Introduction
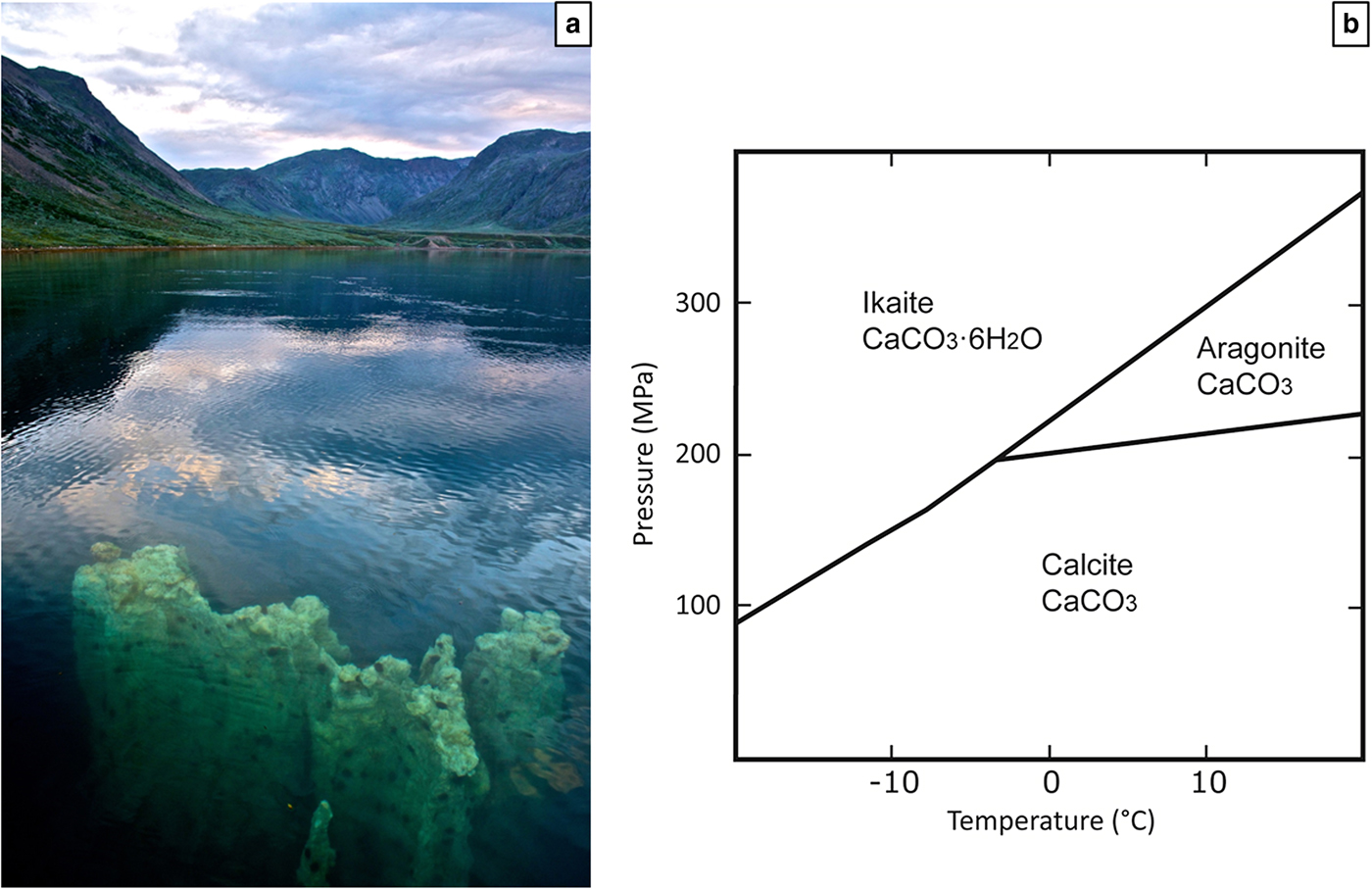
Ikaite (CaCO3·6H2O) was first synthesized experimentally in the early 1800s (Pelouze, Reference Pelouze1831) and later discovered in nature in Ikka fjord, southwest Greenland (Pauly, Reference Pauly1963). Ikka fjord lies at the base of an alkaline magmatic complex and in the cold water of the fjord, ikaite forms up to 20 m high columns (Fig. 1a) which can grow up to 0.5 m per year (Hansen et al., Reference Hansen, Buchardt, Kuhl and Elberling2011). The pressure-temperature stability of ikaite was constrained by Marland (Reference Marland1975) in the system CaCO3–H2O. The stability field (cold temperature and relatively high pressure) for ikaite does not exist on Earth (Fig. 1b), but nevertheless ikaite has been found in sediments, in sea ice and in the form of tufa columns. It is therefore probable that parameters other than pressure and temperature favour formation of metastable ikaite instead of the stable carbonate phase, calcite.
