Introduction
Rat lungworm (Angiostrongylus cantonensis) is a zoonotic, pathogenic nematode and the causative organism for the clinical condition known as neuroangiostrongyliasis or rat lungworm disease. Like most nematodes, A. cantonensis has four distinct larval stages (L1–L4) and a sub-adult stage (L5). Each stage is designated by the number of times the larva molts its cuticle in its lifetime (Wharton, Reference Wharton and Carlow1986). The development from L1 to L3 takes place in an intermediate gastropod host. Development from L3 to L5, as well as reproduction by adult nematodes and production of eggs and L1, takes place in the definitive rat host. The L3 stage is the only stage known to infect vertebrate hosts, including the definitive (rat) host and accidental hosts such as humans (Mackerras and Sandars, Reference Mackerras and Sandars1955). Transmission to humans is thought to be due to ingestion of L3 larvae in infected gastropods, on improperly washed produce (Heyneman and Lim, Reference Heyneman and Lim1967; Wang et al., Reference Wang, Lai, Zhu, Chen and Lun2008) or in contaminated water (Cheng and Alicata, Reference Cheng and Alicata1964; Richards and Merritt, Reference Richards and Merritt1967; Crook et al., Reference Crook, Fulton and Supanwong1971; Howe et al., Reference Howe, Kaluna, Lozano, Torres Fischer, Tagami, McHugh and Jarvi2019). The invasive semi-slug, Parmarion martensi, is an extremely efficient host and is thought to be the primary cause of the increase in severe neuroangiostrongyliasis in Hawaii since its introduction in 2004 (Hollingsworth et al., Reference Hollingsworth, Kaneta, Sullivan, Bishop, Qvarnstrom, da Silva and Robinson2007; Jarvi et al., Reference Jarvi, Howe and Macomber2018). Due to the potential severity of the disease in Hawaii, the prevention and treatment of neuroangiostrongyliasis have a renewed sense of urgency.
At the forefront of prevention and treatment of neuroangiostrongyliasis, research is needed to identify ways to disinfect contaminated produce and water as well as verifying the effects of anthelmintic drugs on A. cantonensis L3 larvae. New technology offers improved methods over those historically used in determining nematode viability. This type of study requires the researcher to determine whether larvae are killed after treatment and are therefore non-infective. If a larva moves under the microscope, it must be alive, and therefore has at least the potential of infectivity; but if it is not moving, there is currently no reliable in vitro assay to determine that the larva is truly dead and therefore non-infective. Sano et al. (Reference Sano, Terada, Ishii, Kino and Hayashi1981) developed a method to study in vitro effects of various chemical substances based on the motility of parasitic nematodes such as A. cantonensis, Angiostrongylus costaricensis and Dirofilaria immitis (dog heartworm). The study documented the paralytic effect of various anthelmintics, such as avermectin B1a, ivermectin, diethylcarbamazine and pyrantel, on A. cantonensis (reviewed in Mentz and Graeff-Teixeira, Reference Mentz and Graeff-Teixeira2003). Terada et al. (Reference Terada, Rodriguez, Dharejo, Ishii and Sano1986) evaluated the in vivo sensitivity of 19 different anthelmintics on A. cantonensis and A. costaricensis using larval motility as a measure of drug sensitivity. They found that hexylresorcinol, bithionol, niclosamide and levamisole significantly decreased the motility of A. costaricensis while A. cantonensis remained active and that piperazine, avermectin B1a and ivermectin significantly decreased the motility of A. cantonensis while minimally affecting the motility of A. costaricensis. Conclusions based on the motility of nematodes can be misleading because inactive or sessile behaviours in nematodes are common regardless of exposure to any substance (Kusnawidjaja, Reference Kusnawidjaja1960; Anderson et al., Reference Anderson, Gould, Ingham and Coleman1979). In addition, nematodes may appear to be immobilized by exposure to sub-lethal doses of a test substance when the test substance itself may not possess a larvicidal or nematocidal effect but may induce paralysis or an anaesthetic effect from which the larvae may recover with time (Chandel, Reference Chandel2002).
The gold standard to determine the efficacy of a substance against A. cantonensis is to infect rodents with substance-treated larvae and monitor for infection (Crook et al., Reference Crook, Fulton and Supanwong1971). Zanini and Graeff-Teixeira (Reference Zanini and Graeff-Teixeira2001) isolated A. costaricensis L3 larvae from slugs (Phyllocaulis soleiformis) by digestion with 30% (w/v) pepsin-0.7% HCl solution for 2 h at 37 °C. The isolated larvae were then treated with 1.5% bleach, vinegar and saturated cooking salt. Treated larvae were then administered into Swiss mice. Mice were euthanized and autopsied, showing that the 1.5% bleach treatment was the most effective, with one adult worm recovered from 1/35 mice. In the vinegar-treated group, 29 adult worms were recovered among 32 mice, and 90 adult worms were recovered from the salt-treated group from 21 mice. Crook et al. (Reference Crook, Fulton and Supanwong1971) isolated A. cantonensis L3 larvae by drowning giant African snails (Achatina fulica). The isolated larvae were then treated with iodine solution (~15 mg L−1 of water), and the treated larvae were then fed to rats. After 21 days, a reduced number of L5 larvae were isolated from the rats infected with iodine-treated larvae compared to controls, but the iodine treatment did not completely kill all larvae.
