Introduction
The genus Cladonia P. Browne represents one of the largest genera of lichen-forming fungi, with more than 400 described species (Ahti Reference Ahti2000). Cladonia species are often major contributors to overall biomass in the ground-layer vegetation in arctic and alpine tundra, in lichen woodlands, on rock outcrops, on heaths, and on peatlands (Lechowicz & Adams Reference Lechowicz and Adams1974). Several Cladonia species, commonly termed reindeer lichens, serve as a winter food source for animals. Other species are found in habitats where higher plants are not competitors, such as wood or burned habitats. These lichens usually develop two distinct kinds of thallus morphology: a horizontal primary thallus (foliose or crustose, largely absent in reindeer lichens) and a vertical secondary thallus called a podetium (fruticose, bearing the hymenia). These thalli are among the most complex and aesthetic in lichens and, not surprisingly, there is a tremendous variation in morphological details, which provides many characters for classification. As frequently found with lichens, the interpretation of phenotypic variation of the thallus has been controversial (Stenroos & DePriest Reference Stenroos and DePriest1998; Stenroos et al. Reference Stenroos, Hyvonen, Myllys, Thell and Ahti2002; Divakar et al. Reference Divakar, Crespo, Blanco and Lumbsch2006; Grube & Hawksworth Reference Grube and Hawksworth2007).
The traditional species circumscription of Cladonia is based on morphological and chemical characters. However, several recent molecular studies have revealed a lack of correlation between morphological and molecular data, and many traditionally delimited species are problematic or even artificial in light of these data (Myllys et al. Reference Myllys, Stenroos, Thell and Ahti2003; Kotelko & Piercey-Normore Reference Kotelko and Piercey-Normore2010; Piercey-Normore et al. Reference Piercey-Normore, Ahti and Goward2010; Pino-Bodas et al. Reference Pino-Bodas, Martín and Burgaz2012a , Reference Pino-Bodas, Rosa Burgaz, Martín and Lumbsch b ). The incongruence between morphological and genetic data is usually attributed either to significant intraspecific variation of the species as a response to environmental conditions, or to genetic recombination (e.g., Fontaine et al. Reference Fontaine, Ahti and Piercey-Normore2010; Kotelko & Piercey-Normore Reference Kotelko and Piercey-Normore2010).
Zeorin-containing red-fruited Cladonia species are conspicuous lichens, two of which were distinguished by Linnaeus (Reference Linnaeus1753). Similar to most other Cladonia species, members of this aggregate usually grow in habitats with a low rate of competition from vascular plants (e.g., on sandy or rocky soils, on thin soil over rock, on bark, or on rotten wood). Currently, the aggregate of zeorin-containing red-fruited and scyphose (cup-forming) Cladonia species consists of five species worldwide, of which four are known from Europe and North America [C. coccifera (L.) Willd., C. deformis (L.) Hoffm., C. diversa Asperges ex S. Stenroos, and C. pleurota (Flörke) Schaer]. The fifth species, C. sinensis S. Stenroos & J. B. Chen (Stenroos et al. Reference Stenroos, Vitikainen and Koponen1994), has a limited distribution in South-East Asia, and was not included in the analysis.
This group of species is characterized by similar chemical patterns (presence of usnic acid derivates and zeorin, occasionally accompanied by porphyrilic acid), and species within this group are delimited morphologically. The size, shape and location of the vegetative propagules on the podetia are traditionally considered as the most important diagnostic characters separating species (e.g., Asperges Reference Asperges1983; Stenroos Reference Stenroos1989). The shape and width of the podetium is another relevant morphological feature commonly used to distinguish the species belonging to this group (e. g., Asperges Reference Asperges1983; Osyczka Reference Osyczka2011; Ahti & Stenroos Reference Ahti and Stenroos2012).
Cladonia coccifera (Fig. 1A & B) is an esorediate species with gradually expanded cups. The surface of the podetium is areolate corticate, covered by bullate and scaly plates. This species had often been confused with C. diversa, C. pleurota or C. borealis (Stenroos Reference Stenroos1989; Osyczka Reference Osyczka2011). Cladonia deformis (Fig. 1C & D) is easy to recognize when well developed. Podetia are usually tall, relatively narrow and farinose sorediate. However, it might also be short-podetiate and then difficult to distinguish from C. pleurota (Osyczka Reference Osyczka2011). Cladonia pleurota (Fig. 1G & H) is morphologically very variable (Stenroos Reference Stenroos1989). Young individuals are usually completely granulose sorediate, but when fertile the surface may turn almost totally corticate and is partly covered by granules or verruculae (Stenroos Reference Stenroos1989). Cladonia diversa (Fig. 1E & F), described by Asperges (Reference Asperges1983), is the most controversial species. The podetia of this species are usually slender and microsquamulose-granulose. Because of its obvious morphological similarity to C. coccifera (esorediate podetia covered by irregular plates and/or granules), the natural status of this species was disputed by Stenroos (Reference Stenroos1989). However, recently Ahti & Stenroos (Reference Ahti and Stenroos2012) became “more convinced that it is an acceptable taxon”.

Fig. 1. Morphology of Cladonia species studied. A & B, Cladonia coccifera (CL179); C & D, C. deformis (CL176); E & F, C. diversa (CL173, topotype); G & H, C. pleurota (CL136). Scales: A, C, E & G=5 mm; B, D, F & H=1 mm. In colour online.
Until now, no comprehensive attempt has been made to assess phenotypically circumscribed red-fruited Cladonia species within a molecular phylogenetic context. However, some species belonging to this aggregate were included in previous studies which focused on the generic phylogeny of Cladonia (Stenroos et al. Reference Stenroos, Hyvonen, Myllys, Thell and Ahti2002), a study of lichen diversity in some Antarctic regions (Lee et al. Reference Lee, Lee, Hur, Andreev and Hong2008), or a DNA-barcoding study of taxonomically diverse lichens in the UK (Kelly et al. Reference Kelly, Hollingsworth, Coppins, Ellis, Harrold, Tosh and Yahr2011). Stenroos et al. (Reference Stenroos, Hyvonen, Myllys, Thell and Ahti2002) examined four species belonging to this aggregate (from 1 to 4 specimens per species) and the possible polyphyly of C. coccifera.
Many phylogenetic surveys using sequence data inferred the evolution of Cladonia at higher taxonomical levels. Myllys et al. (Reference Myllys, Stenroos, Thell and Ahti2003) investigated the genetic diversity of two closely related putative species, C. arbuscula and C. mitis. The analysis involved four markers: ITS rDNA, a group I intron in SSU rDNA at position 1516 (according to Escherichia coli numbering), two introns in β-tubulin gene, and a single intron in the GAPDH gene. Surprisingly, a significant conflict between the four gene regions was detected. According to their conclusions, Myllys et al. (Reference Myllys, Stenroos, Thell and Ahti2003) regarded either incomplete lineage sorting or recombination as the most likely reason for the incongruences among the markers. Recently, Fontaine et al. (Reference Fontaine, Ahti and Piercey-Normore2010) studied the C. gracilis complex by using ITS rDNA and polyketide synthase (PKS) genes and obtained similar results, but they suggest paralogy in both genes as an alternative explanation of the incongruence identified between individual gene trees.
In the present study, inferences from ITS rDNA and an intron-containing portion of the β-tubulin gene were used to explore the genetic diversity of currently recognized zeorin-containing red-fruited Cladonia species. We examined numerous collections of all the four currently accepted zeorin-containing red-fruited Cladonia species known from the European continent and North America. In addition, we address possible explanations for the incongruence between individual gene trees detected in this study, similar to other multilocus studies of Cladonia (Myllys et al. Reference Myllys, Stenroos, Thell and Ahti2003; Fontaine et al. Reference Fontaine, Ahti and Piercey-Normore2010).
