




Introduction
Host switching by parasites can have far-reaching implications for the ecology and evolution of both parasites and their hosts (Harbison and Clayton, Reference Harbison and Clayton2011; Brucker and Bordenstein, Reference Brucker and Bordenstein2012; Brockhurst et al., Reference Brockhurst, Chapman, King, Mank, Paterson and Hurst2014; Auld and Tinsley, Reference Auld and Tinsley2015; Hoberg and Brooks, Reference Hoberg and Brooks2015), and directly impact several contemporary biological challenges, including emerging infectious disease (Morens and Fauci, Reference Morens and Fauci2013), biological invasions (Stricker et al., Reference Stricker, Harmon, Goss, Clay and Luke Flory2016) and conservation of biodiversity (Thompson et al., Reference Thompson, Lymbery and Smith2010; Lymbery et al., Reference Lymbery, Morine, Kanani, Beatty and Morgan2014). Despite this, little is known about the evolutionary patterns of host switching among eukaryotic symbionts of animals. Parasites have historically been assumed to infect groupings of taxonomically similar hosts along a continuum from specialists, that are limited to infecting smaller clades, to generalists that are capable of infecting organisms across a larger clade (Hoberg et al., Reference Hoberg, Brooks, Siegel-Causey, Clayton and Moore1997; Poulin et al., Reference Poulin, Krasnov and Mouillot2011a). These patterns were generally believed to result in concurrent host–parasite phylogenies, either through cospeciation of specialist parasites with their hosts or host switching of generalist parasites to closely-related host clades (Hafner and Nadler, Reference Hafner and Nadler1988; Klassen, Reference Klassen1992). While these processes have been documented in some groups of parasites, the application of molecular techniques has largely refuted them as general rules guiding parasite evolution (Vienne et al., Reference Vienne, Refrégier, López-Villavicencio, Tellier, Hood and Giraud2013): incongruent host–parasite phylogenies are now known to be common, and long-distance host switching appears ubiquitous in the evolution of many parasites. Ecological theory has been applied to explain these observations through a paradigm known as ‘ecological fitting’ (Agosta et al., Reference Agosta, Janz and Brooks2010; Araujo et al., Reference Araujo, Braga, Brooks, Agosta, Hoberg, von Hartenthal and Boeger2015). Simply put, this framework views hosts as resources for symbionts and explains evolutionary patterns of host switching in terms of adaptation towards resource acquisition, but its general applicability to host–parasite relationships requires further empirical exploration (Nylin et al., Reference Nylin, Agosta, Bensch, Boeger, Braga, Brooks, Forister, Hambäck, Hoberg, Nyman and Schäpers2018). Along these lines, and in the context of trophic niche partitioning, parasites have been proposed to colonize new hosts in order to take advantage of previously untapped resources and then specialize on these new hosts to exploit these resources more effectively (Combes and Théron, Reference Combes and Théron2000; Agosta et al., Reference Agosta, Janz and Brooks2010). Constraints against host switching undoubtedly limit the capacity of parasites to exploit all potential hosts, and several hypotheses have been proposed to explain when host switching is most likely to occur. The challenges faced by parasites when establishing in new hosts have been likened to similar processes in free-living systems, such as island colonization, biological invasions and succession (Kuris et al., Reference Kuris, Blaustein and Alio1980). From this perspective, host switching is expected to occur based upon the number of opportunities for colonization events as mitigated by exposure (i.e. propagule pressure; Araujo et al., Reference Araujo, Braga, Brooks, Agosta, Hoberg, von Hartenthal and Boeger2015). As such, it should occur between taxa that share similar physical spaces and high levels of exposure to one another. Host switching is also expected to occur in conjunction with the conformity of new hosts to the fundamental niche occupied by the parasite (Combes and Théron, Reference Combes and Théron2000; Fodor, Reference Fodor2011; Poulin et al., Reference Poulin, Krasnov, Mouillot and Thieltges2011b; Escobar and Craft, Reference Escobar and Craft2016). Similarities in host anatomy and physiology may be important for creating niche overlap in the internal environments of their hosts (Dallas and Presley, Reference Dallas and Presley2014; Huang et al., Reference Huang, Bininda-Emonds, Stephens, Gittleman and Altizer2014; Leung and Koprivnikar, Reference Leung and Koprivnikar2016). Closely-related hosts are likely to possess similar anatomy and physiology, and this may favor host switching within related taxa (Poulin et al., Reference Poulin, Krasnov and Mouillot2011a; Cooper et al., Reference Cooper, Griffin, Franz, Omotayo and Nunn2012; Nylin et al., Reference Nylin, Agosta, Bensch, Boeger, Braga, Brooks, Forister, Hambäck, Hoberg, Nyman and Schäpers2018). For parasites of the gut, diet overlap may also be a particularly important indicator of similarities in internal host habitat due to similarities in resource availability and convergent evolution on host digestive function (Karasov et al., Reference Karasov, Martinez del Rio and Caviedes-Vidal2011; Karasov and Douglas, Reference Karasov and Douglas2013). Exposure and niche overlap might also affect the breadth of hosts that parasitic lineages can infect, in so far that exposure to more types of hosts and adaptation to generalist hosts with less specialized internal environments may favour generalist strategies in parasites. However, at present, hypotheses on the ecological conditions that drive host switching and host specificity remain inadequately tested. A few detailed studies have been conducted but have tended to focus on plant–pathogen systems, parasitoids and a handful of metazoans (primarily Ecdysozoa) infecting other metazoa (Desneux et al., Reference Desneux, Blahnik, Delebecque and Heimpel2012; Vienne et al., Reference Vienne, Refrégier, López-Villavicencio, Tellier, Hood and Giraud2013; Nylin et al., Reference Nylin, Agosta, Bensch, Boeger, Braga, Brooks, Forister, Hambäck, Hoberg, Nyman and Schäpers2018). Beyond blood apicomplexans (i.e. Haemosporida, such as Plasmodium and other malarial agents: Hellgren et al., Reference Hellgren, Pérez-Tris and Bensch2009; Nilsson et al., Reference Nilsson, Taubert, Hellgren, Huang, Palinauskas, Markovets, Valkiūnas and Bensch2016; Svensson-Coelho et al., Reference Svensson-Coelho, Loiselle, Blake and Ricklefs2016), almost nothing is known about evolutionary patterns of host usage in protistan symbionts of vertebrates.