It is the standard practice to challenge rats in order to determine post-treatment larvae viability. However, the use of in vivo studies for every test substance of interest is not practical due to the time, expense and care requirements for the rats, as well as the mandate to minimize live animal use under animal welfare guidelines. Development of an in vitro viability assay would provide the opportunity to evaluate the sensitivity of A. cantonensis to a multitude of test substances or physical treatments. Ferreira et al. (Reference Ferreira, Mendes, Bueno, de-Araújo, Bartholomeu and Fujiwara2015) demonstrated differential staining of Caenorhabditis elegans, a free-living non-parasitic nematode, using propidium iodide (PI). They determined the IC50s of two widely used, broad-spectrum anthelmintics, albendazole and ivermectin, using fluorescence from the bound PI as the indicator of death. PI is a fluorescent stain that is excited by green light (535 nm) and emits red florescence (617 nm). The cationic nature of the PI molecule makes it membrane impermeable (due to the positive charge on the extracellular membrane surface of cells). In order for PI to enter a cell, the integrity of the cell membrane must be sufficiently compromised, i.e. when the cell is dead. Staining occurs when PI intercalates with the DNA non-specifically. After binding to DNA, the fluorescence of PI is enhanced 20- to 30-fold, making the difference between background signal and unbound stain negligible (Gilbert and Ehrenstein, Reference Gilbert and Ehrenstein1984; Zhao et al., Reference Zhao, Oczos, Janowski, Trembecka, Dobrucki, Darzynkiewicz and Wlodkowic2010). Although PI is widely used in culture cell viability assays to identify dead cells (Zhao et al., Reference Zhao, Oczos, Janowski, Trembecka, Dobrucki, Darzynkiewicz and Wlodkowic2010) including cells of C. elegans (Ferreira et al., Reference Ferreira, Mendes, Bueno, de-Araújo, Bartholomeu and Fujiwara2015), the efficacy of PI in A. cantonensis (L3) is unknown. In this study, we evaluate a PI-based in vitro viability assay for L3 A. cantonensis larvae and then validate this assay in a rat model.
Methods
Rats
Wistar IGC outbred laboratory strains of Rattus norvegicus (n = 40) were obtained from Charles River Labs (Raleigh, NC, USA) and housed in individual 21 × 47 × 26 cm polycarbonate shoebox cages (per ILAR 2011) at the USDA-APHIS Wildlife Services National Wildlife Research Center (NWRC) Hawaii Field Station, Hilo, HI, USA. Rats were maintained on Laboratory Rodent Diet 5001 (LabDiet, St. Louis, MO, USA) and provided water ad libitum. Rats were allowed to acclimate for 7–10 days prior to the start of the study. Individual animals were identified with a cage card which identified the animal number. Rats were 7 months old at the start of the study. All procedures in this study were completed in accordance with the USDA-Animal Plant Health Inspection Service-National Wildlife Research Center, which approved this study (QA-2879) (see Ethical standards).
Study design
Each rat received 1 mL of dH2O containing 50 larvae and the uninfected control group received 1 mL of dH2O without larvae. Treatment groups were: (1) live-stained larvae (n = 5 rats); (2) live-unstained larvae (n = 5 rats); (3) killed-stained larvae (n = 10 rats); (4) killed-unstained larvae (n = 10 rats); and (5) uninfected controls (n = 5 rats). All rats were monitored daily, and body weights were recorded weekly. As the spleen is a lymphatic organ, spleen weights were recorded at the time of necropsy to evaluate any difference in mass between groups.
Angiostrongylus cantonensis larvae collection
Parmarion martensi are semi-slugs known to have high infection levels of A. cantonensis in Hawaii (Hollingsworth et al., Reference Hollingsworth, Kaneta, Sullivan, Bishop, Qvarnstrom, da Silva and Robinson2007). Semi-slugs were collected from the University of Hawaii at Hilo campus and immersed in 50 mL dH2O in 50 mL Falcon tubes for 72 h. The bottom 15 mL from each tube was transferred to a culture plate (100 mm × 15 mm) (Howe et al., Reference Howe, Kaluna, Lozano, Torres Fischer, Tagami, McHugh and Jarvi2019) and observations were made under a dissection microscope (Wild Heerbrugg M4A APO) at 10–40× to detect the presence of larvae. Live larvae with S and Q swimming movements attributed to A. cantonensis L3 larvae (Lv et al., Reference Lv, Zhang, Zhang, Steinmann, Zhou and Utzinger2009) were collected using a 20 µL micropipette and transferred into 0.5% pepsin-0.5% HCl (pH 1.41) solution for 30 min at room temperature (Ash, Reference Ash1970; Wang et al., Reference Wang, Chao and Chen1991). Larvae were then transferred into a 100 mm × 15 mm culture plate containing 30 mL of dH2O using a micropipette; this preparation served as the larval stock.
Larvae preparation and staining
Larvae preparations (n = 34) consisted of 50 hand-counted larvae in 100 µL dH2O from the larval stock which were added to 900 µL of dH2O in 1.5 mL Eppendorf tubes. Live larvae replicates (n = 6 stained, n = 6 unstained) were prepared as described above. Killed larvae preparations (n = 11 stained, n = 11 unstained) were made by adding 50 larvae in 100 µL dH2O from the larval stock into 900 µL of 100% methanol, followed by freezing at −80 °C for 30 min (Ferreira et al., Reference Ferreira, Mendes, Bueno, de-Araújo, Bartholomeu and Fujiwara2015). Methanol was removed by centrifugation at 2000 rpm for 5 min, ~900 µL supernatant removed and replaced with 800 µL of dH2O resulting in a final volume of 900 µL. This washing procedure was repeated three times.
PI (Biotium, Freemont, CA, USA) was used to make a 1.25% or 8.7 mm stock solution in dH2O. One hundred microlitres of the stock PI solution was added to each of the 11 tubes/replicates in the stained killed larvae group and to the six tubes/replicates in the stained live larvae group, resulting in a final volume of 1 mL in each tube/replicate. For the unstained larvae groups, dH2O was used instead of PI. All tubes were incubated for 30 min at room temperature. The excess (unbound) PI was removed by following the dH2O washing procedure as described above for removing methanol. For visual confirmation of staining, one tube/replicate from each group was picked at random, centrifuged at 2000 rpm for 5 min and the bottom 100 µL was transferred into individual wells of a black, transparent-bottom view plate (#384, PerkinElmer, Waltham, MA, USA). Imaging was conducted on a high content imaging system (Operetta, Perkin Elmer, USA) with a 20× long working distance lens using brightfield, PI and fluorescein filters.
Viable fluorescent stains Hoechst 33 342 (Life Technologies Corporation, USA), calcein AM (Invitrogen, USA), acridine orange (Acros Organics, USA) and DAPI (Sigma-Aldrich, USA) (Kusnawidjaja, Reference Kusnawidjaja1960; Anderson et al., Reference Anderson, Gould, Ingham and Coleman1979) were tested for their effectiveness in staining live A. cantonensis larvae. Tests were conducted in a 384-well plate (PerkinElmer). Each test involved 30 larvae in triplicate in tap water (100 µL well−1) with final concentrations of 1, 0.8, 0.5, 0.2 and 0.1 mm of each stain. Plates were subject to room temperature incubation for 24 h. Visualization was conducted by fluorescent microscopy (Leica, DMIL LED, Germany) and Operetta.