Materials and Methods
Species sampling and determination
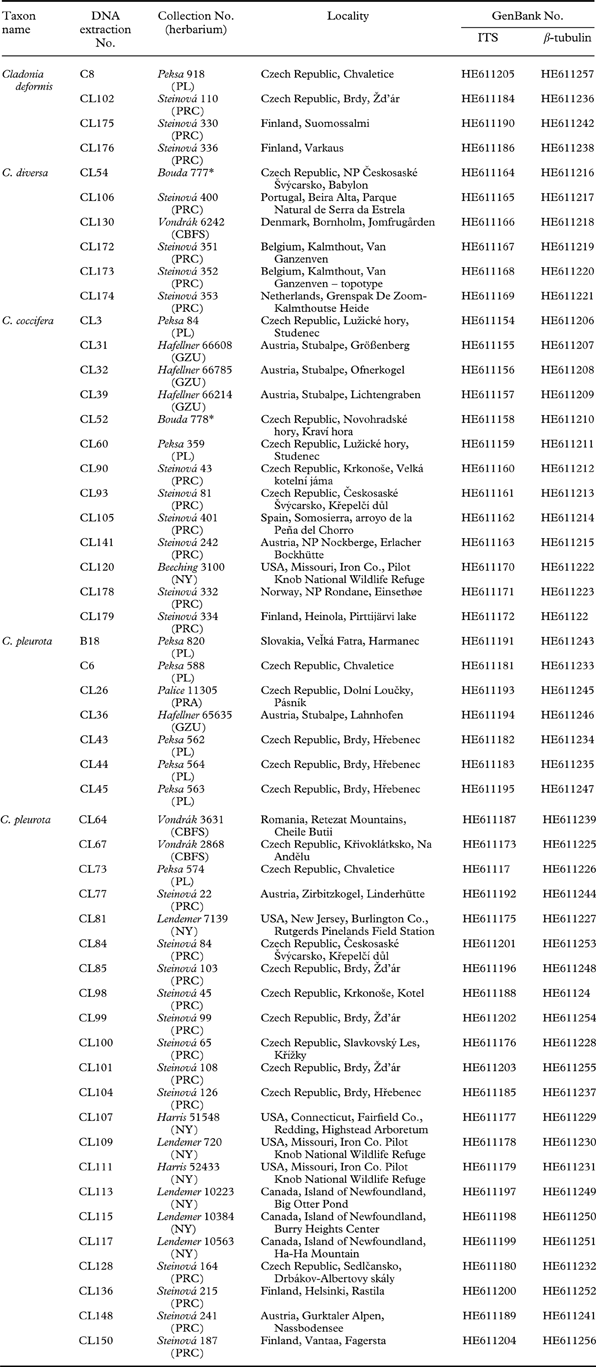
The material for this study was either collected by the authors or obtained from the following herbaria: BG, CBFS, GZU, NY, PL, PRA, PRC, and PRM. A total of 52 samples were collected, largely in Europe (44 specimens); eight collections were made in North America (Table 1). All the specimens were examined by the first author and revised by S. Stenroos and T. Ahti. Patterns in secondary metabolite variation were identified by thin-layer chromatography (TLC) on Merck silica gel 60 F254 pre-coated glass plates in solvent systems A, B and C, according to Orange et al. (Reference Orange, James and White2001). Cladonia crispata and C. squamosa were used as an outgroup, based on the study of Stenroos et al. (Reference Stenroos, Hyvonen, Myllys, Thell and Ahti2002).
Table 1. List of lichen taxa used in this study with collection information and GenBank accession numbers.
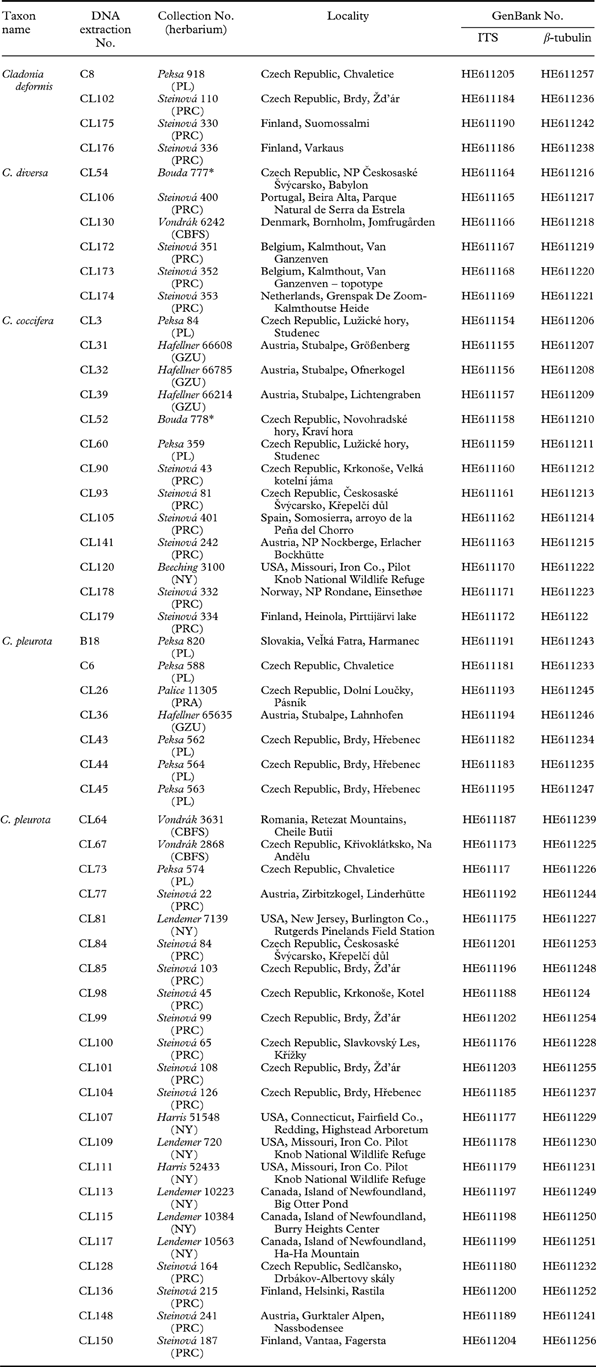
* private herbarium
DNA extraction, PCR, and DNA sequencing
Fine ground lichen material was used for total genomic DNA extraction with the CTAB protocol (Cubero et al. Reference Cubero, Crespo, Fatehi and Bridge1999) or the Invisorb Spin Plant Mini Kit (Invitek). The fungal nuclear ITS region and an intron-containing portion of the β-tubulin gene were amplified with the following primers: ITS1F (Gardes & Bruns Reference Gardes and Bruns1993) and ITS4 (White et al. Reference White, Bruns, Lee, Taylor, Innis, Gelfand, Sninsky and White1990), and Bt3-LM and Bt10-LM (Myllys et al. Reference Myllys, Lohtander and Tehler2001). In most cases, PCR reactions were prepared for a 30 µl final volume containing 4·05 µl double-distilled water, 3 µl 10×Taq polymerase reaction buffer (10 mM Tris; pH 8·3), 1·8 µl MgCl2 (25 mM), 3 µl of 2·5 mM dNTPs, 0·15 µl Taq DNA polymerase, 1·5 µl of each of the 10 mM primers. Amplifications consisted of an initial 2 min denaturation at 95°C, followed by 30 cycles of 1 min at 95°C, 1 min at 54°C (ITS)/51°C (β-tubulin), 1 min at 72°C, and a final extension of 7 min at 72°C.
The PCR products were quantified on a 1% agarose gel stained with ethidium bromide and cleaned either with QIAquick PCR Purification Kit (Qiagen) or JetQuick PCR Product Purification Kit (Genomed), according to the manufacturer's protocols.
Sequencing of PCR products was performed with an Applied Biosystems (New York, USA) automated sequencer (ABI 3730xl) at Macrogen Corp. in Seoul, Korea. The PCR primers were also used for sequencing.
Sequence alignment and model selection
Sequences were initially aligned using CLUSTAL X 1.83 (Thompson et al. Reference Thompson, Gibson, Plewniak, Jeanmougin and Higgins1997) and MUSCLE (Edgar Reference Edgar2004). ITS sequences (comprising ITS1, 5.8S, and ITS2 regions) were aligned on the basis of their rRNA secondary structure information (see below) with MEGA 4 (Kumar et al. Reference Kumar, Nei, Dudley and Tamura2008). For subsequent phylogenetic analyses, the alignments were minimalized to contain the unique sequences only. Alignments can be downloaded at http://botany.natur.cuni.cz/algo/align/03_Cladonia_ITS.nex (ITS) http://botany.natur.cuni.cz/algo/align/03_Cladonia_BT.nex (β-tubulin).
For both ITS and β-tubulin datasets, suitable partitioning strategy and partition-specific substitution models were selected in a multi-step process (Verbruggen et al. Reference Verbruggen, Maggs, Saunders, Le Gall, Yoon and De Clerck2010). Initially, guide trees were obtained by carrying out a second-level maximum likelihood (ML) search on the unpartitioned dataset with an HKY + Γ8 model with TreeFinder (Jobb et al. Reference Jobb, von Haeseler and Strimmer2004) by using the Bayesian information criterion (BIC). Then, the datasets were divided by five (ITS) and six (β-tubulin), respectively, different partitioning strategies. For each partition present in these partitioning strategies, 12 different nucleotide substitution models were evaluated (F81, HKY, GTR, and their combinations with Γ, I, and Γ +I). Subsequently, Bayesian information criterion (BIC) calculations were performed for all potential partitioning strategies, assuming the guide tree and evaluated models for each partition. For both datasets, three partitioning strategies with the best fit to the data (lowest BIC scores) were retained for further analysis. In the next step, the best models of sequence evolution were selected for individual partitions by using the BIC. Finally, the partitioning strategies were re-evaluated using the selected models for particular partitions. This BIC-based model selection procedure selected the following models. For the ITS rDNA dataset, the strategy with 3 partitions was selected: i) ITS1 region (HKY + Γ8), ii) 5.8S rDNA (HKY), and iii) ITS2 region (HKY + Γ8). In the case of the β-tubulin dataset, the strategy with two partitions was selected as the best: (i) first and second codon positions of exon (HKY), and (ii) third codon position of exon and intron region (HKY).