The Blastocystis species-complex (Stramenopile, Phylum: Heterokontaphyta) provides an excellent system for exploring the evolutionary intricacies of specialization, generalization and host switching in a protistan symbiont. Blastocystis spp. are ubiquitous symbionts of metazoa and occur across an extremely broad range of hosts, including, but not limited to, mollusks, annelids and insects, as well as all major groups of vertebrates: actinopterygians, amphibians, mammals, lepidosaurs, archosaurs and testudines (Stensvold et al., Reference Stensvold, Alfellani, Nørskov-Lauritsen, Prip, Victory, Maddox, Nielsen and Clark2009; Alfellani et al., Reference Alfellani, Taner-Mulla, Jacob, Imeede, Yoshikawa, Stensvold and Clark2013; Yoshikawa et al., Reference Yoshikawa, Koyama, Tsuchiya and Takami2016). Sexual reproduction is not known to occur within the Blastocystis species-complex, and members of this genus appear to have colonized their diverse range of hosts clonally. As such, the distribution of different host types across related lineages of Blastocystis spp. should accurately reflect host switching events across its evolutionary history. Although Blastocystis lacks internal species delineations, molecular studies suggest that the Blastocystis species-complex can be broken into several monophyletic lineages, termed subtypes (Scicluna et al., Reference Scicluna, Tawari and Clark2006). Studies on the host specificity of Blastocystis spp. have largely been performed at the level of these subtypes, and found them to be associated with particular, but often broadly divergent, hosts (Alfellani et al., Reference Alfellani, Taner-Mulla, Jacob, Imeede, Yoshikawa, Stensvold and Clark2013). This suggests a rich history of host switching and variation in host specificity both within and between subtypes that is well suited for answering broader evolutionary questions. Moreover, the standardized use of sequences from a single region of the 18S Small-Subunit Ribosomal RNA gene (18S gene) to barcode Blastocystis subtypes (Scicluna et al., Reference Scicluna, Tawari and Clark2006), provides an opportunity to study global evolutionary patterns of host switching and host specificity within this parasite at the level of lineage-specific sequence variants.
Here, we assess the capacity of general ecological processes (i.e. ecological fitting) to explain evolutionary patterns of host usage (i.e. phylogenetically associated motifs of host traits, including host environments, diet and taxonomy) and host specificity (i.e. the breadth of host taxa utilized) within the Blastocystis species-complex using a high-resolution Bayesian phylogenetic tree of partial 18S gene sequences corresponding to the Blastocystis barcode region of this gene (Scicluna et al., Reference Scicluna, Tawari and Clark2006). We use an automated approach to maximize included sequences from the National Center for Bioinformation Technology (NCBI) GenBank (Benson et al., Reference Benson, Cavanaugh, Clark, Karsch-Mizrachi, Lipman, Ostell and Sayers2012); and sample ~20% of all nucleotide sequences attributed to Blastocystis spp. on GenBank. We also supplement these archival sequences by including Blastocystis sequence data we collected from long-tailed macaques (Macaca fascicularis) in Singapore and Bali, Indonesia. We use distance-based statistics to quantitatively assess the marginal effects of several host-associated variables on the structure of our tree. These variables consist of (1) taxonomic patterns of hosts usage (i.e. the families of hosts associated with each unique sequence), which reflect the internal niche space of hosts; (2) the broad dietary patterns (herbivorous, omnivorous, carnivorous) of hosts utilized, which may also reflect internal niche space within hosts; (3) the relative anthropogenic environment (human, captive or domestic, human-associated and wild) of hosts utilized, which likely reflects patterns of exposure across hosts; and, (4) the geographic locations (countries) from which sequences were isolated, which also reflect patterns of exposure across hosts. We further employ distance-based statistics to assess how taxonomic patterns of host usage (anthropogenic environment, host diet and geography) relate to host traits to determine the factors that drive host sharing at the fine-scale of unique-sequence Blastocystis lineages. Finally, we tested hypotheses relating to generalization and specialization of Blastocystis lineages. First, we assessed whether patterns of generalization and specialization, as determined by the number of host families infected, were conserved across lineages and subtypes. We then assessed the role of exposure to more hosts – as reflected by broader geographic distribution and use of ubiquitous human hosts – and, use of omnivorous host, which may present a more generalized internal environment, on host specificity by using a generalized linear model to relate the number of host families infected by each unique-sequence lineage to these traits.
Methods
Macaque sample collection and sequencing
Faecal samples were collected from wild long-tailed macaques (Macaca fascicularis) living across 12 sites in Singapore (N = 77) and 15 sites on Bali (N = 244), Indonesia, as previously described (Lane et al., Reference Lane, Holley, Hollocher and Fuentes2011; Wilcox et al., Reference Wilcox, Lane-Degraaf, Fuentes and Hollocher2015; Klegarth et al., Reference Klegarth, Sanders, Gloss, Lane-deGraaf, Jones-Engel, Fuentes and Hollocher2017). Habitats varied considerably within Bali and between Singapore and Bali, but all sites were subject to interactions between humans and macaques. Sites in Bali contained a variety of landscapes including bamboo forests, wet and dry tropical broadleaf forests, rice agriculture and scrublands. All of these sites were associated with Hindu temples where interaction with humans, including provisioning, occurs. These sites also had variable levels of tourist visitation, and while generally rural, most of these sites contained at least some urbanized habitat (Lane et al., Reference Lane, Holley, Hollocher and Fuentes2011). In contrast, sites in Singapore consisted primarily of edge habitat between highly-developed residential and commercial urban areas and wet broad-leafed tropical forests. Many sites were located within preserved National Parks and subject to regular recreational use by humans. While provisioning of macaques is illegal and subject to heavy fine within Singapore, some provisioning still occurs, and macaques regularly raid anthropogenic food sources, refuse containers and personal affects within Singapore.