Fig. 1. (a) Ikaite column appearing at the surface of the water in Ikka Fjord. Photo: Richard Martin / www.coldzymes.dk. (b) The stability fields of ikaite, calcite and aragonite in the system CaCO3 – H2O. Modified from Marland (Reference Marland1975).
In nature, ikaite has been observed at temperatures between –2°C and 7°C (Huggett et al., Reference Huggett, Schultz, Shearman and Smith2005). Experiments have shown that ikaite remains relatively stable at near-freezing temperatures but decomposes quickly to calcite and water at room temperature (Johnston et al., Reference Johnston, Merwin and Williamson1916). In sediments it has been found that ikaite is pseudomorphically replaced by calcite (Suess et al., Reference Suess, Balzer, Hesse, Muller, Ungerer and Wefer1982; Huggett et al., Reference Huggett, Schultz, Shearman and Smith2005). In Ikka Fjord, ikaite precipitates from a mixture of seawater and spring water which seeps up from the bottom of the fjord. The spring water is a sodium carbonate rich solution with pH 10.16 to 10.5 (at 3.6 ± 0.5°C) and with relatively high concentration (9–25 ppm) of PO43– (Buchardt et al., Reference Buchardt, Israelson, Seaman and Stockmann2001). The composition of the spring water (Table 1) reflects water–rock interaction in the Grønnedal-Íka igneous complex which consists of carbonatite and alkaline rocks, mainly nepheline syenite (Emeleus, Reference Emeleus1964). This igneous complex forms the high ground on the northern side and underlies part of the fjord. Meteoric water precipitating on the complex percolates along fractures in the rocks and reacts with its constituent minerals. The nepheline syenites are rich in nepheline ((Na,K)AlSiO4); and the carbonatite in calcite (CaCO3), a smaller amount of siderite (FeCO3) and apatite (Ca5(PO4)3(F,Cl,OH)). It is probable that reactions involving these minerals lead to high concentrations of Na+, CO32– and PO43– in the spring water.
Table 1. Composition of spring water and seawater.
*From Buchardt et al. (Reference Buchardt, Israelson, Seaman and Stockmann2001); **From Stumm and Morgane (Reference Stumm and Morgan1996).
n.d. – not detected
Dahl and Buchardt (Reference Dahl and Buchardt2006) investigated the mineralogy of the fresh tufa columns as well as debris from the columns found at the bottom of the fjord. The fresh columns are mostly composed of white ikaite crystals with minor aragonite, monohydrocalcite and calcite, whereas debris from the columns was calcite, monohydrocalcite and an unidentified magnesium carbonate (Dahl and Buchardt, Reference Dahl and Buchardt2006). The ikaite columns are porous and harbour active microalgae and cyanobacteria populations (Trampe et al., Reference Trampe, Larsen, Glaring, Stougaard and Kühl2016).