Molecular data and phylogenetic analyses
Possible substitution saturation of both markers studied that would imply a low reliability of phylograms (Lopez et al. Reference Lopez, Forterre and Philippe1999; Muschner et al. Reference Muschner, Lorenz, Cervi, Bonatto, Souza-Chies, Salzano and Freitas2003) was assessed by two different approaches. Firstly, we plotted the uncorrected distances against the corrected distances, determined with the respective model of sequence evolution estimated by the BIC-based model selection as described above (HKY + Γ8 for ITS rDNA and HKY for the β-tubulin dataset). Secondly, the phylogenetic signal present in the data partitions was estimated by ML mapping (Strimmer & von Haeseler Reference Strimmer and von Haeseler1997) using the Tree-puzzle 5.2 program (Schmidt et al. Reference Schmidt, Strimmer, Vingron and von Haeseler2002).
The phylogenetic trees were inferred with Bayesian inference (BI), Maximum Likelihood (ML) and Maximum Parsimony (MP). A Bayesian analysis was implemented using MrBayes version 3.1 (Ronquist & Huelsenbeck Reference Ronquist and Huelsenbeck2003). Two parallel MCMC runs were carried out for 2 million generations, each with one cold and three heated chains. Trees and parameters were sampled every 100 generations. Convergence of the two cold chains was assessed during the run by calculating the average standard deviation of split frequencies (SDSF). The SDSF value between simultaneous runs was 0·004683 in ITS and 0·002003 in the β-tubulin. ML and MP phylograms were obtained using Garli version 2.0, and PAUP version 4.0b10 (Swofford Reference Swofford2002), respectively. The same programs were used for bootstrap analyses. ML analyses consisted of rapid heuristic searches (100 pseudo-replicates) by using automatic termination (the genthreshfortopoterm command set to 100 000). The weighted parsimony (wMP) bootstrapping (1000 replications) was performed using heuristic searches with 100 random sequence addition replicates, tree bisection reconnection swapping, random addition of sequences (the number limited to 10 000 for each replicate), and gap characters treated as a fifth character state. The weight to the characters was assigned using the rescaled consistency index on a scale of 0 to 1000. New weights were based on the mean of the fit values for each character over all of the trees in memory.
The secondary structures of ITS sequences were constructed in order to detect presumed sexual barriers between studied species. The incompatibility for sexual reproduction between species can be ascertained by the presence of compensatory base changes (CBSs; so-called CBS approach) (Coleman Reference Coleman2000; Müller et al. Reference Müller, Philippi, Dandekar, Schultz and Wolf2007). The secondary structures of ITS sequences were constructed using the Mfold computer program version 2.3, (Walter et al. Reference Walter, Turner, Kim, Lyttle, Muller, Mathews and Zuker1994; Zuker Reference Zuker2003), with the folding temperature set to 25°C. The structures were compared with published ITS secondary structures of Cladonia species (Beiggi & Piercey-Normore Reference Beiggi and Piercey-Normore2007). Common secondary structures were created by using RnaViz (version 2; De Rijk et al. Reference De Rijk, Wuyts and De Wachter2003) and used to identify compensatory base changes (CBCs) and hemi-CBCs.
Analyses of hybridization
Two different attempts were used to detect hybridization events in the diversification of Cladonia species. Firstly, incongruence between the ITS and β-tubulin-derived trees was examined using NeighborNet analysis as implemented by the program Splits Tree 4 (Huson & Bryant Reference Huson and Bryant2006). This method provides a visualization of the extent to which a collection of gene trees suggests contradictory taxon relationships. If a collection of gene trees has congruent topologies, consensus networks will be tree-like, and where the relationships are incongruent, the graphs will be net-like (McBreen & Lockhart Reference McBreen and Lockhart2006). To explain the incongruent relationships displayed by a network analysis in terms of reticulation events, a consensus network was constructed.
Secondly, the evidence of hybridization was evaluated by the bootscanning method (Salminen et al. Reference Salminen, Carr, Burke and Mccutchan1995) on the concatenated sequence dataset. We used two different programs to run bootscanning analyses: 1) the alignment was analyzed by SimPlot version 3.5.1 (Lole et al. Reference Lole, Bollinger, Paranjape, Gadkari, Kulkarni, Novak, Ingersoll, Sheppard and Ray1999), using the bootscan option and default settings; 2) several different algorithms (RDP, GENECONV, Chimaera, MaxChi, BootScan, SiScan and 3Seq) were run to determine the presence of recombination using the Recombination Detection Program, Rdp3, v. 3.22 (Martin et al. Reference Martin, Lemey, Lott, Moulton, Posada and Lefeuvre2010). In the case of positive recombination detection, bootstrap support curves were visualized to locate hybrid sequences, and to reveal potential parent sequences present in the alignment.
Results
Secondary chemistry
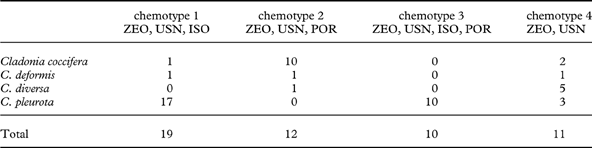
All samples studied contained zeorin and usnic acid as major lichen substances. We detected four chemotypes which differ by the presence/absence of accessory substances isousnic and porphyrilic acids. Isousnic acid (chemotype 1) was present in 19 specimens, porphyrilic acid in 12 specimens (chemotype 2), both were present (chemotype 3) in 10 specimens and neither of the two mentioned (chemotype 4) were found in 11 specimens (Table 2).
Table 2. Number of Cladonia specimens studied of each chemotype
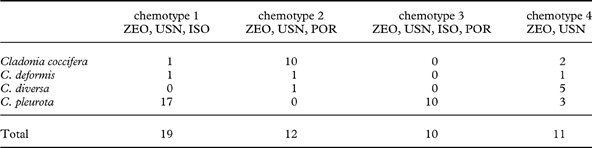
ISO=isousnic acid; POR=porphyrilic acid; USN=usnic acid; ZEO=zeorin
Analysis of the molecular data
Amplification products of the ITS1, 5.8S, and ITS2 regions of the ribosomal rRNA gene were c. 600 bp long, while those of the two exon and one intron regions of the β-tubulin genes were c. 700 bp in length. Although the number of nucleotides analyzed differed accordingly (ITS: 546 bp, β-tubulin: 674 bp), the datasets were comparable in the amount of phylogenetic signal. Although the ITS dataset contained more variable sites (ITS: 44 sites; β-tubulin: 31 sites), the number of parsimony-informative characters was the same for both loci (31). No ambiguous positions that could bias the inference of phylogeny were detected.
Testing the data partitions for substitution saturation (distribution of the uncorrected vs corrected distances) revealed a practically linear correlation, indicating no saturation in both ITS and β-tubulin data (see Appendix 1). Similarly, the results of likelihood mapping demonstrated a strong phylogenetic signal detected in both ITS and β-tubulin loci (89·2% and 94·1% of the fully resolved quartets, respectively).
