are given in table S1. The voucher specimen numbers are used to identify any sequenced individual in the published trees herein and in the GenBank database. All 329 Agrilus records available in the Barcode of Life Database (=BOLD) format, including specimen images (fig. S3) and geo data, are released through a public dataset ‘Agrilus1 329’ available at: dx.doi.org/10.5883/DS-AGRILUS1. These specimens have voucher numbers (column 2 in table S1 in format CNC0000) having the same last four digits, as their BOLD Sample ID (in format CNCCOLVG00000000). The dry-mounted sequenced specimens are deposited in the voucher collections located at the authors’ home institutions. The outgroup was composed of the most likely closely related non-Agrilus Agrilini jewel beetles from the genera Coraebus Gory & Laporte, Meliboeus Deyrolle, Nalanda Thery, and Trachys Fabricius.
DNA isolation, amplification and sequencing
All sequenced specimens were killed and preserved in 96% ethanol and stored at −20 °C until DNA extraction. The samples designated by voucher numbers beginning with CNC were sequenced for the DNA barcoding fragment in the Biodiversity Institute of Ontario, Canada following the standard DNA barcoding protocol (Ivanova et al., Reference Ivanova, de Waard and Hebert2006; Grebennikov et al., Reference Grebennikov, Jendek and Smirnov2017). Additional sequencing of the same specimens for two other DNA markers, as well as sequencing of all samples designated by voucher numbers beginning with “EJ” or “A” was performed in the Laboratory of Molecular Systematics, Palacky University, Olomouc, Czech Republic. For these, genomic DNA was extracted from metathoracic muscles using either the phenol chloroform method or the Qiagen DNeasy® Blood & Tissue Kit (Qiagen Inc.). Extraction yield was measured using a NanoDrop-1000 Spectrophotometer. Three fragments of mitochondrial DNA (mtDNA) were amplified: rrnL fragment using primers 16a and 16b and alternatively the primer ND1A, which is located in the nad1 mtDNA gene; the cox1–3′ fragment using primers Pat and Jerry/JerM, and the cox1–5′ (=DNA barcode) mtDNA fragment using primers LCO1490 K and HCO2198 K (Simon et al., Reference Simon, Frati, Beckenbach, Crespi, Liu and Flook1994; Bocak et al., Reference Bocak, Bocakova, Hunt and Vogler2008). Polymerase chain reaction (PCR) settings and cycle sequencing conditions were those reported by Bocak et al. (Reference Bocak, Bocakova, Hunt and Vogler2008), whereas the PCR products were purified using PCRu96 Plates (Millipore Inc.) and sequenced by an ABI 3130 automated sequencer using the Big Dye Sequencing Kit 1.1. Altogether, 780 new sequences were produced for this study: 166 sequences for rrnL mtDNA, 170 for cox1–3′ mtDNA, and 444 for cox1–5′ (DNA barcode fragment) mtDNA (table S1).
Sequence editing and alignment
Sequences were edited using Sequencher 4.10.1 (Gene Codes Corp.). The length conservative protein coding sequences were aligned using ClustalX 2.1 (Thompson et al., Reference Thompson, Gibson, Plewniak, Jeanmougin and Higgins1997) under default parameter settings and checked for amino acid reading frames to identify and exclude potential pseudogenes from further analyses. The studied group represents a single genus of relatively closely related species and when the nucleotide sequences were translated in amino acid sequence, we identified only a low variability. The pseudogene would be easily identified by the highly divergent amino acid sequence. If any pseudogene is present in the current Agrilus dataset, its mutations have to be synonymous with the amino acid translation and the length of the sequence could not change. Such pseudogene cannot be detected by available methods, but on the other hand, the phylogenetic noise potentially introduced by its presence is negligible.
The length variable rrnL fragment was aligned using Mafft 7 (Katoh & Standley, Reference Katoh and Standley2013) under default parameter settings. The rrnL mtDNA alignment consisted of 883 aligned positions. As all ingroup taxa belong to a single genus, they are highly similar and the maximum number of contiguous non-conserved positions reached eight nucleotides. As such short length variable blocks do not represent a serious problem for alignment, we did not apply any filtering method. When sequences and alignments within these non-conserved blocks were checked with the results of the phylogenetic analysis, we noted that the similar sequences in the non-conservative blocks usually define the cluster of species. The exclusion of such information from the phylogenetic analysis would result in average in worse trees and an increase of false support as demonstrated by Tan et al. (Reference Tan, Muffato, Ledergerber, Herrero, Goldman, Gil and Dessimoz2015).