Buchardt et al. (Reference Buchardt, Israelson, Seaman and Stockmann2001) suggested that the precipitation of ikaite in the fjord was favoured by the cold temperature (< 6°C) and the presence of PO43– in the spring water which is known to inhibit the precipitation of calcite (Burton and Walter, Reference Burton and Walter1986). However, Hu et al. (Reference Hu, Wolf-Gladrow, Dieckmann, Völker and Nehrke2014) experimentally tested the effect of PO43– on synthetic ikaite precipitation in sea ice in experiments with synthetic seawater, and in their study, ikaite was found to precipitate both with and without PO43– in solution. There are several other species know to inhibit calcite growth or nucleation. Magnesium and sulfate are well known calcite growth inhibitors (Berner, Reference Berner1975; Mucci et al., Reference Mucci, Canuel and Zhong1989; Davis et al., Reference Davis, Dove and De Yoreo2000; Nielsen et al., Reference Nielsen, Sand, Rodriguez-Blanco, Bovet, Generosi, Dalby and Stipp2016). The presence of these species in the seawater might therefore favour ikaite precipitation. Further, Nielsen et al. (Reference Nielsen, Sand, Rodriguez-Blanco, Bovet, Generosi, Dalby and Stipp2016) showed that MgSO40 was a particularly effective inhibitor of calcite growth. Meyer (Reference Meyer1984) tested experimentally the effect of several additives on calcite growth and found that a number of elements, e.g. Fe, Zn, Ce, Pb, Co and Mn, strongly inhibited calcite growth. Here we focus specifically on the effect of Mg and SO4 as well as pH on ikaite precipitation in a series of experiments.
Methods
A series of experiments were set up at the Department of Geological Sciences, Stockholm University to simulate the conditions at which ikaite forms in Ikka Fjord. In our experiments, synthetic spring water and either natural or synthetic seawater were mixed. Julabo F-32 and F-25 cooling baths were used to maintain the temperature at 5.00 ± 0.03°C during the experiments. The rate of mixing of Ikka water in seawater was controlled using an Ismatec BVK peristaltic pump. In the experiments we used natural seawater collected near Andøya, Norway which was filtered with Munktelle 00M filters and synthetic seawater prepared to replicate the average seawater composition reported by Stumm and Morgan (Reference Stumm and Morgan1996). The seawater from Andøya was considered a good analogue of seawater in Ikka Fjord for the purpose of our study, because of its similar salinity (33.1‰ and 32.2‰, respectively) and because PO4 concentrations were below detection limits. The composition of synthetic seawater, which was prepared by mixing powders from MERCK in 1 litre of ultrapure deionized water (MilliQ resistivity >18.2 MΩ cm), is shown in Table 1.
In our experiments, we mixed the following solutions: Solution 1 represented the sodium carbonated spring water. It was made with a mixture of 0.1 M Na2CO3 and 0.1 M NaHCO3 in ratios 1:1, 3:1 and 4:1 which represent the range of different spring waters in the columns described by Buchardt et al. (Reference Buchardt, Israelson, Seaman and Stockmann2001). The pH of these solutions were 10.1, 10.6 and 10.7, respectively, at 5°C. We extended the range of pH values beyond those seen at Ikka Fjord by varying the ratios of 0.1 M Na2CO3 and 0.1 M NaHCO3 from 1:15 to 1:0, which gave a pH range from 9.1 to 11.8 at 5°C. Solution 2 represented seawater. We used natural seawater from Andøya, Norway and synthetic seawater (Table 1). In some experiments, either SO4 or Mg was removed. In other experiments, Mg concentration was varied by changing the proportions of MgCl2·6H2O in the mixture. In some experiments, loss in ionic strength due to removal of Mg was compensated by the addition of NaCl. This was done because variation in ionic strength can interfere with calcite precipitation and/or ikaite precipitation (Hu et al., Reference Hu, Wolf-Gladrow, Dieckmann, Völker and Nehrke2014).