Autofluorescence
To determine the extent to which L3 A. cantonensis larvae autofluoresce, dead larvae were prepared as described above and live larvae were treated with 15 mm ivermectin pharmaceutical secondary standard prepared using 2% DMSO-dH2O (Sigma-Aldrich) to reduce their motility for imaging. Lambda scans were performed (n = 10) using a Leica confocal microscope (SPE). Excitation was evoked using a 488 nM laser and subsequent emission was detected through a beam splitter with a reflection/transmission ratio of 30/70. For quantification of the different samples, a dichroic beam splitter was used that cut-off emission at 488 and 635 nM. For comparative analysis, the emissions of different samples were acquired simultaneously and quantified between 525 and 535 nM.
Rat gavage
Rats were sedated with an isoflurane-oxygen vaporizer/regulator in an induction chamber and larvae preparations were gavaged into the distal oesophagus of the rats based on the appropriate assigned treatment groups. Sterile 13 gauge, 90 mm long flexible plastic gavage tubes (Instech Laboratories, Inc., Plymouth Meeting, PA, USA) and 1 mL sterile tuberculin syringes (Norm-ject, Tuttlingen, Germany) were used to deliver the 1 mL A. cantonensis larvae dose or water only dose to each rat.
Necropsy
Rats were held for 6 weeks and humanely euthanized in a CO2 chamber. Up to 2 mL of whole blood samples were collected via cardiac puncture post-euthanasia.
Selected organs (brain, heart, lung and spleen) were dissected and placed into individual culture plates to be examined under a microscope for the presence of adult worms, decaying larvae or L3 stage A. cantonensis. Dissection tools were used for each rat after being disinfected and decontaminated of DNA by a ⩾10 min soak in a 1:10 commercial bleach containing 6.0% (v/v) sodium hypochlorite (Clorox, Oakland CA, USA) (Prince and Andrus, Reference Prince and Andrus1992; Besselsen, Reference Besselsen and Meltzer1995). The heart and lungs of each rat were examined using fine-tip forceps under Olympus or Leica 10–40× dissection microscope for extraction of adult worms. Nematodes recovered were sexed, recorded and washed 3× in cold 1× PBS with 1× protease inhibitor (Halt™ Protease and Phosphatase Inhibitor Single-Use Cocktail EDTA-free, Thermo Fisher Scientific, Waltham, MA, USA) before being placed in pre-weighed tubes containing the same solution on ice. Nematodes were then stored frozen at −80 °C for future studies. Spleen weights were recorded because the spleen is a lymphatic organ and can serve as an indicator of infection. Finally, ~250 mg of bilateral cortex and spinal cord (bregma ~+1.08 to −5.28 mm, and posterior to ~15.00 mm, respectively) (Paxinos and Watson, Reference Paxinos and Watson2007) and right upper lung lobe from each rat were collected into 2 mL screw cap tubes containing 500 mL DNA lysis buffer (0.1 M Tris-HCl, 0.1 M EDTA, 2% SDS) and stored at −80 °C. Tissue samples were stored frozen until DNA was extracted for PCR. DNA extractions and real-time PCR were carried out as described (Jarvi et al., Reference Jarvi, Farias, Howe, Jacquier, Hollingsworth and Pitt2012; Howe et al., Reference Howe, Kaluna, Lozano, Torres Fischer, Tagami, McHugh and Jarvi2019).
Data summary and statistics
The Anderson–Darling normality test was used to determine the distribution of data for each treatment group for spleen weight and difference in body weight between the start of the trial and upon necropsy. Differences in normally distributed data were evaluated with t-tests, Mann–Whitney U tests were used for data not normally distributed with two groups, and Kruskal–Wallace for data not normally distributed evaluating the five treatment groups. All statistics were completed using Minitab 18 (Minitab Inc., 2010).
Results
Rats
The number of male and female rats assigned to each treatment group was: treatment group (1) stained live larvae (3 male/2 female rats); (2) unstained live larvae (2 male/3 female rats); (3) stained killed larvae (5 male/5 female rats); (4) unstained killed larvae (5 male/5 female rats); and (5) uninfected controls (3 male/2 female rats). A total of 35 rats were included in the study.
Larvae and staining
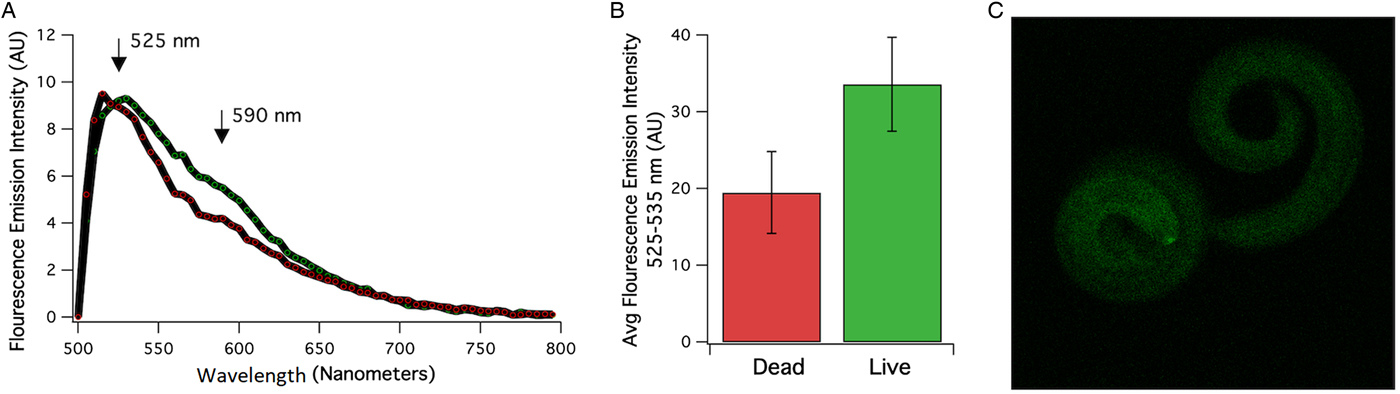
A total of 1700 A. cantonensis larvae were isolated from a single P. martensi which was estimated to have been infected with over 5000 larvae. A lambda scan was conducted to assess the autofluorescent properties that dead and live larvae may display using an excitation laser of 488 nM and the evoked emission was recorded between 500 and 795 nM using confocal acquisition (Fig. 1). The emission spectra of dead and live larvae that were acquired simultaneously in the same field of view which resulted in a maximum emission at ~525 nM and a secondary peak at 590 nM are shown in Fig. 1A. In order to display the entire spectrum between 500 and 795 nM, the emission was evoked and recorded through a beam splitter with a reflection-to-transmission (RT) ratio of 30 to 70%, respectively. The use of an RT beam splitter results in reduced excitation and emission properties. In order to resolve emission differences between live and dead larvae, a dichroic beam splitter was used with bandpass cut-off at 488 and 635 nM. Indeed, the increase in excitation and emission displayed the higher fluorescent properties that live (right) larvae displayed compared to their dead (left) counterparts (Fig. 1B). An example image of dead and live larvae of the acquisitions that were quantified is shown in Fig. 1C.