DNA was extracted from all samples using QIAamp DNA Stool Minikits (Qiagen, Silicon Valley CA). Extracts were used as templates in standard PCR reactions with Taq Polymerase (Bulldog Bio, Portsmouth NH) using the forward primer GCTTATCTGGTTGATCCTGC and the reverse primer GAGCTTTTTAACTGAACAACG (modified from Clark, Reference Clark1997 and Scicluna et al., Reference Scicluna, Tawari and Clark2006, respectively), targeting the barcode region of the 18S rRNA gene. Thermocycler conditions consisted of an initial denature step of 2:00 min at 94°C, followed by 10 cycles of touchdown PCR with a 94°C denature for 30 s, a 30 s annealing period, which started at 60°C and dropped 0.5°C every cycle and a 4:00 min extension phase at 68°C; these 10 cycles were followed by another 20 cycles under the same conditions with the exception of a constant annealing temperature of 55°C. A final extension phase was included; this lasted 4:00 min at 68°C.
Samples were Sanger sequenced twice and sequences were concatenated using Sequencher (Gene Codes Corporation, Ann Arbor MI). All concatenated sequences were confirmed as Blastocystis using BLAST (Morgulis et al., Reference Morgulis, Coulouris, Raytselis, Madden, Agarwala and Schäffer2008) on the National Center for Biotechnology Information (NCBI) GenBank (Benson et al., Reference Benson, Cavanaugh, Clark, Karsch-Mizrachi, Lipman, Ostell and Sayers2012) and the curated archival sequence site PUBMLST: Blastocystis (Jolley and Maiden, Reference Jolley and Maiden2010). Approximately 1–2% of sequences were found to belong to unicellular fungi, through these searches, and discarded at this step. Only previously unreported sequences that were isolated from multiple macaque samples were included in our analysis (Accession Numbers: MN611237, MN611238, MN611239 and MN611240). In total, one previously unreported sequence was included from Singapore, and two previously unreported sequences were included from Bali, Indonesia. MN611237 was an exact match to allele 141 on Pubmlst, but was not available on GenBank at the time we initiated this study.
Archival sequence compilation and sequence processing
Sequences were compiled using an Esearch (Kans, Reference Kans2017) script in Unix tcsh shell to obtain all sequences on NCBI GenBank (Benson et al., Reference Benson, Cavanaugh, Clark, Karsch-Mizrachi, Lipman, Ostell and Sayers2012) identified as belonging to Blastocystis spp. (Taxa ID: 12967), and between 348 and 2400 base pairs in length. These length criteria correspond to the minimum full length of the Blastocystis 18S barcode region, and the maximum length of 18S sequences from Blastocystis found on GenBank, excluding those included within scaffolds of whole-genome assemblies. Sequences were retrieved on 18 December 2020 using the following exact search criteria: ‘txid12967[Organism:exp] AND biomol_genomic[PROP] AND (‘348’[SLEN]:‘2200’[SLEN])’.
These sequences were concatenated with an outgroup sequence from Proteromonas lacertae (Arisue et al., Reference Arisue, Hashimoto and Yoshikawa2003; Noël et al., Reference Noël, Dufernez, Gerbod, Edgcomb, Delgado-Viscogliosi, Ho, Singh, Wintjens, Sogin, Capron and Pierce2005; Scicluna et al., Reference Scicluna, Tawari and Clark2006; Yoshikawa et al., Reference Yoshikawa, Koyama, Tsuchiya and Takami2016), Accession #: U37108, a full set of 18S barcode region sequences from PubMLST: Blastocystis (Jolley and Maiden, Reference Jolley and Maiden2010), and a set of unique sequences isolated from long-tailed macaques of Singapore and Bali, Indonesia (see ‘Macaque Sample Collection and Sequencing’).
Barcode regions were extracted from these sequences using the program Hmmer (Eddy, Reference Eddy1998). The barcode region was chosen for analysis, as it is a ubiquitous sequencing target that provides a reasonable prediction of the Blastocystis subtype (Scicluna et al., Reference Scicluna, Tawari and Clark2006). In brief, the full set of barcode-region sequences from PubMLST: Blastocystis was aligned using the Mafft Q-INS-i iterative alignment program for structural RNAs (Katoh and Standley, Reference Katoh and Standley2013). This alignment was used to build an Hmmer profile and ‘nhmmer’ was used to extract all partial sequences matching this barcode region from the full list of sequences using an e-value cutoff of 10−6. The remaining barcode sequences were then filtered to include only those matching identified barcodes at 99.2 and 100% query cover, effectively allowing two mismatches from a known barcode, using USEARCH 11.0.667 (Edgar, Reference Edgar2010). The Proteromonas lacertae barcode alignment from hmmer was then added back to these sequences, such that the outgroup was exempted from filtering. We restrict ourselves to curated barcodes and close matches to these, as preliminary analyses found very poor tree structure when all sequences with three or more mismatches to barcode sequences were included in our analysis. Sequences were then dereplicated with the USEARCH ‘cluster_fast’ algorithm with a 100% identity threshold and early termination rejection criteria (‘maxrejects’) disabled. Sequences were then realigned using the Mafft Q-INS-i interactive alignment program for structural RNAs. This alignment was converted to Nexus format using the ‘Readseq’ sequence conversion tool (Gilbert, Reference Gilbert2003).
Bayesian tree construction
A Bayesian tree was constructed with MrBayes (Ronquist et al., Reference Ronquist, Teslenko, Van Der Mark, Ayres, Darling, Höhna, Larget, Liu, Suchard and Huelsenbeck2012), using the compiled and processed sequence data. The tree was constructed using a Generalized Time Reversible model with gamma-distributed rate variation and a proportion of invariant sites. We chose a general model for tree construction to minimize the implicit assumptions in our analysis, as less constrained models have been shown to lead to more conservative results (Huelsenbeck and Rannala, Reference Huelsenbeck and Rannala2004). Gaps were treated as missing data. Two runs were specified with 224 chains and 36 400 000iterations per run, with 28 swaps per iteration and no heated chains. A majority-rule consensus tree was created using outputs sampled from every 1000 iterations of each run. The first 25% of samples were discarded from each run as real burn-in.