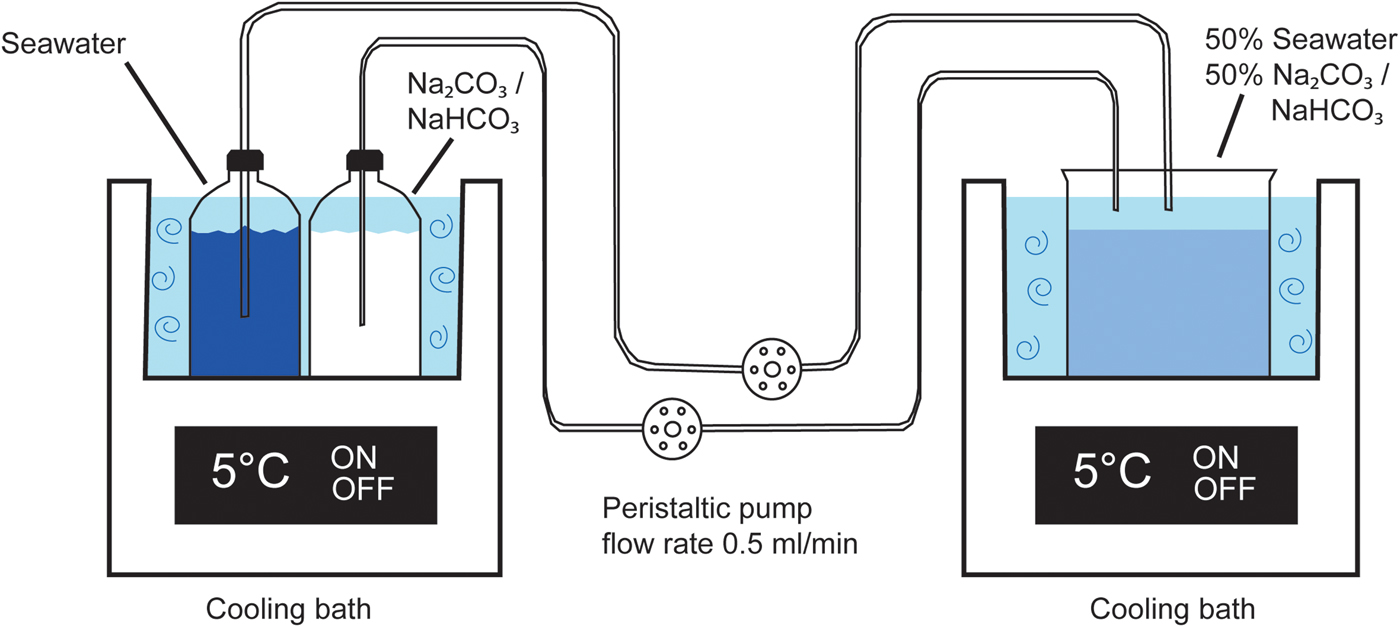
The experiment was set up by placing one flask with the sodium carbonate solution (solution 1) and a second flask with seawater (solution 2) in the first cooling bath maintained at 5°C. Each flask was connected with 1.52 mm Tygon tubing to the peristaltic pump and further to the mixing beaker which was placed in the second cooling bath (Fig. 2). The two solutions were pumped at a rate of 0.48 ± 0.06 ml/min into the mixing beaker until ~300 ml of mixed solution was obtained, at which point the experiment was stopped. The pH of each solution was measured using a HANNA HI-9126 pH meter, a HI-1230B electrode, and the temperature was measured with a HI-7662 sensor after each experiment. The mixed solution was put into a 5°C cold room where it was filtered with Munktell 00 K filters. The precipitate in the filter was dried overnight and collected the next day from the filter paper, weighed, and thereafter stored in a freezer (–18°C). Seeding from calcite remaining from previous experiments was at one point detected. The affected experiments were excluded and thereafter, the beakers were cleaned with weak acid between experiments. Finally, replicate runs of four experiments (1, 2, 4 and 6) gave the same results.
Fig. 2. Experimental set-up. To the left the outlet solutions in the first cooling bath. Solutions are pumped in to a beaker in the second cooling bath. Modified from Stockmann et al. (Reference Stockmann, Tollefsen, Skelton, Brüchert, Balic-Zunic, Langhof, Skogby and Karlsson2018).
The minerals from the precipitate were identified by powder X-ray diffraction (XRD) analysis with an X Pert Pro instrument from PANalytical and the program Data Colllector. The sample supporter was put into a freezer (–18°C) for 10 min before analysis so as to preserve the ikaite crystals. The measurement program used was Absolute Scan 5–70°2θ with a runtime of 11 min. Mineral identification and quantification (Rietveld refinement) of the phases was done with the program HighScore Plus (Degen, Reference Degen, Sadki, Bron, König and Nénert2014).
We obtained scanning electron microscopy (SEM) images of samples from some experiments with a Carl Zeiss Merlin Field-Emission Scanning Electron Microscope using Quorum Technologies PP3000 T Cryo Preparation System at Umeå University and with an Environmental Scanning Electron Microscope with Field Emission Gun of the brand Philips XL-30- ESEM-FEG at Stockholm University, Sweden.