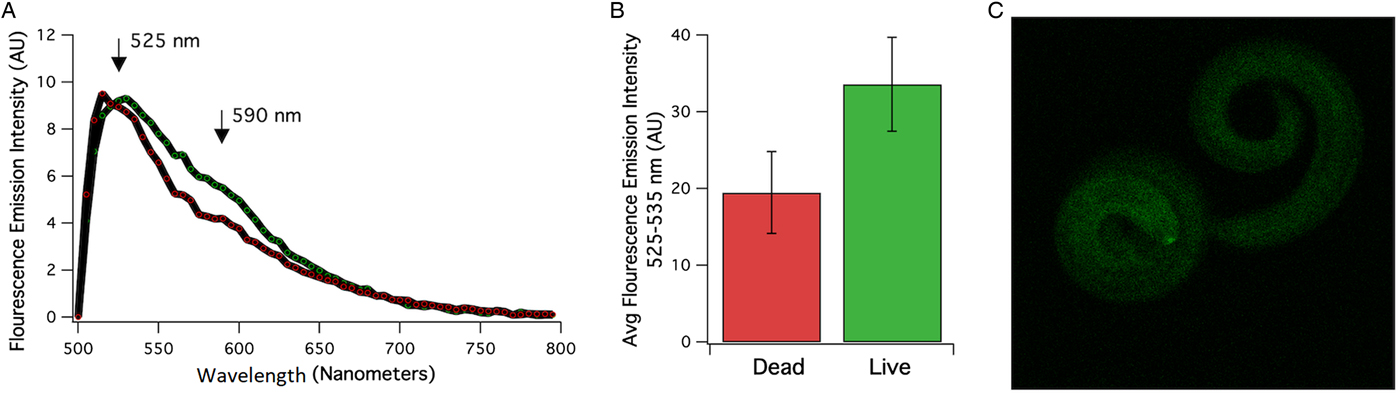
Fig. 1. (A) Averaged emission spectrum of the dead (red) and live (green) larvae. Emission between 500 and 795 nM was detected using a laser scan confocal microscope with excitation at 488 nM and a beam splitter with a reflection/transmission ratio of 30/70. A difference in emission intensity between dead and live larvae was quantified using a dichroic beam splitter and emission was recorded between 525 and 535 nM. (B) Averaged emission intensity at 525–535 nM of live and dead larvae that were recorded in the same field of view (n = 9–11) using dichroic beam splitters at 488 and 635 bandwidths. (C) Examples of fluorescence emitted at 534 nM from live (left) and dead (right worm).
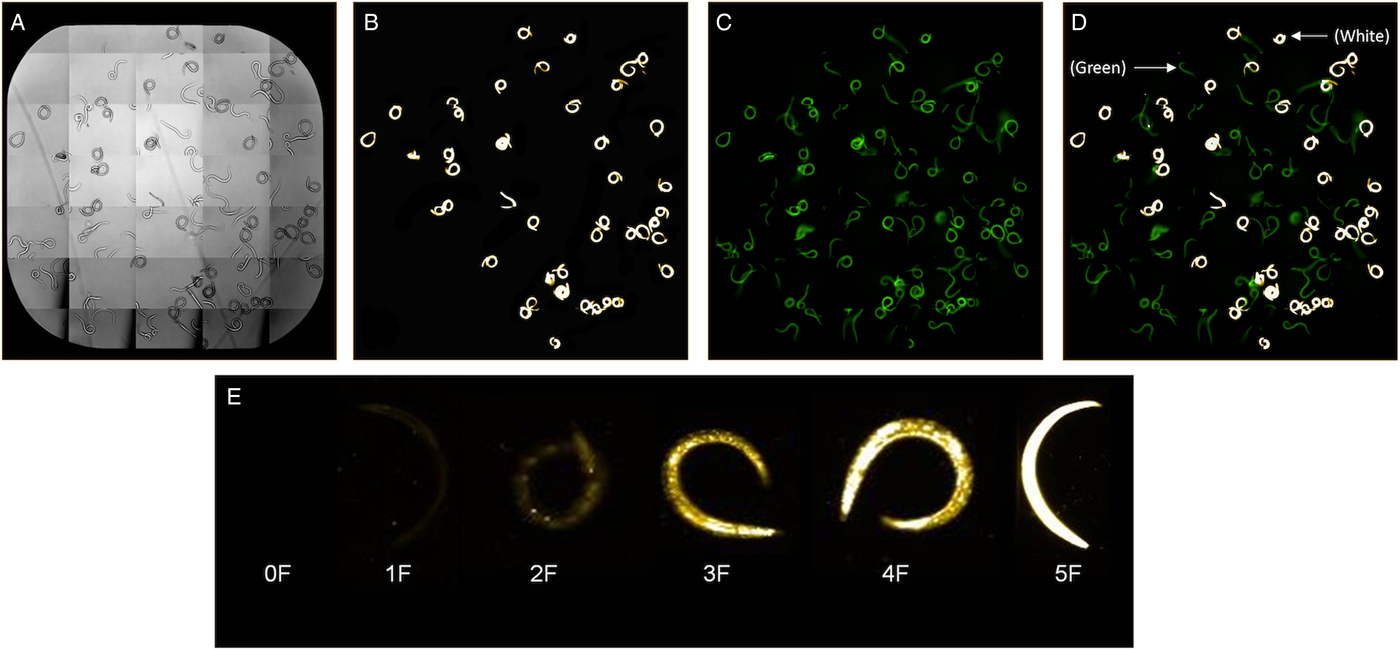
Larvae were prepared and stained as described above and images produced by the Operetta after staining live and dead larvae are shown in Fig. 2. Images were collected using either brightfield (Fig. 2A), PI filter (Fig. 2B) in which excitation occurs at 535 nm and emission at 617 nm, or both PI and fluorescein filters (Fig. 2C) (excitation occurs at 400 nm and emission at 550 nm). The fluorescein filter on the Operetta or use of a filter with fluorescein range on the fluorescent microscope allowed detection of autofluorescent live larvae, which appear green (Fig. 2C). Larvae appear bright yellow or white with the filter for detecting PI-stained larvae using a colour setting of #FFCEOO on the Operetta (Fig 2B and C). Regardless of staining, all live larvae appeared green under the fluorescein filter and all killed larvae appeared white or yellow under the PI filter. Larvae may show varying levels of PI fluorescence over time as they die (Fig 2D and E).