Sequence host data compilation
USEARCH (Edgar, Reference Edgar2010) was used to compile a list of all matching sequences from the full list of all sequences. NCBI GenBank (Benson et al., Reference Benson, Cavanaugh, Clark, Karsch-Mizrachi, Lipman, Ostell and Sayers2012) records were used to obtain information on hosts, and relevant publications were consulted to determine information on hosts and geographic location of isolation whenever necessary. For purposes of our analyses, all hosts were also recorded at the level of family. Host diet was assigned (‘herbivore’, ‘omnivore’, ‘carnivore’) based on the known life history of host species (Dallas and Presley, Reference Dallas and Presley2014; Huang et al., Reference Huang, Bininda-Emonds, Stephens, Gittleman and Altizer2014; Leung and Koprivnikar, Reference Leung and Koprivnikar2016). Hosts were also classified according to relative anthropogenic environment (effectively their environment as defined by a degree of interaction with humans). Four levels of relative anthropogenic environment were used: sequences isolated from Homo sapiens sapiens were classified as belonging to a ‘human’ host environments; sequences isolated from either captive animals or domestic animals were classified as having ‘captive’ host environments; sequences were classified as having ‘Human-Associated’ environments if they were isolated from hosts that live in close proximity to humans, such that interaction with humans contributes substantially to their ecology (e.g. urban animals, provisioned primate populations etc.) or if they were isolated from host populations of previously captive animals that were released by humans; sequences were classified as wild if they were from organisms that lived in non-urban environments and were not otherwise known to be reliant on humans or to have been descendants of captive founders. Finally, we recorded the country from which each sequence was isolated. Information on all reported host families, host association, geographic location, and diet was compiled for each unique sequence as presence–absence data. While all unique sequences were included in our tree construction, we excluded all sequences that lacked information on host taxonomy, host association, or geographic location from any downstream analyses. This includes sequences that were isolated from environmental samples and not isolated from any host. Subtypes were assigned to each sequence using the ‘batch sequence query’ tool on PubMLST: Blastocystis. Sequences were assigned to an ‘exact-subtype’, if they matched a curated allele attributed to a subtype exactly. If sequences failed to match a curated allele exactly, they were assigned a ‘closest-subtype’ on the basis of the most similar allele matched.
Statistical analysis
Data were analysed to assess determinants of tree structure, variation in breadth of host usage across sequences and geographic distribution of sequences. All statistical analyses were run in R Studio 1.1,1456 (RStudio, 2016) using R version 3.5.1 (R Core Team, 2018). We assessed determinants of tree structure using distance-based statistics. The ‘APE’ Package was used to extract distances from our Bayesian consensus tree (Paradis et al., Reference Paradis, Claude and Strimmer2004). The vegan Package (Oksanen et al., Reference Oksanen, Blanchet, Kindt, Legendre, Minchin and O'hara2016) was used for calculating all other distances and distance-based tests with the exception of Mantel and partial-Mantel tests, which were performed in the ‘ecodist’ package to obtain two-tailed P values (Goslee and Urban, Reference Goslee and Urban2007). Jaccard distance metrics were used for all presence–absence data on compiled host information. Euclidean distances were used for count data.
Where applicable, permanova's and generalized linear models used marginal sums of squares to assess the independent impact of each term. Partial-Mantel tests were likewise used to assess partial correlations in multivariate correlations between distance matrices, and are always reported as two-tailed results. Generalized-linear models used quasi-Poisson family error distributions for count data, and as such assumed error-distributions are proportional to the mean. We conduct 17 independent statistical hypothesis tests in this paper. We account for multiple tests by adjusting our alpha-value for accepting statistical significance to 0.003 in accordance with a Šidák correction for 17 multiple tests (Šidák, Reference Šidák1967).
Results
Sequences
We downloaded 7665 sequences from GenBank that matched our search criteria. In total, 2668 sequences met our inclusion criteria and were included in our analysis. This corresponds to ~22% of all 11 864 genomic DNA sequences attributable to Blastocystis on NCBI at the time of our analysis. These corresponded to 369 unique sequences, excluding the outgroup. Of these, 116 were reported more than once and 253 were singletons. On average there were 7.2 reports per an included sequence. Of the 369 unique Blastocystis sequences, 112 coincided with exact barcode sequences, of which 60 were singletons. When unique sequences were combined, they produced a final alignment of 445 base pairs. This alignment included sequences from Subtypes 1–10 and Subtype 13, as well as two sequences matching barcode Allele 114, which lacks a designated subtype.
Tree topology
Overall, we produced a structured tree with high support (Fig. 1): all branches have posterior probabilities (PPs) above 50% and many branches with PPs exceeding 90%. Trees converged with an average standard deviation of split frequencies of 0.003172; well below the recommended 0.01 convergence diagnostic for acceptance. Our tree was overwhelmingly structured by both exact-subtypes (pseudo-F = 229.1; R 2 = 0.96; P = 0.00001) and closest-matched subtypes (pseudo-F = 708.5; R 2 = 0.95; P = 0.00001). Most subtypes formed monophyletic clades, with only a few exceptions: Subtype 1 was paraphyletic with Subtype 2 (PP = 94.2%), the latter of which formed a polyphyly within this clade. Subtype 6 was likewise paraphyletic with Subtype 9, albeit with low support (PP = 51.9%).