We measured the conductivity, salinity and the ion concentrations of the natural seawater from Andøya, Norway (Table 1). The anions were measured with an IC20 Ion Chromatograph from Dionex and the cations with an inductively coupled plasma atomic emission spectrometer with autosampler SPS-5 from Varian.
We used the software PhreeqC Interactive version 3.3.0 (Parkhurst and Appelo, Reference Parkhurst and Appelo2013) for geochemical modelling of the experiments to obtain the pH of the solutions. Saturation indices (SI), where SI = log (IAP/Ksp), with IAP = ion activity product and Ksp = solubility constant, for the carbonate minerals are included in the PhreeqC llnl database, except for ikaite (CaCO3·6H2O) and vaterite (CaCO3), which were added separately, using solubility constants from Bischoff et al (Reference Bischoff, Fitzpatrick and Rosenbauer1993a) for ikaite and Plummer and Busenberg (Reference Plummer and Busenberg1982) for vaterite. The pH was modelled for a mixture of 50% synthetic or natural seawater and 50% synthetic spring water for each experiment (Supplementary Tables S1 and S2, see Supplementary material below).
Results
The first series of experiments were designed to determine if either Mg or SO4 controlled ikaite precipitation by excluding them from the synthetic seawater mixture. In these experiments (Table 2), ikaite precipitated when we used natural seawater (experiment 1), synthetic seawater (exp. 2) and synthetic seawater with SO4 removed (exp. 3), whereas calcite precipitated when we used synthetic seawater with Mg removed (exp. 4) and synthetic seawater with Mg removed and NaCl added to maintain ionic strength (exp. 5).
Table 2. Summary of ikaite experiments.
Temperature experiments at 5°C. Syn = synthetic.
ik – ikaite; cal – calcite; vat – vaterite; arg – aragonite; nq – nesquehonite; mhc – monohydrocalcite; n.p. – no precipitate.
* pH was calculated with PhreeqC.
** Mg in mmol/kg. With Andøya seawater s.d. ±0,5, with synthetic seawater s.d. ±0.02.
*** Minimum amount of precipitate.
# Weighted profile. R-factor (Rwp) was between 8–14% except samples 29 R wp = 16%, 32 R wp = 37% and 34 R wp = 20%. R-factor values from Toby (Reference Toby2006).
The second, third and fourth series of experiments were designed to find threshold concentrations of Mg required for ikaite formation to occur at different pH values. In these experiments, Mg concentration was increased stepwise (Table 2). The threshold Mg concentrations were 2.46–4.92 (±0.02), 4.92–7.38 (±0.02) and 7.38–9.84 (±0.02) mmol/kg for the second third and fourth series, respectively (Table 2). The final precipitates at Mg concentrations higher than these threshold values were either ikaite alone (exp. 8, 9, 12, 20 and 25: Fig. 3a), with calcite (exp. 7, 17a, 17b, 18 and 19: Fig. 3b) or with traces of nesquehonite (exp. 13). The final precipitates consist of calcite at lower Mg concentrations, either alone (exp. 10, 14, 15, 21, 22 and 24; Fig. 3e and f), with aragonite (exp. 11 and 23) or with traces of monohydrocalcite (exp. 16). Both ikaite and either calcite or aragonite were found in the final precipitates in two experiments run at the threshold Mg concentration (exp. 6a and 6b: Fig. 3c).
Fig. 3. SEM images of experiments. (a) Ikaite crystals from exp. 12b. (b) Ikaite crystals from exp. 17. (c) Calcite and/or ikaite from exp. 6b. (d) Aragonite and/or calcite with spherulite morphology from exp. 35. (e) Calcite with smooth rhomb shape from exp. 24. (f) Calcite crystals from exp. 14.