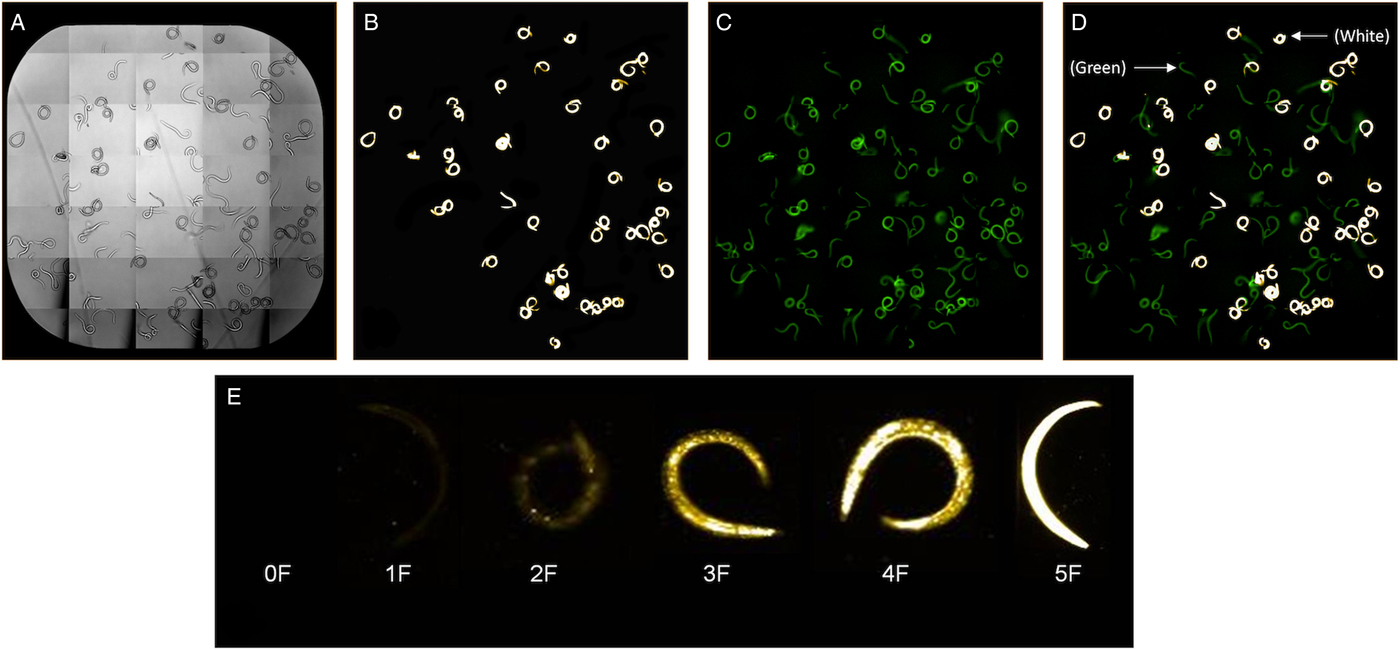
Fig. 2. Larvae visualization on the Operetta High-Content Imaging System under brightfield (A), propidium iodide (PI) filter (B), fluorescein filters (C) and PI and fluorescein filters (D) using a PI channel colour setting of #FFCE00. Killed larvae after PI staining appear bright white and autofluorescence of live and killed non-PI stained larvae appear green under the fluorescein filter (D). Live larvae may show varying levels of PI fluorescence over time as larvae die and categorization of PI stained larvae from 0F with no fluorescence to 5F with maximum fluorescence is shown in (E).
No staining occurred in larvae exposed to calcein AM for 24 h (data not shown), and only moderate staining occurred when larvae were exposed to Hoechst 33 342 for 24 h of incubation. Strong staining occurred when larvae were exposed to acridine orange, but there were also high levels of background fluorescence from the unbound stain. Additionally, all the larvae appeared dead after 12 h of incubation with acridine orange.
Body and spleen weights
Pre-treatment body weight of rats (prior to gavage) was recorded at the start of the trial and recorded weekly thereafter for 5 weeks, and post-euthanasia body weight was recorded at week 6. Body weight consistently increased over the course of the trial. We conducted a Kruskal–Wallis test to evaluate the overall change in body weight between treatments and found no significant differences (P = 0.931) when testing included both males and females (data not shown). Since male weight was consistently greater than female weight, we completed subsequent analysis within each treatment group on males and females separately. We calculated the per cent change in body weight as: (post-euthanasia weight minus pre-treatment weight) divided by pre-treatment weight ×100. Within each treatment, we found a significant difference between males and females (P < 0.001) in per cent change in body weight, with males having greater body weight increases than females (data not shown).
Spleen weight data, which included males and females, had a normal distribution, so one-way analysis of variance and t-tests were conducted. Significant differences were found among groups (P = 0.035) and individual comparison t-tests were run on all possible combinations of groups to locate the significant comparison(s). Only the spleen weights of the control group vs unstained live group were significantly different (P = 0.039), with values for the control vs stained live (P = 0.074) and the unstained killed vs stained live (P = 0.077) group comparisons trending towards significance. In all cases, mean spleen weights were greater in the live (stained or unstained) groups than in those of the killed or control groups. To determine if spleen weight was correlated with body weight, we evaluated spleen weight as a percentage of body weight as: (spleen weight divided by post-euthanasia body weight) × 100. A Mann–Whitney U test was used since only females' data (not males' data) were normally distributed. Females showed a significantly higher spleen weight in proportion to their body weight than males (P < 0.001) even though mean spleen weight of males was significantly greater (2.809) than the mean spleen weight of females (2.399) (P < 0.001).