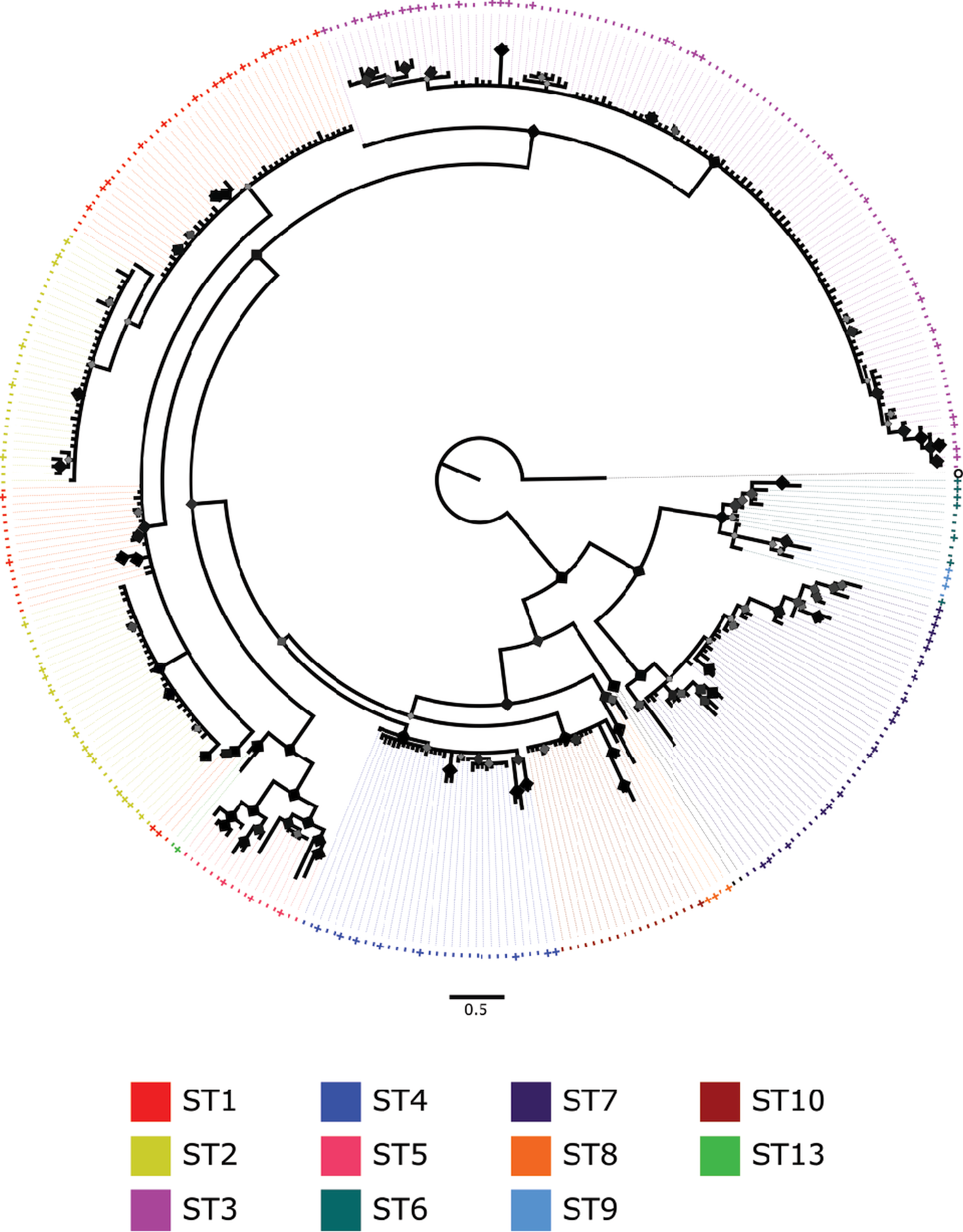
Fig. 1. Bayesian tree of unique Blastocystis 18S gene barcode sequences colour-coded by subtype (abbreviated ST in legend). Sequences matching exact-subtype reference alleles are denoted with ‘ + ’ signs. ‘O’ denotes the outgroup represented by Proteromonas lacertae. Posterior probabilities (PPs) are indicated by diamond shapes on nodes with smaller lighter diamonds representing lower PPs (lowest ~50%) and larger darker diamonds representing higher PPs (maxing at 100%).
Overall tree topology did coincide with occurrence in specific taxa of hosts (Fig. 2A). Specific clades were observed predominantly to infect Suidae, Bovidae, non-human primates, and fowl. Sequences infecting humans were observed across most of the tree, with the exception of two clades (ST5 and ST10) that specialized on a variety of even-toed ungulate (Artiodactyla) families. Two small subclades of ST1 and ST3 were likewise observed exclusively in non-human primates. Isolates from different relative anthropogenic environments were observed across the tree (Fig. 2B), although most sequences were from either human or captive animal sources, and several blocks of sequences were observed that occurred in only humans and only captive animals. Geographic patterns of occurrence were also reflected by tree structure (Fig. 2C). In general, sequences and clades were widely distributed across continents; however, some small clades of sequences were observed predominantly in particular countries, such as France, Japan, and the Philippines. Sequences were overwhelmingly collected from omnivorous hosts, but several small clades infecting herbivores were observed in several parts of the tree, as was one large clade (ST10) occurring strictly in herbivores (Fig. 2D).
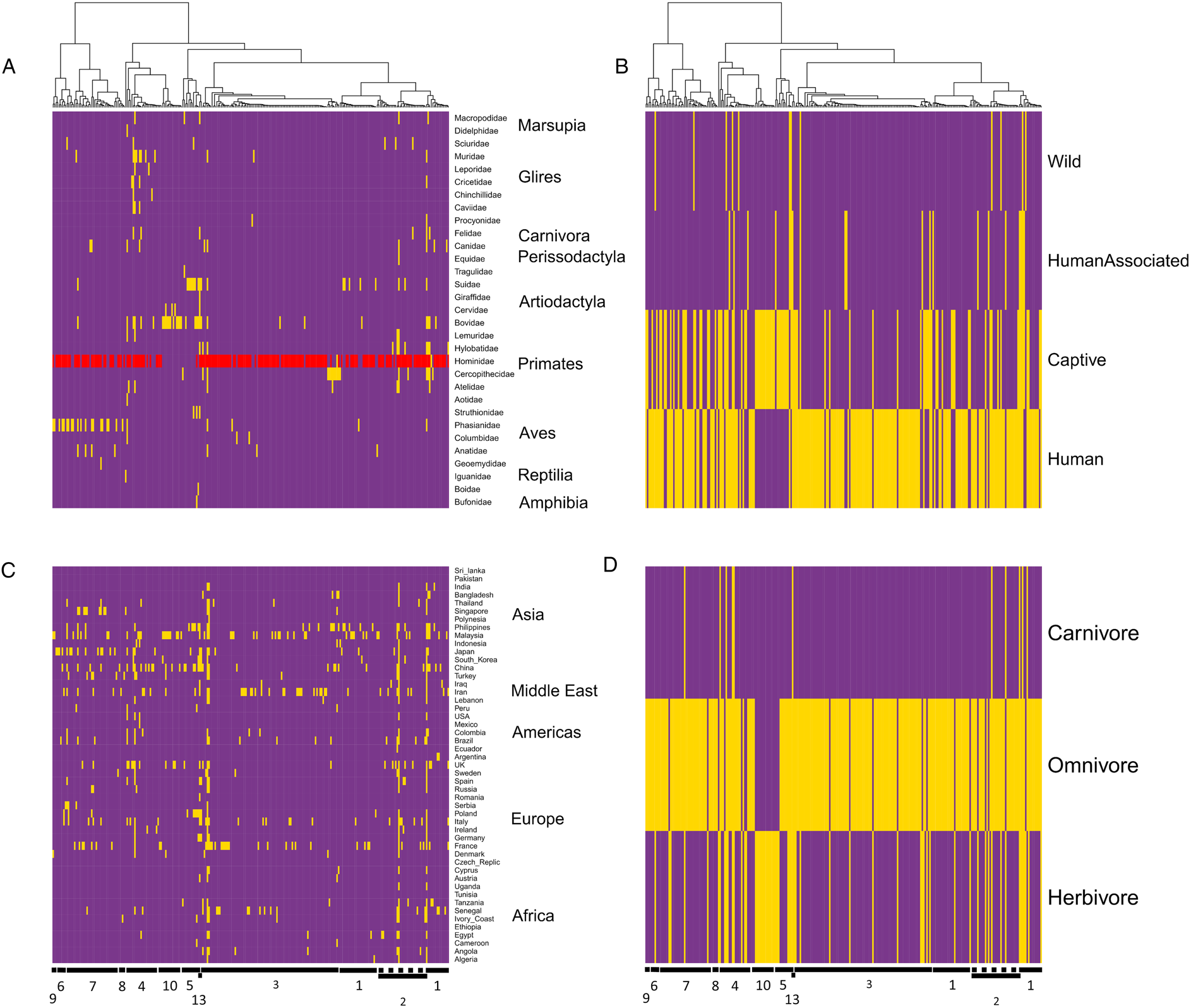
Fig. 2. Heatmaps of the Blastocystis phylogeny in relation to distributions of reports by host: (A) families; (B) anthropogenic environment; (C) country; (D) diet. Yellow blocks denote the presence of a sequence in each particular grouping, and purple blocks indicate the absence of sequences in respective groupings. As Hominidae contain both human and non-human primates, sequences that were isolated from at least one human are denoted in red, where-as sequences only isolated from non-human members of this family (i.e. ‘great apes’) are denoted in yellow. Note that the presence of a sequence from a human host does not preclude its presence in a non-human Hominidae as well. Host families are grouped by broader taxonomic positions (orders, infraorder or class). Countries are grouped according to broader geographic regions.
Patterns of host usage
A multi-way PerMANOVA was used to assess how patterns of host usage (as represented by patterns of host taxonomy, relative anthropogenic environments of hosts, countries of sequence isolation and host diets) related to a tree structure. Patterns of host-family usage (pseudo-F = 1.773; partial-R 2 = 0.13; P = 0.00242) and country of isolation (pseudo-F = 2.193; partial-R 2 = 0.228; P = 0.00003) were significant predictors of the tree structure. These relationships were further assessed using partial-Mantel tests to correlate phylogenetic distances to Jaccard distances calculated from patterns of host usage. Similar to the previous analysis, host