The protein coding cox1–3′ and cox1–5′ mtDNA fragments formed matrices of 853 and 659 aligned positions, respectively. The Kimura 2-parameter genetic distances among Agrilus sequences reached up to 22.0, 28.9, and 32.6%, in rrnL, cox1–3′, and cox1–5′ mtDNA, respectively.
Three maximum likelihood (ML) analyses each utilizing a different dataset
Phylogeny was inferred using ML optimality criterion, as implemented in RAxML 8.1 (Stamatakis, Reference Stamatakis2014). When applicable, each dataset (see below) was partitioned by genes and, alternatively, by gene and codon positions for protein-coding fragments. The nucleotide substitution model applied for all gene fragments was set to GTR + I + G, as suggested by the AIC criterion in jModelTest 2.1.2 (Darriba et al., Reference Darriba, Taboada, Doallo and Posada2012), with all partitions unlinked. Confidence intervals were determined with 1000 non-parametric bootstrap replicates (Felsenstein, Reference Felsenstein1985) utilizing the rapid bootstrap option under the GTRGAMMA substitution model. Recovered internal Agrilus clades were compared with the species-groups as defined by Jendek & Grebennikov (Reference Jendek and Grebennikov2011) and by Alexeev (Reference Alexeev1998, as subgenera).
All three fragment-specific datasets differ notably in the number of terminals. To address this data inequality, three separate analyses, each utilizing separate datasets, were created. Analysis 1 utilized the densest and least inclusive three-fragment dataset containing 124 terminals (including five non-Agrilus outgroups). The average representation was 2.36 fragments per terminal, while no terminal was represented by less than two fragments. In this analysis, all Agrilus species (and in some cases geographically distant conspecific populations and/or populations collected from different host plants) were represented by a single terminal. Analysis 2 utilized the least dense and most inclusive three-fragment dataset containing 475 terminals (including non-Agrilus five outgroups), many of them represented by the DNA barcode fragment only (although the majority of species had at least one additional fragment sequenced for at least a single individual) with the average representation 1.64 fragments per terminal. Analysis 3 utilized the one-fragment (DNA barcode) dataset containing 725 terminals (including 14 Coraebus outgroups). For this analysis, our newly generated DNA barcodes were supplemented with those publicly available from the BOLD database (http://www.boldsystems.org, accessed on 13 July 2017, their list is in table S2) and merged in the 475-taxa dataset.
Species delimitation analyses
The morphology-based identification of Agrilus specimens resulted in a total of 85 named Agrilus species, plus 15 unnamed provisional species. The morphologically defined species originated from Europe and the Mediterranean Region (~40 spp.), Eastern Asia (~40 spp.) and North America (~20 spp.). In some cases, an unidentified specimen clustered on the trees with presumably conspecific individuals with similar DNA sequences; provisional species names for such specimens are given in brackets in fig. S1.
To investigate an algorithm-based species delimitation in Agrilus, putative species were estimated alternatively using 2 and 3% DNA barcoding thresholds, as implemented in Species Identifier 1.7.7 (Meier et al., Reference Meier, Shiyang, Vaidya and Ng2006). The morphology-based species definitions were separately tested by using uncorrected pairwise distances among Agrilus sequences for all three sequenced fragments. Further, we identified putative species entities by implementing the Bayesian Poisson Tree Processes (bPTP) model for species delimitation (the bPTP server at species.h-its.org). The algorithm delimits putative species from the tree with branch lengths representing the number of substitutions (Zhang et al., Reference Zhang, Kapli, Pavlidis and Stamatakis2013) and provides posterior probabilities of all descendants under a node representing a single species. We ran separate analyses for the trees inferred using ML criterion and utilizing three datasets, each constituted by a single DNA fragment (cox1–3′, cox1–5′, and rrnL). The highest numbers of individuals and morphologically pre-defined species were represented in the cox1–5′ (DNA barcode) dataset with 444 sequences and 94 morphologically delimited species (named or unnamed); the rrnL dataset contained 166 sequences and 69 species, while the cox1–3′ dataset contained 170 sequences and 75 species.