Adult worm isolation
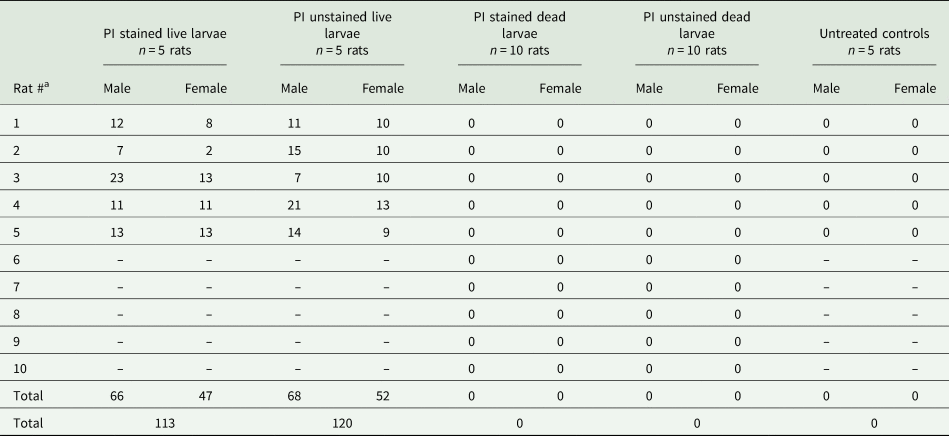
Rats were euthanized at approximately 6 weeks' post-gavage (days 42–43). On the 40th day after gavage, one of the female rats from the live-unstained group was found dead in the cage. This rat exhibited normal weekly body weight gains and appeared healthy and active 24 h prior to its demise. No signs of external trauma or bleeding were observed. This rat was not eliminated from the study because it was only 2 days away from the completion of the study. This rat was stored in −20 °C freezer until the day of necropsy and 23 adult worms (both male and female) were collected from the pulmonary artery of this particular rat. Adult A. cantonensis were isolated from the hearts and lungs of all rats receiving live (stained or unstained) larvae (Table 1). No significant difference in the number of adult worms was found between the PI stained live group (113 adults/~250 larvae, 45.2%) and the PI unstained live group (120 adults/~250 larvae, 48%) (P = 0.797). A total number of 234/~500 (46.8%) adult A. cantonensis were isolated from the rats that received live larvae. The mean number of adult male worms isolated per individual rat was 13.4, and 9.9 per adult female worms. No significant differences were observed between the total number of male vs female adult worms isolated (P = 0.094). Adult A. cantonensis were not found in any group that received killed larvae.
Table 1. Counts of male and female adult A. cantonensis recovered from study animals 42–43 days post-gavage with 50 nematodes
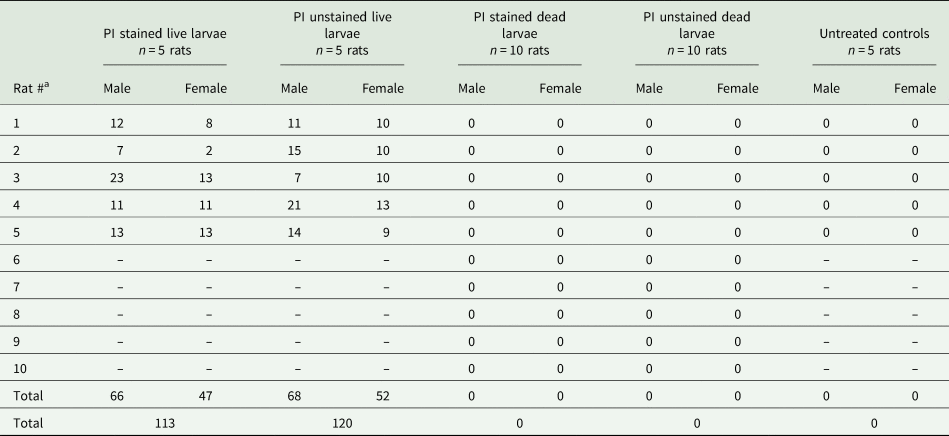
Angiostrongylus cantonensis were only found in the live larvae (stained and unstained) treatment groups. Untreated controls received only dH2O.
a Respective to treatment group.
Effects of A. cantonensis on lungs and brain
In previous studies, granulation was observed in the A. cantonensis-infected rats in the upper right lung lobe (Jarvi et al., Reference Jarvi, Quarta, Jacquier, Howe, Bicakci, Dasalla, Lovesy, Snook, McHugh and Niebuhr2017). This granulation pattern was observed in all rats that received live larvae but was not seen in rats that received killed larvae or in the control rats.
Examination of the brain revealed blood clots and damaged vessels in 8/10 (80%) of the rats that received live larvae (both stained and unstained; data not shown); however, all of the rats that received killed larvae had brain tissue that appeared healthy. Eggs were seen during lung examination of 3/5 live PI stained group as well as 1/5 of the live unstained group, whereas eggs were not seen in the lungs of rats that received killed larvae. L1 larvae were observed in the feces of 2/10 rats that received live larvae (from one rat in each stained and unstained live groups). L1 larvae were not observed in the feces of the rats that received killed larvae.