The fifth and sixth series of experiments were designed to find threshold pH values required for ikaite formation to occur at different concentrations of Mg. In the fifth series, Andøya seawater (Table 1) was used. This gave a Mg concentration of 22.80 ± 0.50 mmol/kg in the mixed solution. In the sixth series, synthetic seawater was used and the Mg concentration of the mixed solution was 14.76 ± 0.02 mmol/kg. In both series, pH was increased stepwise by increasing the proportion of 0.1 M Na2CO3 in solution 1. The threshold pH values at and above which the final precipitates were ikaite were 9.32 and 9.69. The final precipitates were calcite below this threshold, either alone (exp. 34) or with aragonite (exp. 27, 28 and 35: Fig. 3d). In one experiment conducted using Andøya seawater at comparatively low pH (8.62), no precipitate was formed. The final precipitates were ikaite above this threshold either alone (exp. 30, 31 and 32), with calcite (exp. 36 and 37) or with an amorphous phase (exp. 33). In one experiment at the threshold pH value (exp. 29) the final precipitate was monohydrocalcite together with both calcite and ikaite.
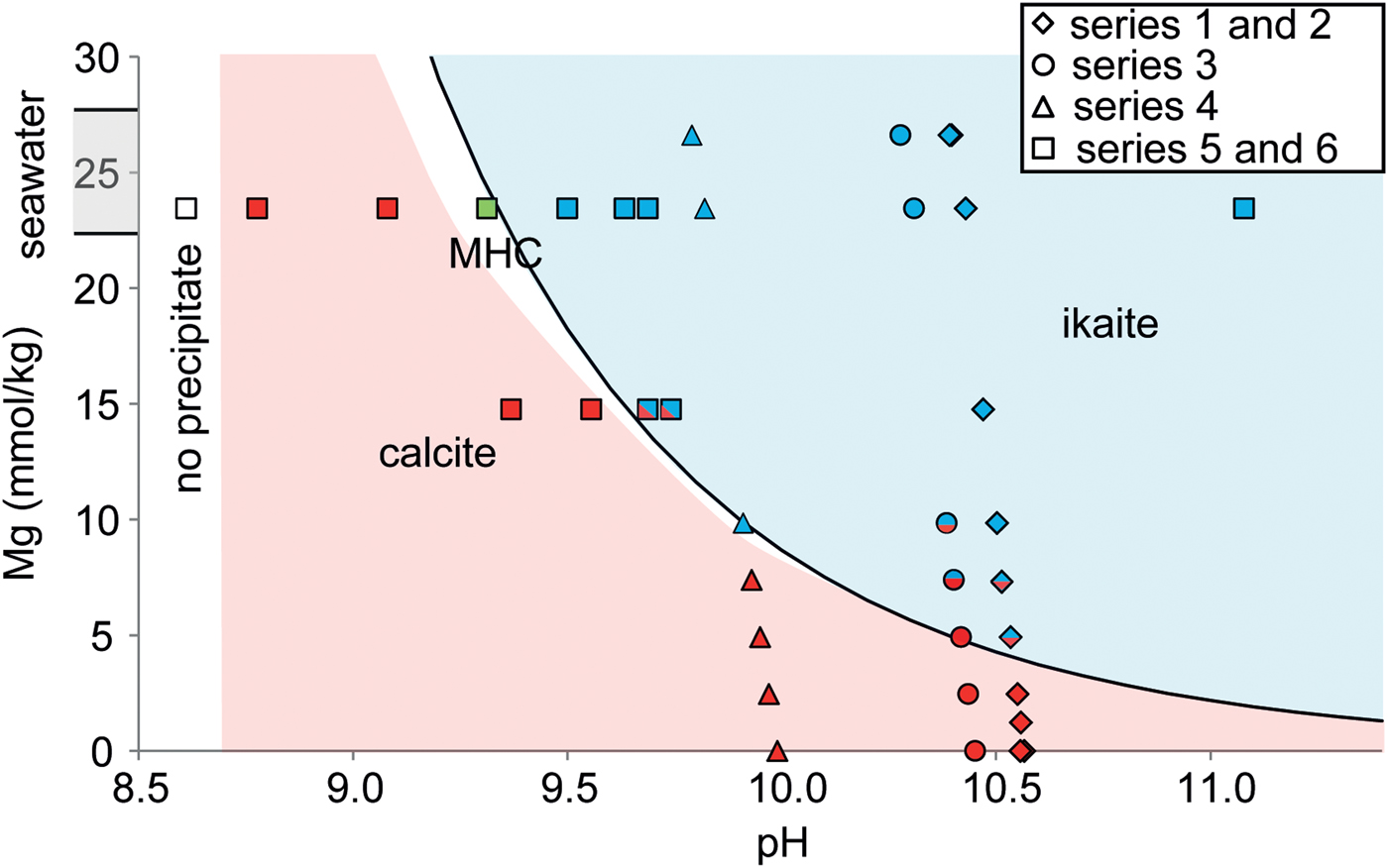
By combining our results, we were able to construct a kinetic stability diagram for ikaite and calcite precipitation as a function of pH and Mg concentration at 5°C (Fig. 4). The pH was calculated using PhreeqC because the measured pH from the experiments was not representative due to rapid precipitation in the supersaturated mixed solution, which lowered its value rapidly. Ikaite was found to precipitate at high Mg concentration and high pH (Fig. 4), i.e. the threshold Mg concentration required for ikaite precipitation to occur is lower at higher pH. Our results also suggest monohydrocalcite precipitation at lower pH and higher (seawater) Mg concentration. The kinetic boundary has been set at the appearance of ikaite in the precipitate. The line which bounds the field within which ikaite precipitation occurs follows a power law:
where C is the concentration of Mg in mmol/kg.