Results
Considering that analysis 1 utilized a pruned dataset with a lesser fraction of missing data (as compared with analysis 2), its topology is reported herein and discussed in more detail, while analysis 2 (fig. S1), although generally congruent, is mentioned only for comparative purposes.
ML analyses of the three-fragment datasets (analyses 1 and 2) recovered monophyletic Agrilus with bootstrap support (BS) of 65% (fig. 4) and 60% (fig. S1), respectively. Subsequent basal-most splits within Agrilus are conflictingly resolved in both analyses and have negligibly low statistical support with SB often <10%. Both analyses consistently recovered six morphologically pre-defined species-groups (fig. 4), although in most cases these clades did not have a well-supported sister clade (with the exception of the convexicollis- and cyanescens species-groups, which together formed a strongly supported clade with BS 93%, fig. 4).
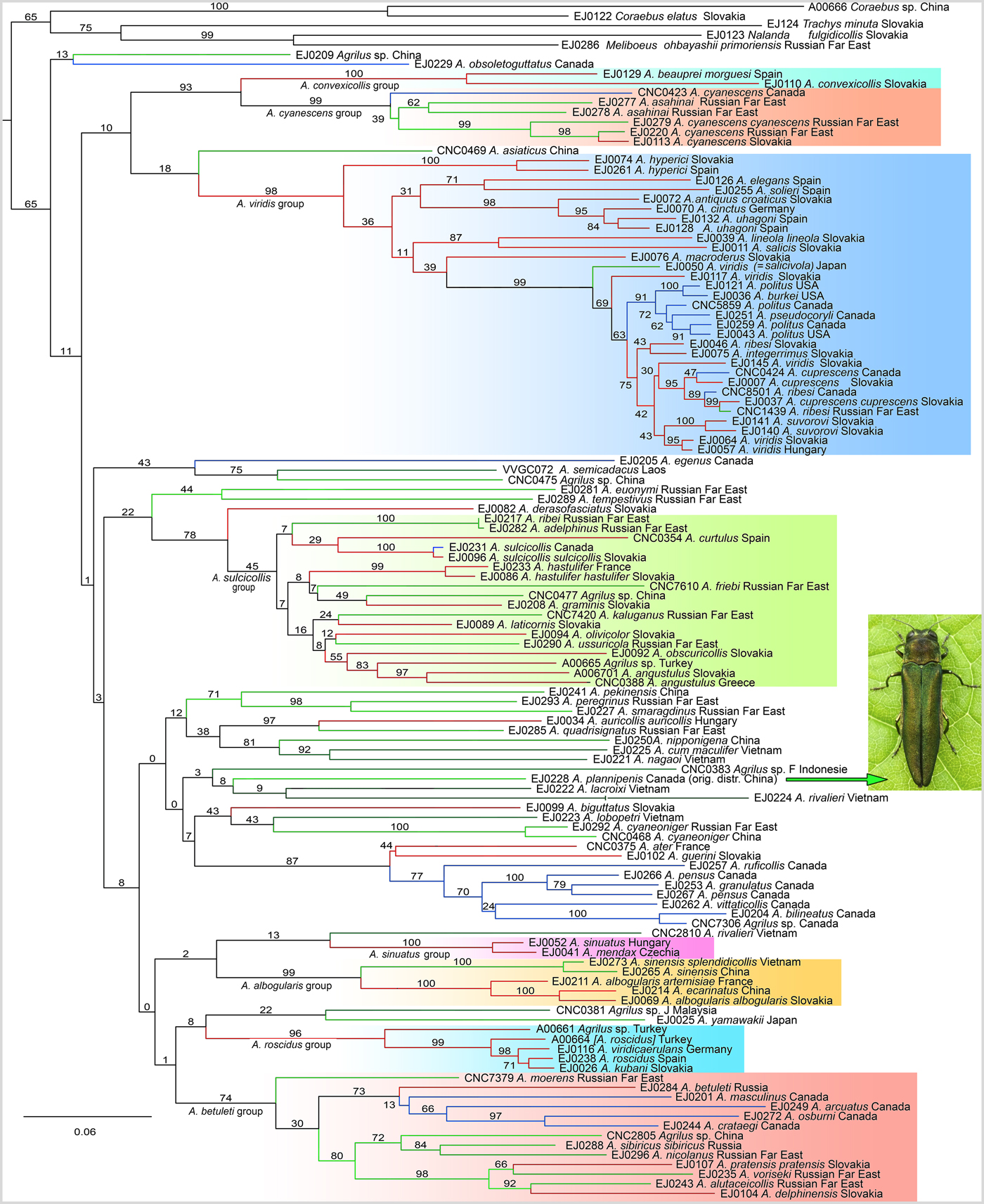
Fig. 4. The phylogram of the genus Agrilus recovered by Analysis 1. Numbers at internodes are bootstrap values; species groups are highlighted in different background colors; colors of ingroup branches correspond to geographical units (red: Europe, green: Asia, blue: North America).