taxonomy (r = 0.166; P = 0.00002) and countries of origin (r = 0.0570; P = 0.00006) had weak partial positive correlations to tree structure. Anthropogenic environment (r = −0.106; P = 0.00178) and diet (r = −0.086; P = 0.00022) had weak negative partial correlations to tree structure, likely resulting from reversal of sign due to high-collinearity with taxonomy and weaker effect size. A multi-way PerMANOVA was used to assess drivers of taxonomic patterns in host usage among lineages of the same unique sequences. Patterns of host family usage were strongly correlated to patterns of relative anthropogenic environment (pseudo-F = 25.17; partial-R 2 = 0.16; P = 0.00001) and also significantly correlated to patterns of host diets (pseudo-F = 10.69; partial-R 2 = 0.051; P = 0.00001) and countries of sequence isolation (pseudo-F = 2.48; partial-R 2 = 0.16; P = 0.00001). Partial-Mantel tests with the same general hypotheses likewise showed a strong partial correlation of anthropogenic environment (r = 0.779; P = 0.00001), and a moderate partial correlation of host diet (r = 0.38, P = 0.00001) with clusters of host families infected by each unique sequence. A PerMANOVA was also used to test for differences in taxonomic patterns of host usage between closest-subtypes: we found that taxonomic patterns of host usage differed significantly between closest-subtypes (pseudo-F = 7.82; R 2 = 0.23; P = 0.00001).
Patterns of host specificity: generalization and specialization
To assess whether generalization and specialization were evolutionarily conserved traits, we tested for a correlation between phylogenetic distance and differences in the number of families infected across all unique sequences using a Mantel test and found a significant relationship (r = 0.14; P = 0.00096), demonstrating that breadth of host usage was phylogenetically conserved. We likewise found a significant difference between closest-subtypes and the number of families infected (Kruskal-Wallis χ 2 = 32.125; P = 0.0007). To assess whether the presence in a ubiquitous host, dietary generalist or wide geographic dispersion influenced breadth of host usage, we used a generalized linear model to test for the effects of human host usage, omnivorous host usage and the number of countries from which a sequence had been reported on the number of host families infected, with the number of reports included as a blocking covariate. The number of countries from which a sequence had reported was positively correlated to the number of families from which a sequence had been isolated (t-value = 9.592; P < 0.00001, partial-R 2 = 0.19), but no relationship was found between presence–absence in human hosts or omnivorous hosts and the number of host families infected. We then used a generalized linear model to further assess the relationship between the number of countries from which a sequence had been reported and anthropogenic environment, again using the number of reports as a blocking covariate. Infections in humans (t-value = 7.198; P < 0.00001; partial-R 2 = 0.30) and captive animals (t-value = 9.195; P < 0.00001; partial-R 2 = 0.38) were both strongly and significantly associated with broader geographic distribution; no association was found between infections in human-associated or wild animals and broader geographic distribution.