Real-time PCR
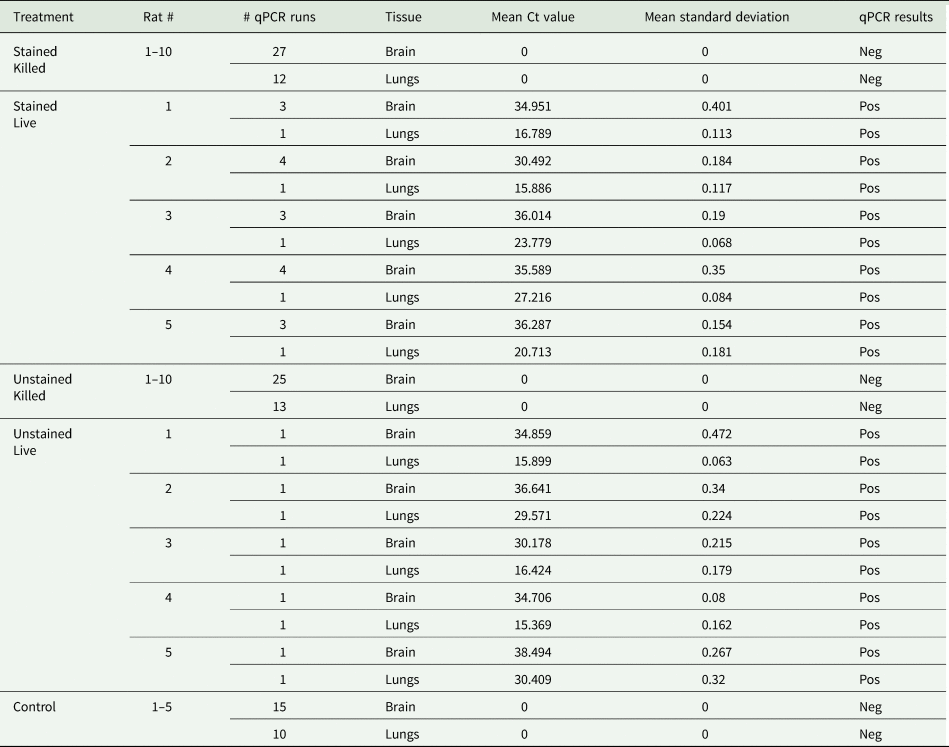
Brain and lung tissues from rats that received live larvae were positive for A. cantonensis DNA, whereas brain and lung tissues from rats that received killed larvae were negative for A. cantonensis DNA (Table 2). Samples were run in triplicate and data were included only when the standard deviation among replicates was <0.5. The cycle threshold (CT) was set to 0.25 throughout all runs. Results of the PCR reactions of the stained killed group (n = 10), the unstained killed group (n = 10) and the control group (n = 5) are consolidated in Table 2, as all tests were negative.
Table 2. Angiostrongylus cantonensis DNA detection by real-time PCR in the brain and lungs of rats post-euthanasia
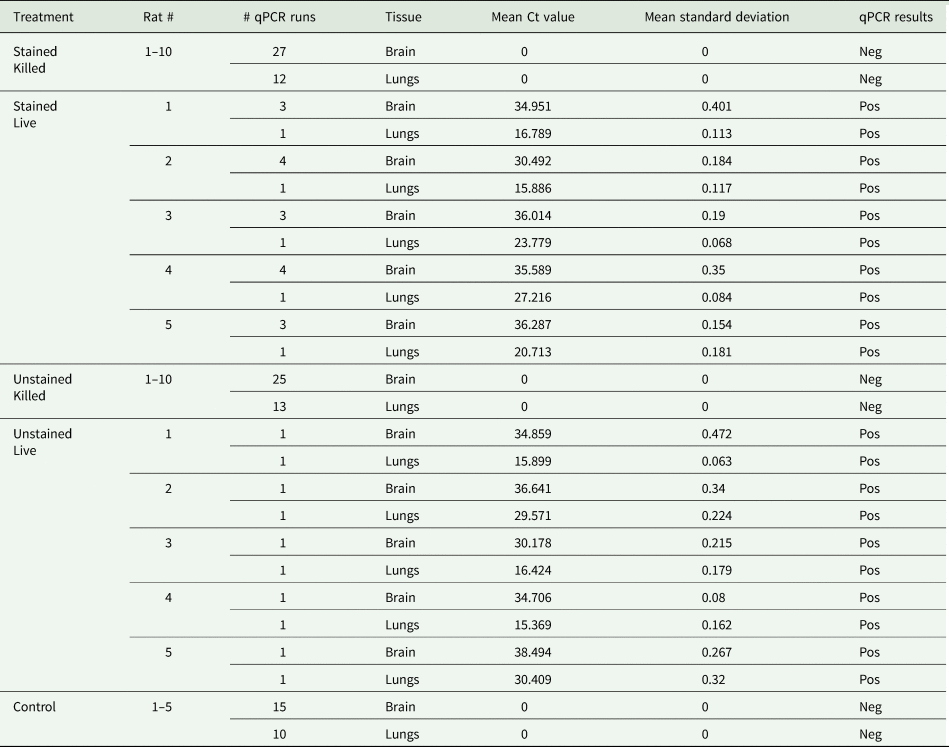
All reactions were run in triplicate.
Discussion
Rat lungworm is an emerging and neglected tropical disease which is spreading throughout Asia, the Caribbean, Africa, Hawaii (USA), and has been recently reported from the continental USA in Florida, Louisiana, Texas, Oklahoma and Tennessee (Teem et al., Reference Teem, Qvarnstrom, Bishop, Alexandre, Carter, White-Mclean and Smith2013; Hammoud et al., Reference Hammoud, Nayes, Murphy, Heresi, Butler and Pérez2017; Howe and Jarvi, Reference Howe and Jarvi2017; Jarvi et al., Reference Jarvi, Quarta, Jacquier, Howe, Bicakci, Dasalla, Lovesy, Snook, McHugh and Niebuhr2017; Stockdale-Walden et al., Reference Stockdale Walden, Slapcinsky, Roff, Mendieta Calle, Diaz Goodwin and Stern2017; Kim et al., Reference Kim, Wong, Curry, Yeung, Hayes and Cowie2018). Thorough studies of the efficacy of anthelmintics on A. cantonensis may produce better treatment and/or management strategies for neuroangiostrongyliasis. A previous study investigating the in vitro efficacy of anthelmintics on A. cantonensis larvae was based on the motility of the larvae after exposure to the test substances (Terada et al., Reference Terada, Rodriguez, Dharejo, Ishii and Sano1986). Using lack of motility as a marker for death is risky, as larvae may appear less active or inactive after exposure to a substance. But lack of motility may or may not be a result of the test substance (Kusnawidjaja, Reference Kusnawidjaja1960; Anderson et al., Reference Anderson, Gould, Ingham and Coleman1979; Terada et al., Reference Terada, Rodriguez, Dharejo, Ishii and Sano1986), nor does lack of motility always indicate mortality; hence, a reliable marker of death is required to ensure larvae are dead and therefore non-infective. Attempts were made to stain A. cantonensis larvae with calcein AM and Hoechst 33 342, two vital fluorescent stains, to determine efficacy; neither stain was successful when incubated with live larvae. The thick, collagenous larval cuticle makes it difficult for vital stains to penetrate and to come in contact with the interior cells (Anderson et al., Reference Anderson, Gould, Ingham and Coleman1979; Cox et al., Reference Cox, Staprans and Edgar1981). Acridine orange was also tried but resulted in high background fluorescence, and the larvae appeared dead by 12 h incubation. This study describes the use of PI to stain live and dead L3 A. cantonensis larvae (with live and dead unstained larvae as controls), with assay validation in rats via infection challenges. The PI stain was only observed in larvae killed by freezing in methanol, with no live larvae taking up the stain. Adult worms and A. cantonensis DNA were found in the heart, lung or brain tissues of rats gavaged only with stained or unstained live larvae.