The dichotomies corresponding to individual species and small groups of species are often statistically well-supported with BS >70% (fig. 4). Many morphologically pre-defined species were rendered paraphyletic by representatives of other nominal species. Thus, for example, A. asahinai from the Russian Far East is nested inside of A. cyanescens (fig. 4); the latter is a morphologically coherent species with a native Palaearctic distribution (also introduced to North America). Perhaps the most notable incongruence among the morphologically pre-defined species is depicted by the strongly supported (BS 99%) clade consisting of A. viridis (Linnaeus) (specimen EJ0050) and its sister-clade containing, in addition to the same nominal species, specimens of seven other species.
Depending on the method, the number of putative species identified using the cox1–5′ dataset varied between 104 and 171 (table 1 and fig. S2). The lowest number of putative species was recovered when the 3% uncorrected genetic distance threshold was applied, while the highest number was recovered when the putative species was inferred using bPTP (tables 1 and S3; same species always designated with the same letter of the combination of letters, numbers designate merged subsets of individuals). The number of putative species inferred from the rrnL and cox1–3′ datasets varied between 65 and 79, and between 78 and 85, respectively (tables 1 and S3).
Table 1. Comparison of the algorithmic and morphological identifications of Agrilus. Details in Supplementary table S3.

Discussion
Limitations of the analyses
This work is the first and perhaps too bold of an attempt to uncover the internal phylogenetic structure of the largest animal genus by using only three mitochondrial fragments sequenced somewhat inconsistently for ~3% of the known species diversity. The reported results herein, therefore, do not match in their clarity and statistical support those obtained in more thoroughly executed analyses of other mega-diverse animal genera (i.e. Maddison, Reference Maddison2012 on Bembidion Laterille; Breeschoten et al., Reference Breeschoten, Doorenweerd, Tarasov and Vogler2016 on Onthophagus Latreille). Besides, however, releasing sequence data and thus making them publicly available, our results are informative enough to draw a few preliminary conclusions.
First genus-wide DNA reference library of Holarctic Agrilus
The currently reported analysis is based on inexpensive and rapidly obtainable DNA sequences of three short mitochondrial fragments. The recent efforts to document and quantify diversity using molecular data generally follow two main directions. In the minimalistic approach, the short cox1–5′ mtDNA fragment (=DNA barcode) is mass-sequenced and deposited in the Barcode of Life Data System, which at present contains data on over 250,000 nominal animal species (barcodeoflife.org; Hebert et al., Reference Hebert, Ratnasingham and deWaard2003; Pentinsaari et al., Reference Pentinsaari, Hebert and Mutanen2014a). Alternatively, phylogenetic trees are normally produced using a higher number of markers (such as the entire mitochondrial genome) and might later be combined with densely sampled barcode databases (Bocak et al., Reference Bocak, Barton, Crampton-Platt, Chesters, Ahrens and Vogler2014; Zhou et al., Reference Zhou, Frandsen, Holzenthal, Beet, Bennett, Blahnik, Bonada, Cartwright, Chuluunbat, Cocks, Collins, de Waard, Dean, Flint, Hausmann, Hendrich, Hess, Hogg, Kondratieff, Malicky, Milton, Morinière, Morse, Mwangi, Pauls, Gonzalez, Rinne, Robinson, Salokannel, Shackleton, Smith, Stamatakis, StClair, Thomas, Zamora-Muñoz, Ziesmann and Kjer2016; Linard et al., Reference Linard, Crampton-Platt, Moriniere, Timmermans, Andujar, Arribas, Miller, Lipecki, Favreau, Hunter, Gomez-Rodriguez, Barton, Nie, Gillett, Breeschoten, Bocak and Vogler2018). Here, we combine with satisfactory results the DNA barcode data (444 fragments of cox1–5′ mtDNA) and two additional mtDNA fragments (166 fragments of rrnL and 170 fragments of cox1–3′ mtDNA) in the single dataset capable of resolving ~100 species, i.e. ~3% of Agrilus diversity. Although comparable to the 2.3% density of DNA samples available for the whole of Coleoptera (Bocak et al., Reference Bocak, Barton, Crampton-Platt, Chesters, Ahrens and Vogler2014), this sampling density is still scanty, particularly considering the geographical and biological spread of the genus Agrilus. With all these limitations, our results are first of their kind for the Holarctic representatives of the genus Agrilus and as such are the best available for the tree-based identification of unknown congeneric samples.