Discussion
Here we use an archival data approach to build a highly inclusive tree of 18S barcode region sequences from Blastocystis spp. to evaluate evolutionary patterns of host usage. We report a tree that is structured by shared taxonomic patterns of hosts infected and geographic regions of isolation. While taxonomic patterns of hosts used, were correlated to tree structure, these patterns were not constrained to groupings of closely related hosts, but were strongly correlated to shared anthropogenic environments and shared geographic locations. These results are consistent with ecological fitting and suggest that shared patterns of exposure contribute to sharing of hosts at the level of individual strains. While individual strains and subtypes are subject to phylogenetic constraints on the patterns of hosts that they infect, these patterns may, but do not necessarily, reflect shared host ancestry or conserved internal environments linked to diet. Degrees of generalization and specialization were also phylogenetically constrained and differed between subtypes. We observe a link between more generalist strains and broader geographic distribution, which is coupled with an observed link between broader geographic distribution and the capacity to infect humans and animals kept by humans across Blastocystis strains. These results suggest that humans and their practices in animal husbandry have helped to distribute associated Blastocystis strains around the globe, providing an abundant supply of potential new hosts, and creating circumstances favourable for host generalization. Results related to both host switching and generalization support the ecological fitting paradigm and indicate that the capacity of Blastocytis to colonize new host taxa is determined by a mixture of propagule pressure and adaption to the unique internal environments of particular hosts.
Our results must be interpreted with a few caveats. First, we use only the barcode region of the Blastocytis 18S gene. While this allows for a broad sampling of Blastocystis sequences from a range of hosts, it provides a far from exhaustive look at Blastocystis host usage. Countless studies characterize Blastocystis using additional amplicons (Santín et al., Reference Santín, Gómez-Muñoz, Solano-Aguilar and Fayer2011), and these may yield somewhat different patterns of host usage, particularly as amplicon regions used likely correlated to study organism. Second, our filtration criteria exclude sequences from several subtypes and purported subtypes that lack curated allele sequences on PubMLST (Stensvold and Clark, Reference Stensvold and Clark2020). While our results should be reliable in terms of the sequences we do sample, more complex patterns may occur in what we could not sample. Limited sampling in general and the restrictions of our analysis have likely biased our analysis toward more generalist strains. Conversely, we likely overestimate specificity by missing strains in rare hosts. The goals of this paper are to assess broad patterns of host usage, and our results should not be interpreted as a strict guide to host range in the Blastocystis species-complex per se.
The weight of exposure appears to be a major contributing factor to host switching in Blastocystis. This is in keeping with long-standing reports of zoonotic Blastocystis transmission to humans on the basis of close exposure to a taxonomically diverse range of other hosts (Parkar et al., Reference Parkar, Traub, Vitali, Elliot, Levecke, Robertson, Geurden, Steele, Drake and Thompson2010; Cian et al., Reference Cian, El Safadi, Osman, Moriniere, Gantois, Benamrouz-Vanneste, Delgado-Viscogliosi, Guyot, Li, Monchy and Noël2017), and reports of reverse zoonosis resulting from close contact between humans and other animals (Ramírez et al., Reference Ramírez, Sánchez, Bautista, Corredor, Flórez and Stensvold2014; Wang et al., Reference Wang, Owen, Traub, Cuttell, Inpankaew and Bielefeldt-Ohmann2014a). Within Subtypes 1 and 3 we find clades that predominantly occur in humans, but that occasionally occurs in a range of unrelated domestic animals – including, dogs, cats, cows, pigs, chickens and ducks – suggesting reverse-zoonotic colonization of these animals by these strains as a result of high-propagule pressure. Broadly speaking, these findings are in accordance with other studies that report exposure as a driver of similarities in parasite community assembly across relatively unrelated hosts (Dallas and Presley, Reference Dallas and Presley2014; Huang et al., Reference Huang, Bininda-Emonds, Stephens, Gittleman and Altizer2014), and mathematical models that have implicated propagule pressure as a major force favouring colonization of new hosts by parasites regardless of their evolutionary relationship to previous hosts (Araujo et al., Reference Araujo, Braga, Brooks, Agosta, Hoberg, von Hartenthal and Boeger2015). We also note that propagule pressure has been shown to be one of the most important and consistent contributing factors in the ability of animals and plants to invade new environments, and recolonize old environments following disturbance (Holle and Simberloff, Reference Holle and Simberloff2005; Lockwood et al., Reference Lockwood, Cassey and Blackburn2005; Simberloff, Reference Simberloff2009). In this light, our findings suggest a commonality in the ecological principles governing colonization of new environments by parasites and free-living organisms.
We found more limited evidence for shared internal host niche space as an important requisite for the movement of Blastocystis between hosts. We had hypothesized that convergent traits in the digestive tracts of herbivores, omnivores, and carnivores would lead to more similar internal environments within these hosts and that these would, in turn, facilitate conserved evolutionary patterns of host usage within these groups. We find limited evidence for this in that taxonomic patterns of host usage do correlate to diet, even when controlling for other factors, and in Subtype 10, which was reported exclusively in herbivorous even-toed ungulates (Artiodactyla) within the Infraorder Pecora. However, the extremely small marginal effect size of diet in explaining strain-specific patterns of infection, and the narrow range of hosts from which Subtype 10 has been reported, make extrapolation from these findings dubious. As Blastocystis spp. seem capable of colonizing diverse niche space within the large intestines of any given host (Moe et al., Reference Moe, Singh, Howe, Ho, Tan, Chen, Ng and Yap1997; Li et al., Reference Li, Deng, Li, Cao, Li and Yan2013; Fayer et al., Reference Fayer, Elsasser, Gould, Solano, Urban and Santin2014; Wang et al., Reference Wang, Bielefeldt-Ohmann, Traub, Cuttell and Owen2014b), the small-scale differences in internal gut microenvironment or broad anatomical differences, such as gut length, associated with host adaptation to diets (Karasov et al., Reference Karasov, Martinez del Rio and Caviedes-Vidal2011; Karasov and Douglas, Reference Karasov and Douglas2013) may be unimportant to the survivability of Blastocystis spp. within hosts. Internal host environments have the potential to be far more homogenous with respect to many important determinants of ecological range (Rollins et al., Reference Rollins, Bryant and Montandon1984; Clarke and Rothery, Reference Clarke and Rothery2008; Vester et al., Reference Vester, Burke, Dikeman, Simmons and Swanson2008; Yang et al., Reference Yang, LaMarca, Kaminski, Chu and Hu2017) than free-living environments, and this may diminish the applicability of ecological theory to the cross-host transmission of Blastocystis and other parasites. Alternatively, the broad categories we use to classify diet in this study may have failed to capture the important differences in nutritional, physical and chemical environments that influence differential survival and colonization capacity of Blastocystis species across hosts. Despite some convergence, the digestive environments of omnivorous humans, pigs, and fowl, for example, are quite different (Kararli, Reference Kararli1995). As dietary intakes have certainly been shown to influence the assemblies of both prokaryotic (David et al., Reference David, Maurice, Carmody, Gootenberg, Button, Wolfe, Ling, Devlin, Varma, Fischbach and Biddinger2014) and eukaryotic (Crompton, Reference Crompton1987; Wilcox and Hollocher, Reference Wilcox and Hollocher2018) components of vertebrate-associated gut communities, the importance of finer-scale dietary intakes cannot be ruled out.