Fig. 4. Phase diagram for ikaite and calcite precipitation as a function of pH and Mg concentration in solution at 5°C. The blue symbols represent ikaite precipitations, the red symbols calcite precipitation and the green symbol monohydrocalcite (MHC).
Secondary electron and back-scattered electron images revealed euhedral ikaite crystals from all samples in the ikaite stability field (Fig. 3a –c; Fig. 4). Calcite aggregates were spherulitic (Fig. 3d) or smoothed (Fig. 3e) when Mg was present in the mixture. Calcite crystals were only euhedral (Fig. 3f) in the sample precipitated from a solution that contained no Mg.
Discussion
In our experiments removing SO4 from the seawater had no effect on ikaite precipitation (Table 2; exp. 3). In contrast, our experimental results show that both Mg concentration and pH control ikaite precipitation (Table 2; Fig. 4). Ikaite precipitated in solutions with high pH values and Mg concentrations. In experiments with natural seawater, (Mg concentration of mixed solution = 22.80 ± 0.50 mmol/kg), ikaite precipitation required pH > 9.3 (Table 2; exp. 29).
Geochemical modelling using PhreeqC verified that most carbonates were supersaturated (Table S1), implying that some phases did not precipitate for kinetic reasons. One exception is nesquehonite, which although undersaturated, precipitated in exp. 13, but this might simply reflect that the solubility constant of nesquehonite is not well constrained in the PhreeqC database.
Several findings from our study suggest that a primary kinetic factor which allows ikaite formation is that Mg2+ inhibits calcite nucleation and/or growth: (1) Scanning electron microscope images reveal a progression from euhedral ikaite crystals precipitated from solutions with high pH and high Mg concentration (Fig. 3a,b) to calcite aggregates with spherulite morphology (Fig. 3d), smooth rhomb structures (Fig. 3e) and ultimately euhedral forms (Fig. 3f) precipitated from a solution that contained no Mg. (2) The amount of precipitates obtained was generally higher for ikaite (>0.08 g) and lower for calcite and aragonite (<0.08 g). This could imply that ikaite nucleation and growth is kinetically favoured but might also reflect the higher molecular weight of ikaite because of its water content. (3) Calcite formation was triggered by seeding in two experiments. (4) Calcite typically precipitates together with aragonite which is known to precipitate when calcite nucleation and growth is inhibited by the presence of Mg2+ (Deleuze and Brantley, Reference Deleuze and Brantley1997).