Agrilus diversity: species-groups and their relationships
Even though this study gives the first insight in the molecular phylogeny of a high number of Agrilus species, the statistical support for the basal-most dichotomies is virtually absent (figs 4 and 5). This is likely due to the DNA saturation effect, especially in the third positions of the protein-coding fragments. The robustly supported clades are few in the number of species usually containing 10 or fewer species. Even with these limitations, our topology (fig. 4) includes all major Agrilus species-groups of the Northern temperate zone and we suppose that inclusion in the analysis of an additional sample should, in most cases, lead to its reliable placement among the most closely related organisms (Bocak et al., Reference Bocak, Barton, Crampton-Platt, Chesters, Ahrens and Vogler2014; Zhou et al., Reference Zhou, Frandsen, Holzenthal, Beet, Bennett, Blahnik, Bonada, Cartwright, Chuluunbat, Cocks, Collins, de Waard, Dean, Flint, Hausmann, Hendrich, Hess, Hogg, Kondratieff, Malicky, Milton, Morinière, Morse, Mwangi, Pauls, Gonzalez, Rinne, Robinson, Salokannel, Shackleton, Smith, Stamatakis, StClair, Thomas, Zamora-Muñoz, Ziesmann and Kjer2016; Linard et al., Reference Linard, Crampton-Platt, Moriniere, Timmermans, Andujar, Arribas, Miller, Lipecki, Favreau, Hunter, Gomez-Rodriguez, Barton, Nie, Gillett, Breeschoten, Bocak and Vogler2018).
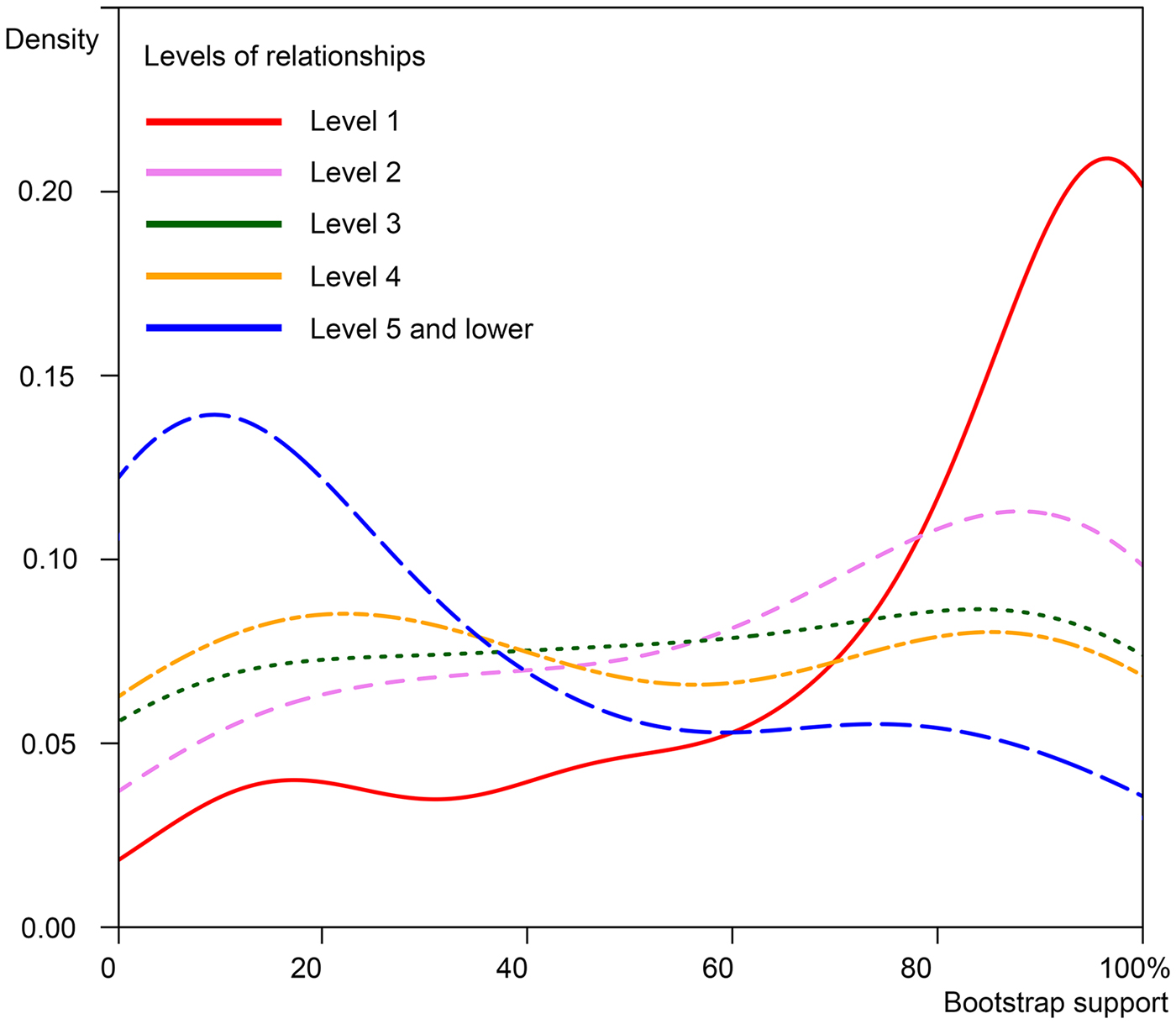
Fig. 5. Densities of bootstrap support for various levels of relationships in the phylogram depicted in fig. 4. Level 1 represents the sister-pairs; level 2 represents the sister-pair and an additional single species or another sister pair; further levels represent a clade of the previous level plus an additional species or a clade of the same or lower level.