Phylogenetic analyses
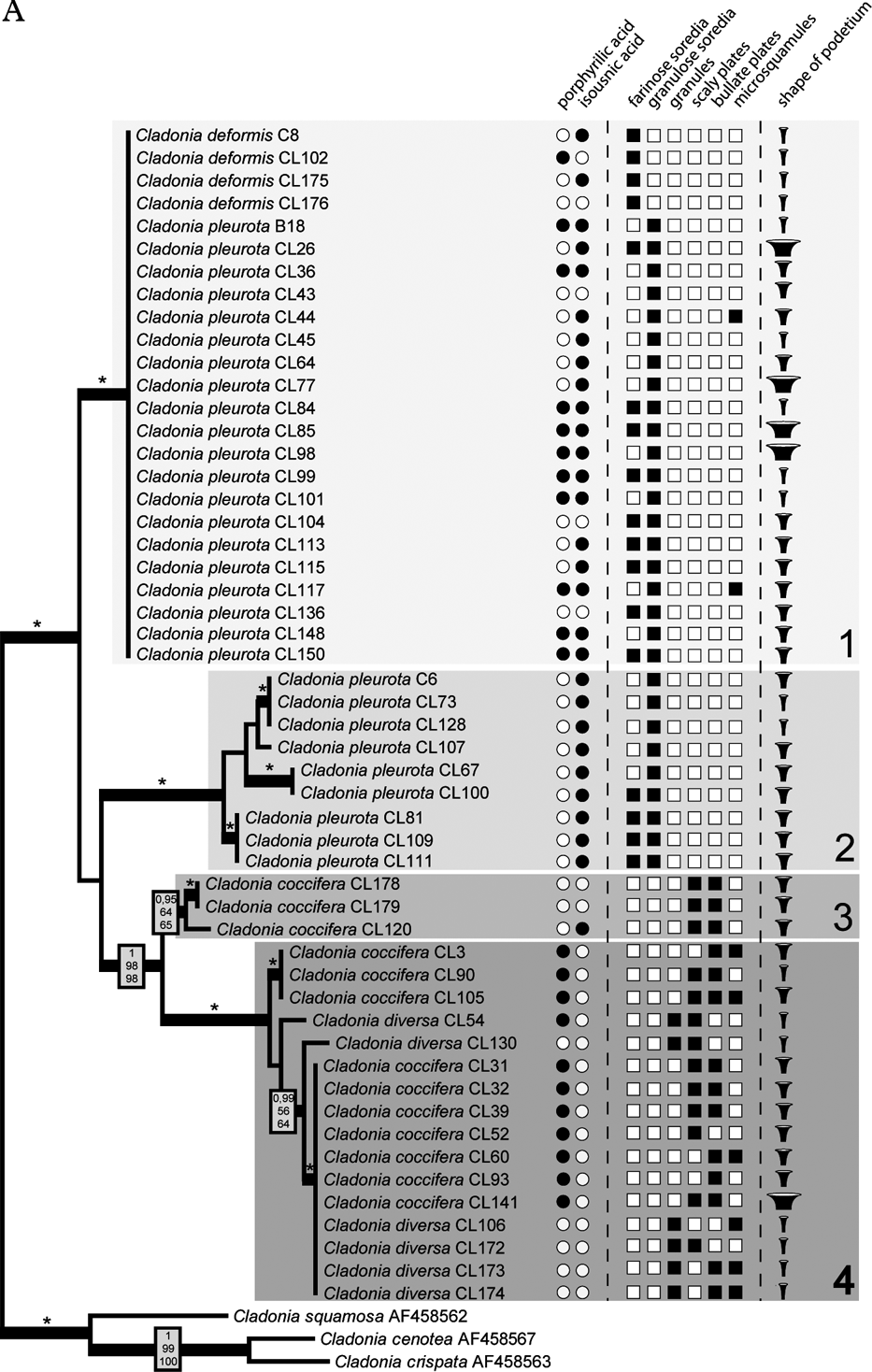
The Bayesian, MP and ML analyses yielded trees with similar topology. Figure 2 shows the phylogram obtained from the Bayesian analysis. It revealed three well-supported (#1, #2, and #4) and one moderately supported (#3) lineages. Lineage #1 comprised 24 identical sequences belonging to C. deformis and C. pleurota. Cladonia pleurota strains also formed lineage #2. In contrast, lineage #4 contained sequences belonging to both C. coccifera and C. diversa, and lineage #3 comprised three C. coccifera strains.
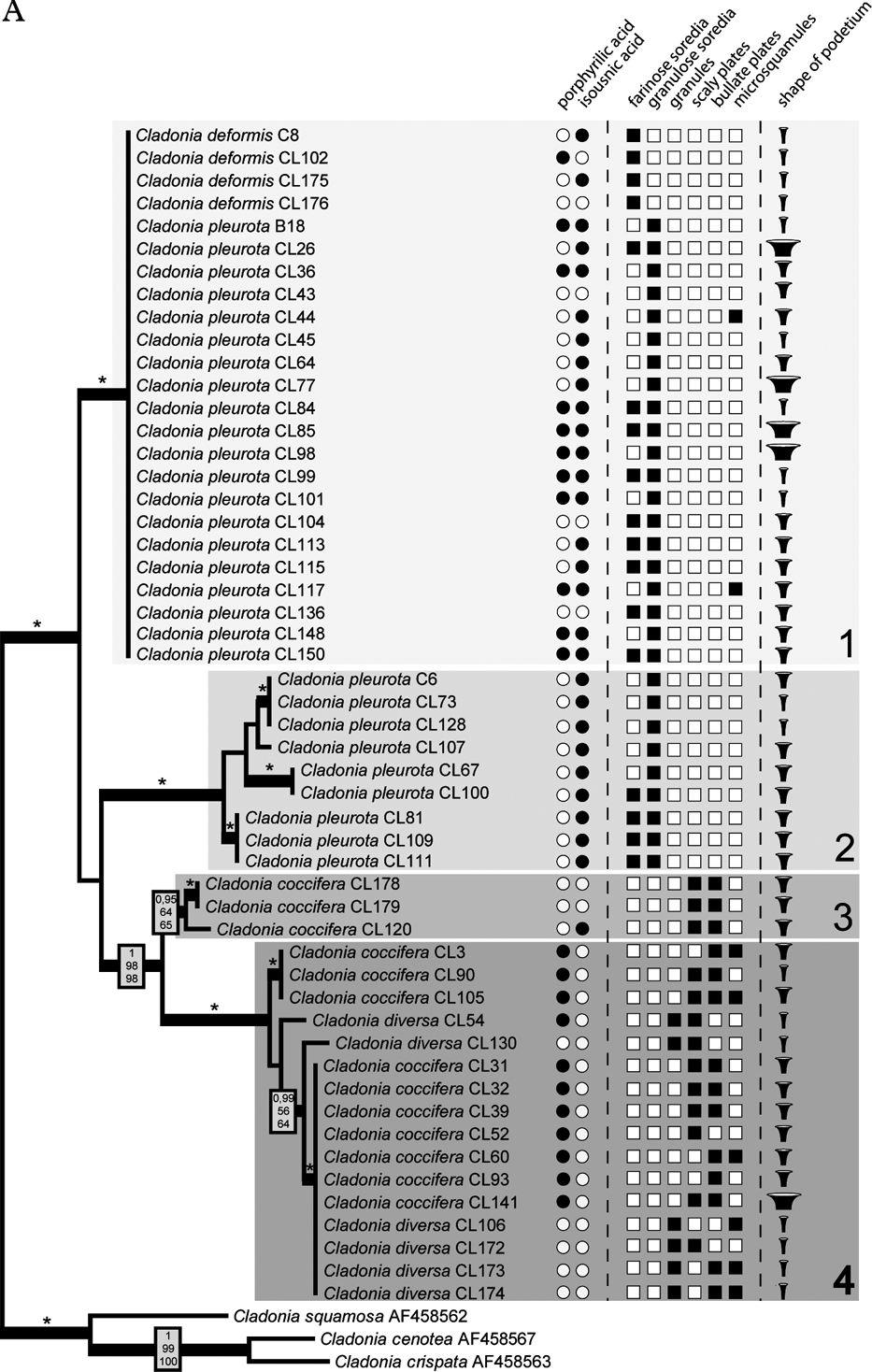
Fig. 2. Comparison of Bayesian topologies based on the β-tubulin (A) and ITS rDNA (B) datasets, together with observed chemical and morphological characters. For the analyses, an HKY + Γ8 model for ITS1 and ITS2 regions, and HKY model for 5.8S rDNA, three codon positions and exon of β-tubulin gene was used. Values at the nodes indicate statistical support estimated by three methods: MrBayes posterior node probability (top), maximum likelihood bootstrap (in the middle) and maximum parsimony bootstrap (bottom). Thick branches represent nodes receiving PP support ≥0·90; asterisks (*) indicate statistical support 1/100/100. Only values receiving PP support ≥0·90 are shown. Species affiliation to four β-tubulin clades (including the corresponding sub-clades on the ITS rDNA tree) is indicated. Scale bar: estimated number of substitutions per site. ![]() , displays narrow, slender podetia;
, displays narrow, slender podetia; ![]() , moderately broad cups;
, moderately broad cups; ![]() , extremely broad podetia.
, extremely broad podetia.

Fig. 2. Continued
Compared to the β-tubulin gene tree, ITS phylogenetic analysis inferred a clearly different topology. Three of the four well-resolved lineages of β-tubulin phylogeny were not resolved, but separated into different and distantly related clades (Fig. 2). Lineage #1 was separated into four lineages (#1a, #1b, #1c, and #1d). Whereas lineages #1a, #1c, and #1d contained both C. pleurota and C. deformis specimens, lineage #1b comprised only the C. pleurota strains. Lineage #4 from the β-tubulin gene tree formed lineages #4a and #4b in the ITS phylogram. Although receiving low support, they were obviously unrelated. Clade #4a was composed of all the analyzed C. diversa strains, whereas lineage #4b contained sequences belonging to C. coccifera. Finally, lineage #3 was split into two lineages: unsupported lineage #3a and lineage #3b containing only one sequence. Lineage #2 was the only one that was inferred with high statistical support by both ITS and β-tubulin phylogenetic analyses.
Since ITS and β-tubulin phylogenies were obviously not congruent, concatenated analysis was not performed.