There were five treatment groups included in this study. The purpose of the stained dead larvae treatment was to verify that the method used to kill the larvae resulted in non-infective larvae. The stained live treatment served to determine if the PI interfered with the infection and development in the rat. No significant differences were seen in the number of adult worms isolated from the PI-treated live larvae (n = 113) compared with the unstained live larvae group (n = 120 adult worms) confirming that the stain had no effect on worm development in the rats. The unstained dead larvae treatment demonstrated that the technique used to kill the larvae was effective in the absence of PI. The live unstained treatment showed that larvae do not lose virulence during processing. The untreated control group received 1 mL of dH2O with no larvae to monitor for cross-contamination and to compare organ health and body mass with the other groups. PI was effective in differentially staining dead larvae and did not interfere with normal larval development in rats.
For this study, 1700 A. cantonensis L3 larvae were isolated and exposed to 0.5% pepsin-HCl (pH 1.41) solution (artificial gastric juice) for 30 min. Larvae were exposed to an acid solution in order to kill any soil nematodes present in the slug, as soil nematodes cannot survive acid exposure (Matute, Reference Matute2013), and to mimic any unknown physiological or biochemical effects that may be triggered in A. cantonensis L3 larvae by gastric acid when ingested by a mammalian host. L3 larvae were killed using 100% methanol and freezing at −80 °C for 30 min. It should be noted that larvae were still alive and motile when water or 70% isopropyl was used in place of methanol. After freezing, methanol was replaced with dH20 to avoid any methanol-associated complications in rats.
While the images we produced on the Operetta after staining dead larvae with PI resulted in bright white larvae, some variation in staining has been noted over time in other studies as the larvae begin to die. Based on observation, the larvae appear to take up the stain from the inside, starting with the gastrointestinal tract. As cells throughout the larvae die at different rates they seem to take up the stain differentially, resulting in a range of staining. However, this study did not assess the physiological state or infectivity of partially stained larvae and thus it cannot be determined whether such larvae are alive or infective. When fluorescent microscopy is used for imaging, since the peak emission of PI is 617 nm, the PI-stained larvae are expected to appear red. The colour setting on the Operetta was selected as bright yellow or white to enhance contrast. The cuticle of nematodes is composed of collagen, which is known to have autofluorescence between 420 and 510 nm (Cox et al., Reference Cox, Staprans and Edgar1981; Croce and Bottiroli, Reference Croce and Bottiroli2014; Page et al., Reference Page, Stepek, Winter and Pertab2014). The use of the fluorescein filter allowed us to visualize peak autofluorescence of A. cantonensis larvae which, in combination with the PI filter, allowed another mechanism for distinguishing live vs dead larvae.
At the end of the study, we found no significant differences in body weight between treatment groups; however, the male weight was consistently greater than the female weight, which is typical of Wistar rat strains (https://www.criver.com/products-services/find-model/wistar-igs-rat?region=3611). Analysing males separately from females, we did find a significant difference in the change in body weight between pre-treatment and post-euthanasia weights with the males increasing more in body mass than females. This could be due to increased food consumption and faster growth rate by the males.
The mean spleen mass of rats that received live larvae was greater compared to groups that received killed larvae (stained and unstained) and controls, but significant differences were only observed between the control vs unstained live groups. While mean spleen weight was significantly greater in males than in females, interestingly, spleen weight as a percentage of body weight was significantly greater in females than in males. However, this finding does not appear to be experimentally-related as this was also observed in the control group.
From our previous studies, we observed that granulation of the upper right lung lobe is a sign of A. cantonensis infection in rats (Jarvi et al., Reference Jarvi, Quarta, Jacquier, Howe, Bicakci, Dasalla, Lovesy, Snook, McHugh and Niebuhr2017). Similar granulations were seen in all the rats that received live larvae (stained and unstained), but not in rats that received killed larvae (stained or unstained) in this study. The upper right lung lobe is the first lobe to receive blood from the heart and may serve as a ‘filter’ to the rest of the lungs.
We detected adult worms in the pulmonary arteries, eggs in the lungs and L1 larvae in the feces of some of the rats receiving live larvae (both stained and unstained), but not in any of the rats receiving dead larvae. This suggests that PI does not interfere with the development and reproduction of A. cantonensis larvae in rats, and that staining with PI provides a valid assay for determining death in A. cantonensis larvae. This validated assay will allow us to evaluate the effects of anthelmintics on L3 larvae in vitro, determine which commercially available vegetable washes are most effective in killing larvae, and determine what effect other treatments such as heat, freezing, exposure to UV light and irradiation may have on L3 A. cantonensis.
Acknowledgements
The authors would like to thank Ghee Tan and John Cordova for helpful suggestions towards the development of the assay.
Financial support
This work was supported by the Hawaii County Council (especially Council members Eileen O'hara, Jen Ruggles, Tim Richards, Karen Eoff, Maile David, and Sue Lee Loy), USDA-APHIS Wildlife Services National Wildlife Research Center Hawai`i Field Station, the Hawai`i State Legislature, and the Daniel K. Inouye College of Pharmacy.
Conflict of interest
None.
Ethical standards
All animal procedures were conducted according to the Guidelines of the American Society of Mammologists for the use of mammals in research (Sikes et al., Reference Sikes and Gannon2011) and following approved Institutional Animal Care and Use Committee protocols (USDA NWRC QA-2879) and (University of Hawaii) exempt TEX 18-007. This study was approved by the University of Hawaii Institutional Biosafety Committee.