The main Agrilus intrageneric lineages detected herein are marked on the tree (fig. 4) and correspond to the taxonomically recognized species-groups (or subgenera), shown in table S4 (Alexeev, Reference Riedel, Sagata, Suhardjono, Tänzler and Balke1988; Jendek & Grebennikov, Reference Jendek and Grebennikov2011). The sister-group relationship between the convexicollis- and cyanescens species-groups agrees well with the expectations based on the study of adult morphology. The viridis species-group (fig. 4) corresponds to three species-groups defined by Jendek & Grebennikov (Reference Jendek and Grebennikov2011) and includes widely distributed Palaearctic or Nearctic xylophagous species attacking pioneer trees like Salix, Populus, Betula, Alnus, Rubus, and Corylus. Nearctic members of this group (A. politus and A. burkei) are biologically most similar to the Euro-Siberian A. viridis and have likely dispersed from Asia to North America, as many other herbivorous beetles (Sota et al., Reference Sota, Bocak and Hayashi2008). The sulcicollis species-group is morphologically well defined and contains many species in the Palaearctic and Oriental regions developing on Quercus. This group is further distinct by its members having pronounced sexual dimorphism. The clade of the albogularis species-group is also well defined ecologically and morphologically comprising rhizophagous species of desert and semi-desert areas developing on Compositae, Amaranthaceae and Nitrariaceae. It is, therefore, fair to say that the herein recovered topology (fig. 4) presents the first but still very imperfect glimpse on the internal phylogenetic structure of the North Hemisphere Agrilus.