Hybridization tests
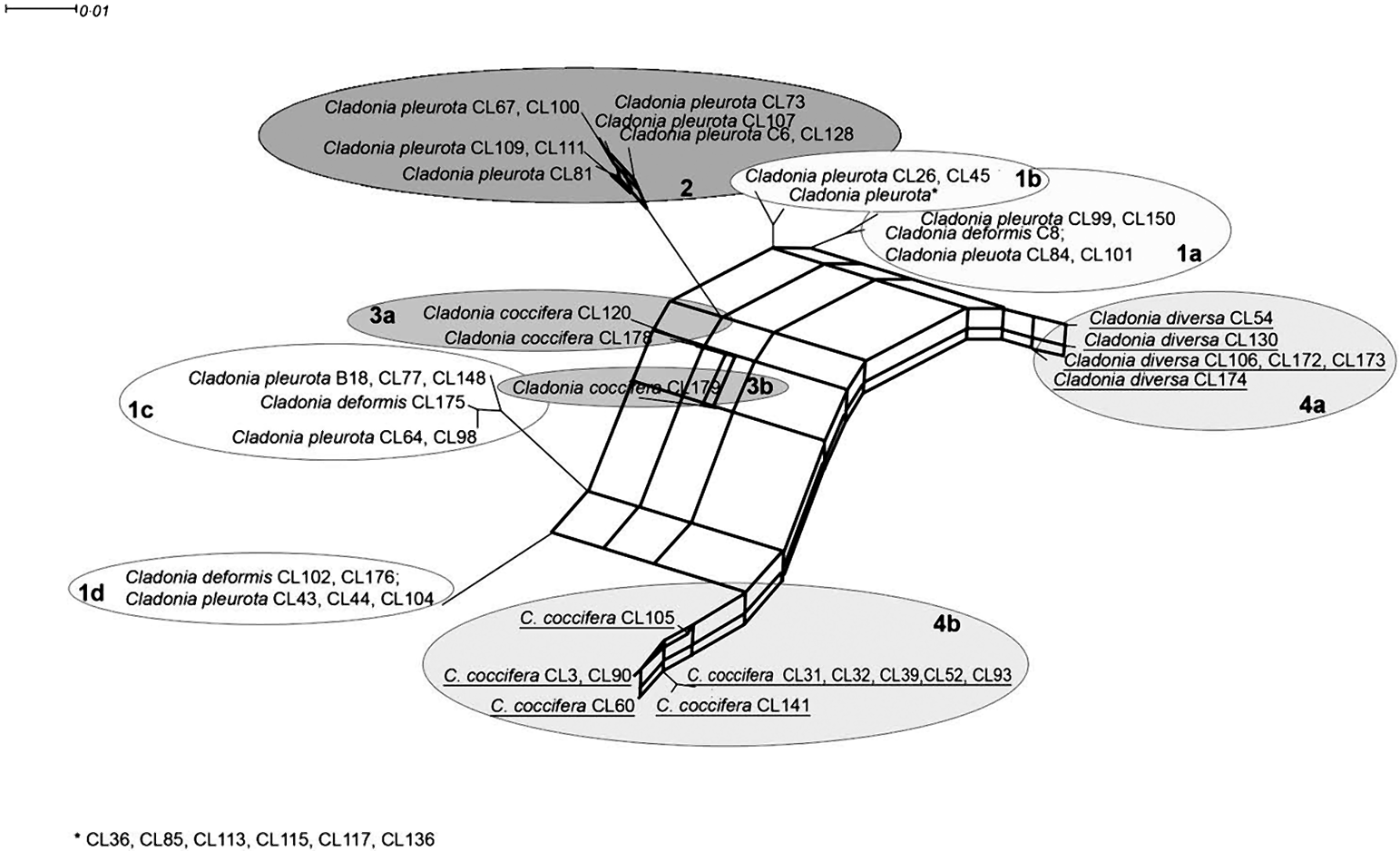
A visual comparison of ITS and β-tubulin phylograms indicated a discrepancy in relationships among some taxa in both markers. For example, C. diversa formed a highly supported monophyletic clade together with some C. coccifera strains in the β-tubulin tree (lineage #4), but it created a separated lineage #4a in the ITS phylogram (which was, however, not statistically supported). A consensus network constructed from the trees obtained from the Bayesian analysis of the β-tubulin gene and ITS suggested contradictory taxon relationships. This network (Fig. 3) explains the conflict between source-tree topologies as a consequence of the hybridization event. Based on the investigation of concatenated datasets, the Phi test did find statistically significant evidence for recombination (P=1·1×10–7).
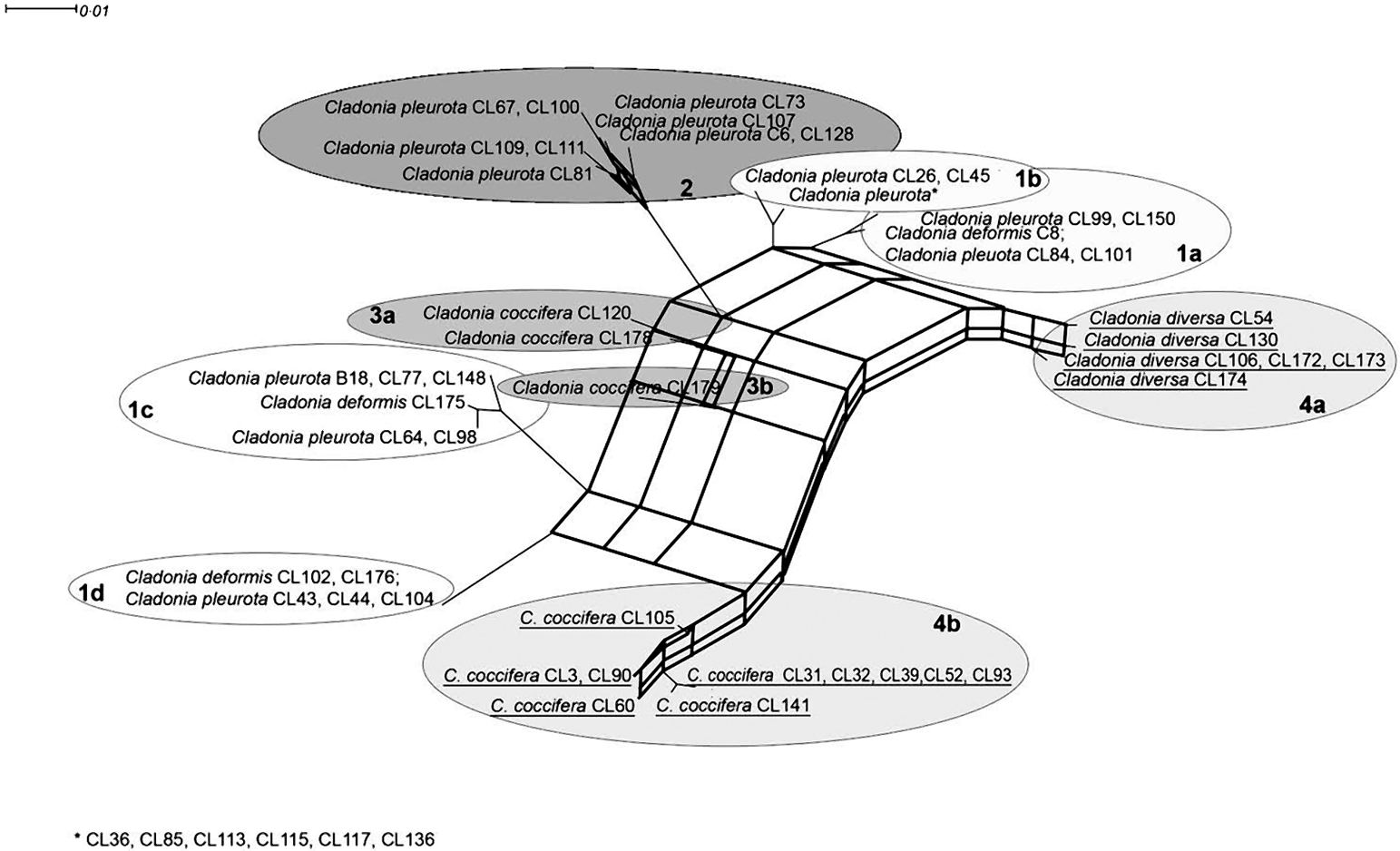
Fig. 3. Consensus network inferred from Bayesian trees from ITS and β-tubulin data. Thick lines represent hybridization events. Taxa involved in hybridization events are underlined. Scale bar shows the estimated number of substitutions per site.