Host taxonomy, insofar as it is an indicator of overall host similarity (Pedersen and Davies, Reference Pedersen and Davies2009; Nylin et al., Reference Nylin, Agosta, Bensch, Boeger, Braga, Brooks, Forister, Hambäck, Hoberg, Nyman and Schäpers2018), maybe a better indicator of niche overlap within-host microenvironments than diet. However, the taxonomic patterns of host usage that we observed are ambiguous with regard to the information that they provide on the internal host environment. That is to say, that while our tree was significantly structured around taxonomic patterns of host usage, these patterns of host usage were not typically reflective of the evolutionary relationships between families of hosts: the clade containing Subtypes 6, 7 and 9 sequences occurred in humans and birds – which are separated by more than 300 million years of evolution (Kumar and Hedges, Reference Kumar and Hedges1998; Hedges and Kumar, Reference Hedges and Kumar2004) – but was rare in non-human mammals, despite certain exposure between domestic mammals and the domestic fowl that host this clade. These types of patterns, which occur throughout the tree, suggest that the shared phylogenetic history of hosts is not an essential influence on the niche space available within these hosts. Overall then, the significant correlation between conserved taxonomic patterns of hosts and tree structure would seem to reflect an important role for historical contingency (i.e. adaptation to ancestral hosts) in the evolutionary patterns of host usage within the Blastocystis species-complex. That is, Blastocystis spp. would appear to more easily infect hosts that they have infected previously, regardless of the evolutionary relationships between these hosts. These findings are in keeping with previous reports of host switching in other systems, and theoretical proposals that suggest retention of adaptation to plesiomorphic (i.e. ancestral) hosts as an important driver of future host-switching events (Agosta et al., Reference Agosta, Janz and Brooks2010). While the overall commonality of this phenomenon is hard to assess, this finding is also consistent with the retained capacity of many human parasites to utilize unrelated animal hosts as reservoirs (Esch and Petersen, Reference Esch and Petersen2013; Šlapeta, Reference Šlapeta2013; Franco et al., Reference Franco, Simarro, Diarra and Jannin2014; Gürtler and Cardinal, Reference Gürtler and Cardinal2015). While we find evidence that Blastocystis spp. are under evolutionary constraint with respect to available hosts, the basis of this constraint remains unresolved. We note, however, that Blastocystis has the smallest reported genome within its phylum and that these reductions are consistent with trends in loss of genome size among other parasites as they become more reliant on host functions (Denoeud et al., Reference Denoeud, Roussel, Noel, Wawrzyniak, Da Silva, Diogon, Viscogliosi, Brochier-Armanet, Couloux, Poulain and Segurens2011). Examinations of these losses and the manner in which Blastocystis has become reliant on host functions may provide additional insights into the ecological determinants of niche space and niche breadth within this parasite.
Overall, our analytical approach demonstrates the utility of Blastocystis for the study of host–parasite evolution and questions of host specificity in particular. To a large extent, studies on host switching and host specificity have focused on plant–insect systems, parasitoids systems and to a lesser extent, metazoan parasites of vertebrates (Desneux et al., Reference Desneux, Blahnik, Delebecque and Heimpel2012; Vienne et al., Reference Vienne, Refrégier, López-Villavicencio, Tellier, Hood and Giraud2013; Nylin et al., Reference Nylin, Agosta, Bensch, Boeger, Braga, Brooks, Forister, Hambäck, Hoberg, Nyman and Schäpers2018). The omnipresence of Blastocystis spp. across higher animals (i.e. coelomates) makes it an excellent candidate organism for studying questions surrounding the ecology and evolution of host usage within microbial eukaryotic symbionts of metazoans. At present, little is known regarding evolutionary patterns of host usage among eukaryotic parasites, commensals and mutualists. However, elucidating these patterns remains important to a wide range of fields, including prevention of emerging infectious disease, host-associated microbiome research, and wildlife conservation (Thompson et al., Reference Thompson, Lymbery and Smith2010; Harbison and Clayton, Reference Harbison and Clayton2011; Brucker and Bordenstein, Reference Brucker and Bordenstein2012; Morens and Fauci, Reference Morens and Fauci2013; Brockhurst et al., Reference Brockhurst, Chapman, King, Mank, Paterson and Hurst2014; Lymbery et al., Reference Lymbery, Morine, Kanani, Beatty and Morgan2014; Auld and Tinsley, Reference Auld and Tinsley2015; Hoberg and Brooks, Reference Hoberg and Brooks2015; Stricker et al., Reference Stricker, Harmon, Goss, Clay and Luke Flory2016). The application of macroscopic free-living ecological theory has been proposed (Agosta et al., Reference Agosta, Janz and Brooks2010; Araujo et al., Reference Araujo, Braga, Brooks, Agosta, Hoberg, von Hartenthal and Boeger2015; Nylin et al., Reference Nylin, Agosta, Bensch, Boeger, Braga, Brooks, Forister, Hambäck, Hoberg, Nyman and Schäpers2018) as a solution to problems in our contemporary understanding of host switching and host specificity. Our findings provide support for this proposal and the paradigm of ecological fitting in a protistan parasite of vertebrates.
Acknowledgements
This research utilized High-Performance Computing Resources provided by the Notre Dame Center for Research and Computing and the New York University Abu Dhabi ‘DALMA’ High-Performance Computing Cluster. We also extend our thanks to Georgia-Leigh Hewitt for her assistance in compiling data on individual Blastocystis sequences.
Financial support
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. This research was supported by funds from the College of Science at the University of Notre Dame and the (United States) National Science Foundation Research Experience for Undergrads (NSF REU) Program.
Conflicts of interest
None.
Ethical standards
This research utilized non-invasive sampling of macaques in accordance with accepted principles of animal welfare and with the approval of the University of Notre Dame IACUC (protocol IDs 07-001, 14-002 and 14-05-1835).