Limits of Agrilus species identification
The algorithmic DNA-based delimitation of species was proposed either as an identification tool when the taxonomic identity of unknown sample is inferred from the genetic distance to the identified reference samples (Hebert et al., Reference Hebert, Ratnasingham and deWaard2003; www.bold.org), or as a tool for identification of putative species for further studies when biological diversity is evaluated, but where the taxonomical identification is impossible or cost ineffective (Pons et al., Reference Pons, Barraclough, Gomez-Zurita, Cardoso, Duran, Hazell, Kamoun, Sumlin and Vogler2006; Vogler & Monaghan, Reference Vogler and Monaghan2007; Monaghan et al., Reference Monaghan, Wild, Elliot, Fujisawa, Balke, Inward, Lees, Ranaivosolo, Eggleton, Barraclough and Vogler2009). To develop such tools to the genus Agrilus, we compared two methods: (1) comparison of uncorrected pairwise distances (Meier et al., Reference Meier, Shiyang, Vaidya and Ng2006) and (2) the species delineating by application of the Bayesian Poisson Tree Process (bPTP; Zhang et al., Reference Zhang, Kapli, Pavlidis and Stamatakis2013).
The 3% genetic divergence was often considered as a limit for the delineation of a putative species using the barcoding marker (Hebert et al., Reference Hebert, Ratnasingham and deWaard2003; Blaxter, Reference Blaxter2004; Smith et al., Reference Smith, Woodley, Janzen, Hallwachs and Hebert2006); however, limits as low as 1% difference were also proposed (Ratnasingham & Hebert, Reference Ratnasingham and Hebert2007). We tested 2 and 3% thresholds and found that the levels of agreement between morphology and distance-based delimitations varied with individual species clades. The methods using genetic divergence generally overestimated the number of species. The algorithmic analysis of the DNA barcode dataset from 95 morphologically identified species indicated the presence of 104 and 115 species, using 3 and 2% threshold, respectively (table 1). Application of these thresholds for other mtDNA fragments generally estimates a lower species number, which is likely due to the lower genetic variability of these fragments (tables 1, S3).
Similarly, the bPTP analysis generally overestimated the number of species (tables 1, S3). The bPTP tends to over-split species-level clades into a higher number of putative species and, therefore, the analysis of the barcode dataset indicated the presence of 171 species, compared with 95 identified using traditional morphology. With the application of both methods, conspecific populations having distant geographic origin were identified as separate putative species, similar to the results of other authors (e.g., Bergsten et al., Reference Bergsten, Bilton, Fujisawa, Elliott, Monaghan, Balke, Heindrich, Geijer, Herrmann, Foster, Ribera, Nulsson, Barraclough and Vogler2012; Li et al., Reference Li, Gunter, Pang and Bocak2015). The dense sampling of populations from the whole range is the only way to identify cases where geographic distance is responsible for increased genetic divergence within a population group that is still connected by gene flow. Concerning our results, we cannot recommend a single threshold for species delimitation in Agrilus.
We found numerous method-dependant disagreements in the species identifications. Several species were represented by multiple unrelated clades. Although we cannot exclude misidentifications and introgressions (Pentinsaari et al., Reference Pentinsaari, Mutanen and Kaila2014b), the recovered relationships might at least in some cases indicate hidden diversity. For example, A. viridis reared from Tillia (fig. S1) represents a distant lineage in regard to the rest of this species. Such a distant position was inferred already by previous authors (Pentinsaari et al., Reference Pentinsaari, Mutanen and Kaila2014b) and ascribed to introgression. As various distant populations of A. viridis from Tillia show genetic diversification, cryptic diversity might be the case here. The current concept of A. viridis makes this taxon the most polyphagous assemblage with 49 species-level host plants (Jendek & Poláková, Reference Jendek and Poláková2014). Additionally, A. viridis has an extremely large geographic range and various populations of adults active in different periods from April to September and in altitudes ranging from sea level to about 1600 m (Jendek & Grebennikov, Reference Jendek and Grebennikov2011). All this suggests that this nominal species might perhaps be better split into many. Prior to any taxonomic actions, however, much denser sampling should be analysed. Another example is A. ribesi, which is represented by three distantly related populations (fig. 4): one from Slovakia, one from the Russian Far East (which might represent an introduced population from Europe), and one from Canada, which was reported as an invasive species (Jendek et al., Reference Jendek, Grebennikov and Bocak2015). The third example is formed by two terminals of A. angustulus from Greece (fig. S1), representing generically dissimilar lineages and separate species. Both genetic introgression and underestimated species diversity might account for the observed results.