The presence of the recombination event was also examined by two tests. Rdp3 analysis detected two hybridization events within both ITS and β-tubulin loci, which led to two hybrid lineages: #4a (all C. diversa strains) and #4b (some C. coccifera strains) (Fig. 3). The recombination was detected by three different tests implemented in Rdp3: MaxChi (P=0·0084), Chimaera (P=0·0030) and 3Seq (P=0·0006). These hybrid lineages formed highly supported monophyletic clade #4 in the β-tubulin tree. Based on the bootscanning analysis conducted by the SimPlot program, we discovered the probable ancestral lineages of both hybrids. In fact, only one parent lineage was unequivocally inferred for both hybrids. In the case of C. diversa (#4a), lineage #1a was identified as ancestral, whereas in the second case (#4b), the ancestral lineage was found to be lineage #1c. The second ancestral lineage in both cases of hybridization was unknown; however, SimPlot identified a hybrid as the ancestor of the other one, and vice versa. From this, we concluded that both of the hybrid lineages had a common, as yet unknown, parent lineage.
ITS1 and ITS2 secondary structure
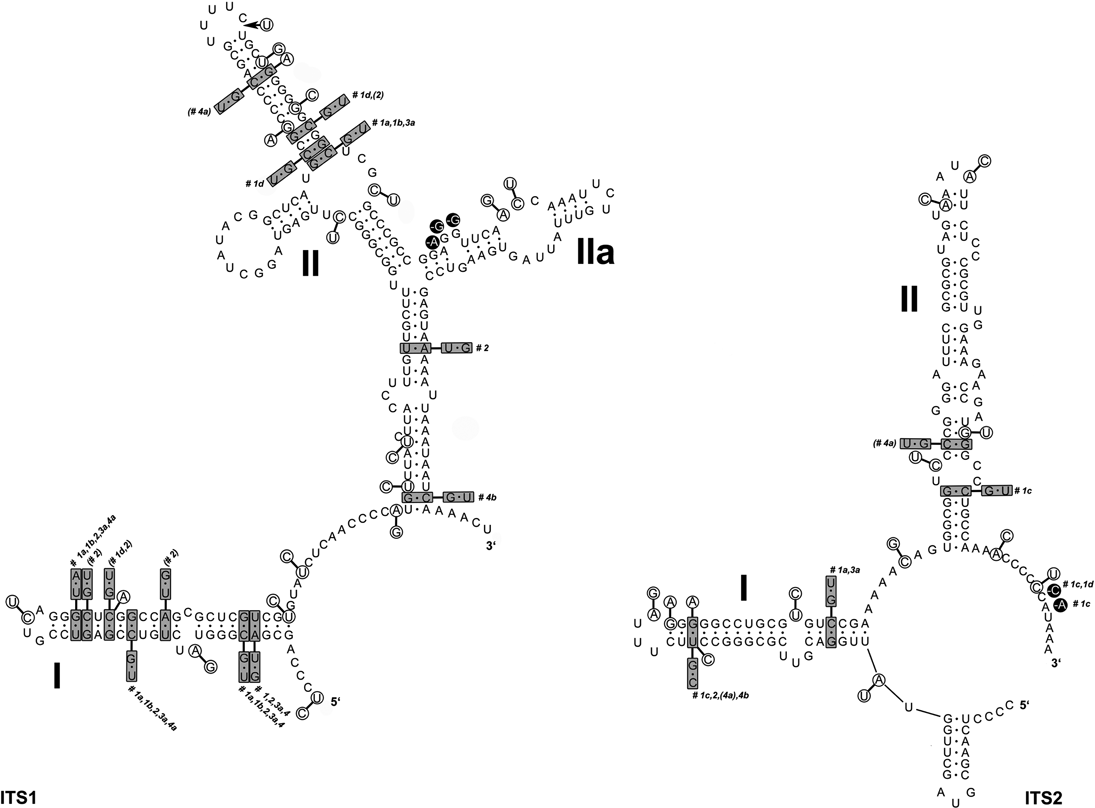
A common organization of ITS1 and ITS2 secondary structures was found in all Cladonia strains. The ITS1 secondary structure comprised two main paired regions (helices I and II), with two additional lateral helices (IIa and IIb) on helix II. Helix I was more divergent than helix II. The ITS2 structure was more conserved than that of ITS1. It contained three paired regions (helices I, II, and III). Helix I was identical in all the specimens examined, whereas helices II and III showed a small degree of divergence. The ITS secondary structures were compared with the lineages inferred in the ITS phylogram (Fig. 4) to check the occurrence of compensatory base changes (CBCs, nucleotide changes at both sides of the paired bases) and hemi-CBCs (change on only one side of the nucleotide pair, but still preserving pairing), according to Coleman (Reference Coleman2000, Reference Coleman2003). We revealed no CBC and 161 hemi-CBCs in all the lineages. The number of hemi-CBCs varied from zero (#3b) to six (#2) between the different lineages. Altogether, 15 hemi-CBC sites were identified in both the ITS regions. ITS1 contained 11 hemi-CBCs, of which seven were located in helix I. Four hemi-CBCs identified in ITS2 were situated in helices II and III. The highest number of hemi-CBCs (seven) was determined between lineages #1d–1a, #1d–4a, #1c–1a, #1c–2, and #1c–4a. In contrast, no hemi-CBC was identified between lineages #1a–4a.
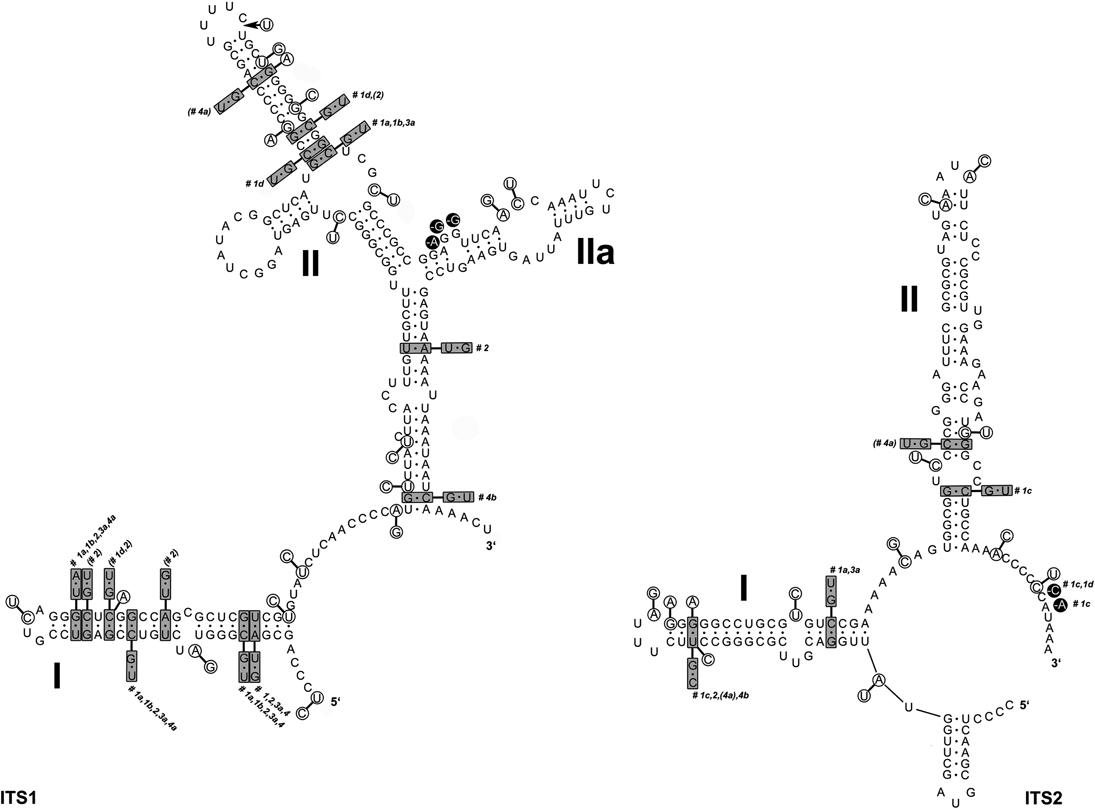
Fig. 4. Secondary structure models of ITS1 and ITS2 transcripts of Cladonia coccifera (DNA extraction number CL93) derived from comparison of 9 zeorin-containing red-fruited Cladonia lineages. Base changes between the different Cladonia genotypes are indicated: the base pairs marked in grey boxes indicate hemi-CBCs; single base changes are marked in circles. The numbers next to the boxes (#1a–4b) specify the Cladonia clades in which the base changes occurred (see Figs 2 & 3).