Another common case of a conflict between DNA-based species limits and morphologically defined species are clades containing a single species with genetic divergence markedly higher than one might expect in a group of interbreeding populations with an effective gene flow. These include A. cyanescens with several genetically distinct populations from Russia, Canada and Central Europe (figs 4m S1, table 1), which are genetically very distant (17.3 and 8.1% in cox1–5′ and cox1–3′, respectively). The geographic distance precluding effective gene flow is one possible explanation; however, a much denser sampling of Siberian populations is needed to test this hypothesis. Two populations of A. hyperici from the western Mediterranean and from Central Europe are also genetically quite distant to each other (7.9 and 6.3% in cox1–5′ and cox1–3′, respectively) and they were recovered as separate species in algorithmic delimitations (table S). Also A. uhagoni Abeille de Perrin consists of two notably dissimilar lineages, both developing on Genista and both from Spain (fig. 4). Either the genetically diversified populations of A. uhagoni with an independent evolutionary history may sympatrically co-occur in Spain, or these samples are misidentified due to the variability of the diagnostic morphological characters. High genetic diversity was also identified in Canadian and US populations of A. politus (Say) (fig. 4), but without any sign of the geographic structure. Their maximum genetic distance reached 5.5, 5.2 and 4.4%, in cox1–5′, cox1–3′, and rrnL, respectively. A few other similar examples might be detected on our topologies (such as A. albogularis Gory, A. roscidus Kiesenwetter, A. cyaneoniger Thomson, figs 4, S1, S2).
Many of the above-mentioned species were recovered as paraphyletic in our analyses by having other morphologically delimited species nested within them. A delayed lineage sorting is a possible explanation for the observed conflicts between morphology- and DNA-based species delimitation. The species rendering other species paraphyletic include: (1) A. antiquus croaticus Abeille de Perrin and A. cinctus (Olivier) nested within A. uhagoni; (2) A. pseudocoryli Fisher and A. burkei Fisher nested within A. politus; (3) A. suvorovi Obenberger nested within A. viridis; (4) A. perisi Cobos nested within A. angustulus; (5) A. viscivorus Bílý and A. kubani Bílý nested within A. roscidus; (6) A. pensus Horn nested within A. anxius Gory; (7) A. ecarinatus Marseul nested within A. albogularis. Many of these cases might suggest imperfect taxonomy. The taxonomic status of A. burkei is unclear and sometimes it is considered intraspecific with A. politus. The only character to distinguish A. ecarinatus is its small body size. We can hypothesize that A. kubani and A. viscivorus are in fact independent species having different host plants, Loranthus europaeus and Viscum album, respectively. In other cases we are unable to account for the observed conflict and, therefore, they require further investigations.
Concluding remarks
The comprehensive phylogeny of Agrilus remains unattainable due to the extreme diversity of this clade, practical rareness of species, inaccessibility of many regions and the shortage of trained human capacity to document such enormous biological diversity. Nevertheless, the herein released DNA database of several hundred samples and about 100 species provides the first dataset likely capable to identify an unknown Agrilus specimen from the northern temperate zone of the Globe. This has significant practical utility, since considering the history of Agrilus invasions (Hoebeke et al., Reference Hoebeke, Jendek, Zablotny, Rieder, Yoo, Grebennikov and Ren2017), this region contains the highest number of economically important and potentially harmful species. Our results convincingly demonstrate that, unlike the minimalistic DNA barcode approach, the multi-marker phylogenetic analysis has a notably higher potential to place an unknown sample among the phylogenetically closest and taxonomically identified relatives. Although morphological misidentifications and sequencing errors cannot be excluded, the relatively high number of disagreement between morphology- and DNA-based species limits calls for closer and more detailed investigation of such cases. Overall, and at the very least, our work is the first attempt to tackle the phylogenetic structure of what currently is the largest conventionally accepted genus of organisms on Earth, the Agrilus jewel beetles.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0007485318000330
Acknowledgements
Individuals listed in table S1 made available Agrilus specimens for this study. Renata Bilkova (Olomouc, Czech Republic) provided technical support in the laboratory. This project was partly funded by the grant CIGA No. 20174312 of the Czech University of Life Sciences Prague, Faculty of Forestry and Wood Sciences, by the Czech Science Foundation (grant P506/11/1706 to LB) and the Faculty of Science, UP Olomouc (grant IGA-PrF to IK). Karen McLachlan Hamilton and Bradley Sinclair (both Ottawa, Canada) critically read an earlier draft of this paper.