Discussion
Phenotypic and genetic variability of red-fruited Cladonia species
The four species considered in this paper are chemically very similar but differ morphologically. The most obvious differences characterizing these species are the size and shape of the podetium, the character of the podetium surface, and the character and size of the vegetative propagules. However, three of these species were shown to be polyphyletic. Only Cladonia diversa formed a monophyletic group in the ITS phylogeny, although it was not supported statistically. Traditionally, this species was regarded as a member of the C. coccifera group. Stenroos (Reference Stenroos1989) doubted the status of C. diversa and found that its total variation is still obscure. Recently, Ahti & Stenroos (Reference Ahti and Stenroos2012) accepted the species as a valid taxon. All the specimens studied have slender, narrow scyphi and their surfaces are covered by microsquamules, irregular plates and granules (Fig. 2). Furthermore, we can also confirm its preference for sandy substrata (out of six specimens, four were collected from sandy dunes, one from a sandstone), as previously reported by Asperges (Reference Asperges1985), Christensen & Johnsen (Reference Christensen and Johnsen2001), Hasse (Reference Hasse2005), and Osyczka (Reference Osyczka2009). Also, chemical patterns are consistent with other sources (e.g. Osyczka Reference Osyczka2011; Ahti & Stenroos Reference Ahti and Stenroos2012). James (Reference James, Smith, Aptroot, Coppins, Fletcher, Gilbert, James and Wolseley2009) proposed porphyrilic acid to be a stabile compound of this species, but according to our results this cannot be confirmed (porphyrilic acid was detected in only one specimen of C. diversa).
The specimens morphologically identified as C. coccifera were distributed in three lineages (#3a, #3b, #4b). Two of these (#3a and #3b) were phylogenetically distant from lineage #4b. Ten specimens representing lineage #4b showed wide morphological variability (Fig. 2), particularly in the features of the vegetative propagules and the shape of podetia. The surface of podetia in some specimens was largely covered with scaly and bullate plates, whereas microsquamules dominated on podetia of other specimens studied. The shape also varied from narrow to very broad cups. In contrast, lineages #3a and #3b contained specimens that were morphologically more uniform and consistent with the traditional delimitation (however, only three C. coccifera specimens were inferred in these lineages). According to our results, clade #4b differed chemically from clades #3a and #3b. Whereas specimens involved in the lineage #4b are characterized by the presence of porphyrilic acid and the absence of isousnic acid, the three specimens from the other two lineages lacked porphyrilic acid. Interestingly, whereas these specimens were collected in Norway and Missouri, the specimens from the lineage #4b were sampled from Central Europe and Spain. This suggests a possible geographical pattern in the distribution (and also chemical variation) of these two lineages, which should be studied more carefully with wider sampling.
Although C. deformis is regarded as a distinct species, the specimens were found in three lineages: #1a, #1c, as well as #1d together with C. pleurota. These two species are traditionally distinguished by the size and shape of the podetium, and by the size of the soredia. Cladonia deformis usually forms elongated farinose-sorediate podetia, whereas C. pleurota is characterized by shorter and more coarsely sorediate podetia. The four studied specimens of C. deformis did not show any uniform chemical pattern. In one case (specimen Cl102 from the Czech Republic) we detected porphyrilic acid, which has not been reported for this species previously.
On the basis of the findings of Stenroos et al. (Reference Stenroos, Hyvonen, Myllys, Thell and Ahti2002), C. pleurota appeared to be a monophyletic species. However, our results with a more detailed sampling of the species disprove the monophyly of this taxon. Specimens of C. pleurota as currently understood are spread across five lineages (#1a, #1b, #1c, #1d, and #2). Although the specimens were studied thoroughly, we did not detect any morphological criteria that would properly characterize any of these lineages (Fig. 2). The podetial surface of all specimens used for the analysis was covered with granulose soredia, sometimes accompanied by farinose soredia in varying amounts. The shape of podetia exhibited considerable variation (from very narrow to short and extremely broad cups) that, however, does not correspond with the genetic diversity of the material studied. Discordance between the high morphological variation and sequence data has recently been discussed by Pérez-Ortega et al. (Reference Pérez-Ortega, Fernández-Mendoza, Raggio, Vivas, Ascaso, Sancho, Printzen and de Los Ríos2012), with respect to vagrant forms in Cetraria aculeata. However, the induction of growth variation as suggested for vagrant vegetative offspring cannot apply in Cladonia. It seems that, on the contrary, there are at least some chemical patterns or tendencies which may help us to define some clades. Lineage #2 comprises nine samples with identical chemical characteristics (chemotype 1: presence of isousnic acid and absence of porphyrilic acid). Porphyrilic acid appears to be a constant substance in samples of clade #1c, but with only six specimens studied we refrain from drawing taxonomic conclusions.
Discordance among gene-tree genealogies
In our study, we analyzed two commonly used and well-established molecular markers for Ascomycota: ITS rDNA and an intron-containing portion of the β-tubulin gene. The strong conflict between the ITS and β-tubulin topologies seems to be a recurrent phenomenon already found by other authors investigating Cladonia phylogenies (Myllys et al. Reference Myllys, Stenroos, Thell and Ahti2003). Moreover, it could in fact represent a more widespread phenomenon than generally anticipated, as it has also been found in other lichen groups (e. g., Ertz et al. Reference Ertz, Miądlikowska, Lutzoni, Dessein, Raspe, Vigneron, Hofstetter and Diederich2009). Phylogenetic incongruences among genes can occur for many reasons, including the presence of pseudogenes, gene paralogy, horizontal gene transfer, incomplete lineage sorting (ILS), and hybridization. Alternatively, the presence of hyphae from more species growing together in the same podetium could be another possible explanation for the conflict (Kotelko & Piercey-Normore Reference Kotelko and Piercey-Normore2010). We can clearly rule this out in our dataset, as we have unambiguous signals with the ITS primers.
Pseudogenes are dysfunctional relatives of known genes that have lost their protein-coding ability or are otherwise no longer expressed in the cell (Vanin Reference Vanin1985). Their base compositions are different from those of functional genes, and they evolve very rapidly (Buckler et al. Reference Buckler, Ippolito and Holtsford1997). Pseudogenous clones are characterized by occasional deletions in genes and spacers, by increased non-synonymous mutations in the otherwise almost identical rRNA-coding regions (Grimm & Denk Reference Grimm and Denk2008; Harpke & Peterson Reference Harpke and Peterson2008), or by low predicted secondary structure stability in ribosomal genes or spacers (Buckler et al. Reference Buckler, Ippolito and Holtsford1997). However, the almost identical 5.8S rRNA sequences (only one substitution was detected), absence of long-branching artefacts in the phylograms, and the absence of deletions suggest the non-pseudogenous nature of any of the ITS sequences analyzed. In addition, conserved sequences of 3′ and 5′ ends also suggest that they are not pseudogenes.
Horizontal gene transfer (HGT) is the transfer of genes across species. This mechanism is well known mainly in bacteria, but it also occurs in the evolution of Eukaryota (Keeling & Palmer Reference Keeling and Palmer2008; Khaldi et al. Reference Khaldi, Collemare, Lebrun and Wolfe2008; Marcet-Houben & Gabaldon Reference Marcet-Houben and Gabaldon2010). However, an HGT event is an unlikely source for the conflict between tree topologies in our dataset because it would imply a recent HGT event between two eukarya lineages, which is rare (Won & Renner Reference Won and Renner2003) and generally involves the transfer of introns (Rot et al. Reference Rot, Goldfarb, Ilan and Huchon2006).
Gene paralogy occurs if a gene in an organism is duplicated to occupy two different positions in the same genome and can also be responsible for the conflict between two phylogenies. Although paralogs of the β-tubulin gene are known from different groups of fungi (e.g., Begerow et al. Reference Begerow, John and Oberwinkler2004; Corradi et al. Reference Corradi, Kuhn and Sanders2004; Msiska & Morton Reference Msiska and Morton2009), they have not been reported from lichenized Ascomycota. Moreover, in our case, intragenomic variation should be found in β-tubulin sequences to explain the incongruence in our dataset by potential gene paralogy. However, we did not find an ambiguous signal in the β-tubulin sequences.
Incomplete lineage sorting, also called deep coalescence, is a phenomenon that can cause conflicting gene and species trees. ILS represents the incomplete random sorting of alleles at many loci independently due to short intervals between divergence events (Blanco-Pastor et al. Reference Blanco-Pastor, Vargas and Pfeil2012). ILS has been reported in many different groups of organisms, more likely in those species which have a large population size and a short time between divergences (e.g., Morando et al. Reference Morando, Avila, Baker and Sites2004; Jakob & Blattner Reference Jakob and Blattner2006; Pollard et al. Reference Pollard, Iyer, Moses and Eisen2006). This process is difficult to distinguish from interspecific hybridization and both may even occur simultaneously (Seehausen Reference Seehausen2004; Meng & Kubatko Reference Meng and Kubatko2009). Although several methods distinguishing these two evolutionary processes have been recently proposed (e.g., Holland et al. Reference Holland, Benthin, Lockhart, Moulton and Huber2008; Bloomquist & Suchard Reference Bloomquist and Suchard2010), many independent loci are needed for their implementation and it is difficult to uncover multiple reticulation events (Blanco-Pastor et al. Reference Blanco-Pastor, Vargas and Pfeil2012).
Since we are not able to distinguish ILS and hybridization, we assume both could be responsible for the incongruence in our dataset. Here we propose the phylogenetic consequences of these two scenarios.
The presence of ILS would indicate that zeorin-containing Cladonias spp. probably diverged relatively recently (Leache & Fujita Reference Leache and Fujita2010). The ITS and β-tubulin phylogenies would represent gene trees, which would not correspond with the species tree. To be able to describe the phylogenetic relationships within this group, even in the presence of incomplete lineage sorting, it will be necessary to study more loci (e.g., Knowles & Carstens Reference Knowles and Carstens2007), which will definitely be within reach with the ongoing Cladonia genome project hosted at Duke University.
Interspecific hybridization is regarded as one of the major factors responsible for conflicts among different loci (e.g., Taylor et al. Reference Taylor, Jacobson, Kroken, Kasuga, Geiser, Hibbett and Fisher2000; Fehrer et al. Reference Fehrer, Gemeinholzer, Chrtek and Braeutigam2007; Ertz et al. Reference Ertz, Miądlikowska, Lutzoni, Dessein, Raspe, Vigneron, Hofstetter and Diederich2009). Similar incongruences have been detected in other lineages of Cladonia (Myllys et al. Reference Myllys, Stenroos, Thell and Ahti2003; Fontaine et al. Reference Fontaine, Ahti and Piercey-Normore2010; Kotelko & Piercey-Normore Reference Kotelko and Piercey-Normore2010). We assume hybridization should be considered as an important mechanism, possibly influencing the evolution of the lichen genus Cladonia, resulting in reticulate evolution that may contribute to the species diversification. Although hybridization is not yet known in lichens, it has been proved to occur in most fungal phyla (e.g., Brasier et al. Reference Brasier, Kirk, Pipe and Buck1998, Reference Brasier, Cooke and Duncan1999; Xu et al. Reference Xu, Vilgalys and Mitchell2000; Craven et al. Reference Craven, Blankenship, Leuchtmann, Hignight and Schardl2001a , Reference Craven, Hsiau, Leuchtmann, Hollin and Schardl b ).
If the effect of hybridization is considered, only one parent lineage could be identified in both hybridization events. The second ancestral lineage is unknown, which could have two alternative explanations: 1) the parent lineage is extinct and could therefore not be detected; 2) we did not sample and analyze the parent lineage. To better understand the species concept and delimitation in the group of zeorin-containing red-fruited Cladonia lichens, it will be important to address the question of frequency of hybridization events more carefully in the future. The suggested hybridization could represent either an exceptional ancient event or a common ongoing process. Sexual compatibility/incompatibility between two organisms can be detected by a comparison of the secondary structure of the ITS (so-called CBS approach). The presence of compensatory base changes (CBSs) indicates incompatibility for sexual reproduction between species (Coleman Reference Coleman2003; Müller et al. Reference Müller, Philippi, Dandekar, Schultz and Wolf2007). In our case, the absence of CBCs reveals that there are presumably no reproduction barriers among the species studied, and hence, we can conclude that the second alternative is more feasible.
Species circumscriptions in Cladonia
Similarly to other recent studies focusing on Cladonia (Fontaine et al. Reference Fontaine, Ahti and Piercey-Normore2010; Kotelko & Piercey-Normore Reference Kotelko and Piercey-Normore2010; Pino-Bodas et al. Reference Pino-Bodas, Rosa Burgaz and Martin2010, Reference Pino-Bodas, Martín and Burgaz2012a ), our investigations clearly revealed the incongruence between the phylogenetically inferred lineages and traditional, morphologically and/or chemically delimited species. In fact, we were not able to find any phenotypic feature to unambiguously define the lineages in most cases (except Cladonia diversa, and chemical patterns in the lineages #2 and #4b).
In general, there are two alternatives for interpreting this incongruence. The phylogenetic units could be either regarded as populations of a morphologically variable species or accepted as ‘cryptic’ or incipient species.
The question of how to treat the ‘cryptic’ species within the traditionally defined nominal species has been discussed by many authors, advocating two different attitudes. Some authors (e.g., Kotelko & Piercey-Normore Reference Kotelko and Piercey-Normore2010) have suggested a more conservative attitude in maintaining the traditional delimitation of the species, even when they are not supported by molecular data. They have argued for the possible implications of these species for ecophysiological studies, and more generally, for the detection and preservation of rare or unusual species. Conversely, other authors (Grube & Kroken Reference Grube and Kroken2000) have proposed applying the phylogenetic species concept and thus defining the well-supported phylogenetic lineages as cryptic species. They mentioned that morphological characterization of the species is often facilitated after finding cryptic lineages with molecular data, as it may then become apparent which characters are significant. They also claimed that the knowledge of cryptic species is useful in investigations at fine scales of taxonomic resolution, such as for interpretations of ecophysiological differences and microhabitat preferences (Grube & Kroken Reference Grube and Kroken2000).
Considering the findings in this study, we adopt the second opinion, understanding the well-supported phylogenetic lineages as separate species, but without describing them formally for the time being. One lineage indeed consisted of morphologically well-characterized specimens of C. diversa, traditionally recognized as a species. Moreover, C. coccifera samples belonging to clade #4b shared identical chemical characteristics (chemotype 2; presence of porphyrilic acid and absence of isousnic acid) and appeared to differ chemically from the other C. coccifera strains (lineages #3a and #3b). It is therefore very likely that the other lineages, even if morphologically indistinguishable, also represent separate species. Zeorin-containing red-fruited Cladonia species have wide morphological and ecological amplitudes, and thus, the correlation between phylogenetically separated lineages and different phenotypic characters should be studied more comprehensively in the future.
We thank Ondřej Peksa, Jan Vondrák, František Bouda, Ana Rosa Burgaz, Toby Spribille, and Thilo Hasse for providing samples, and Teuvo Ahti for the revision of the material and valuable comments. We are very grateful to Zdeněk Palice for helpful discussions and Jiří Malíček for his assistance in taking photographs of the lichens studied. Two anonymous reviewers helped to improve the manuscript considerably. This study was financially supported by project No. 126608 of the Charles University Foundation (GA UK).
Appendix 1. Analysis of substitution saturation
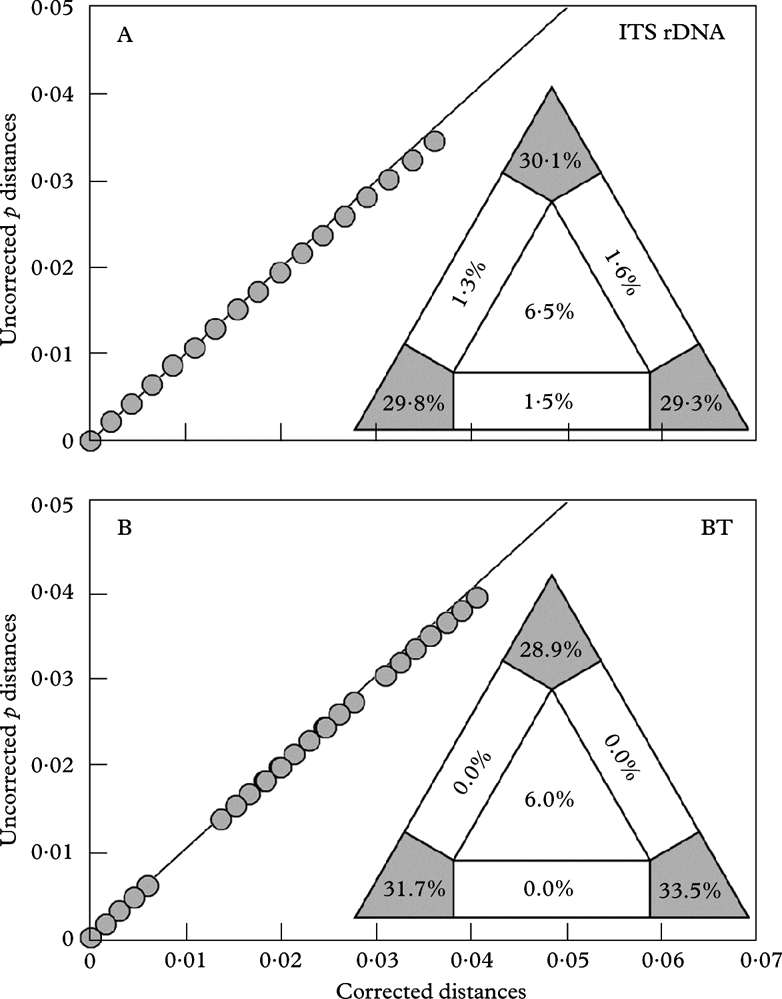
The graphs visualize the saturation of the ITS rDNA and β-tubulin datasets by plotting ML-corrected distances against uncorrected p-distances. Corrected distances are calculated using models estimated by PAUP/Modeltest for each specific data partition. A, analysis of ITS rDNA sequences; B, analysis of β-tubulin sequences. The triangles in the lower right of the graphs illustrate likelihood mapping results. The values in the panels indicate proportion of fully resolved (corners), partially resolved (along the sides), and fully unresolved quartets (in the centre).