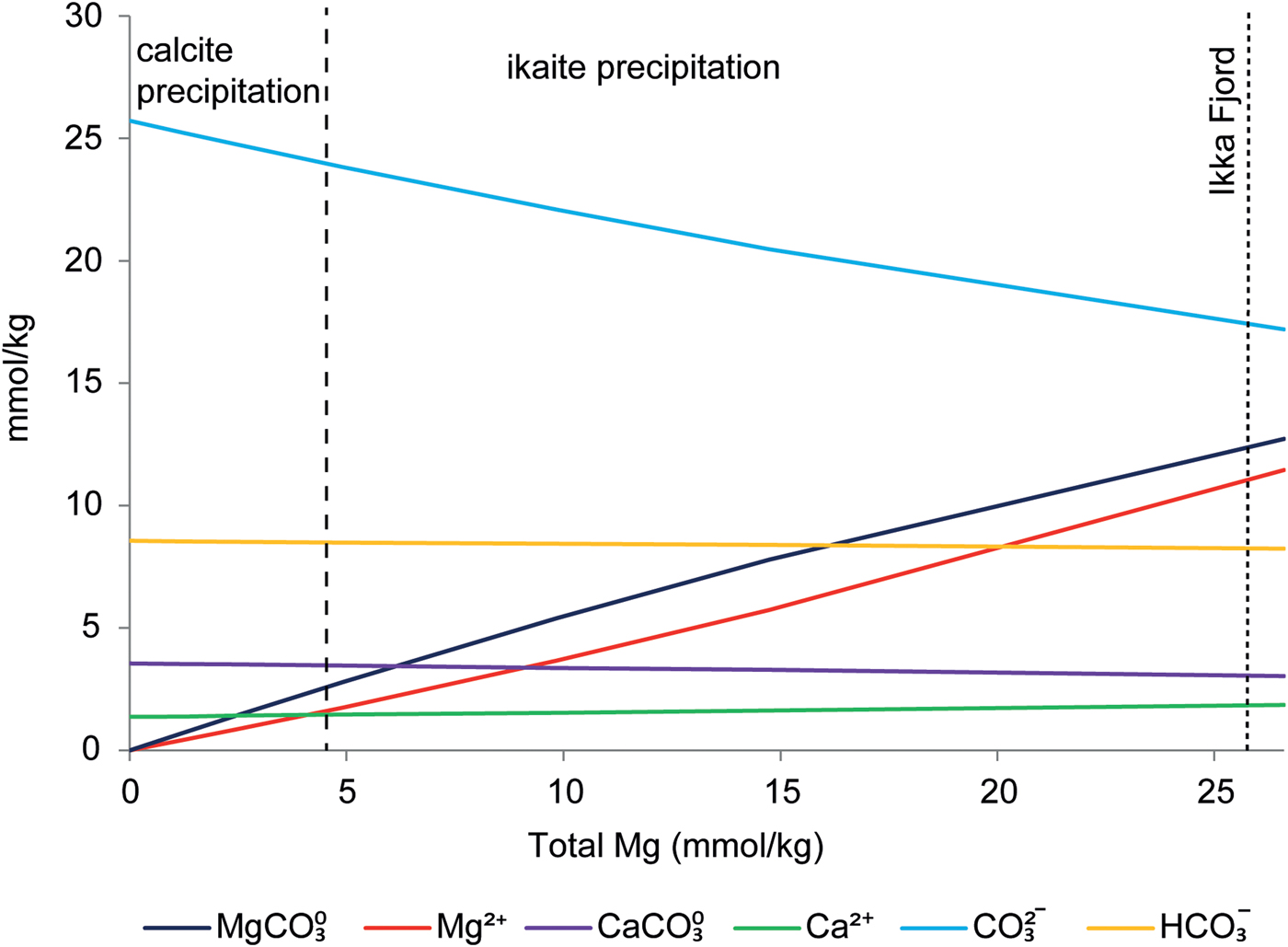
Magnesium inhibiting calcite nucleation has been described previously (Mucci et al., Reference Mucci, Canuel and Zhong1989, Deleuze et al., Reference Degen, Sadki, Bron, König and Nénert1997 and Nielsen et al., Reference Nielsen, Sand, Rodriguez-Blanco, Bovet, Generosi, Dalby and Stipp2016). Nielsen et al. (Reference Nielsen, Sand, Rodriguez-Blanco, Bovet, Generosi, Dalby and Stipp2016) showed experimentally that SO42–, Mg2+ and MgSO40 inhibit calcite growth. The inhibiting effect of SO42– and MgSO40 on calcite cannot explain ikaite precipitation in our study, because ikaite precipitation occurred even after removal of SO4 from the synthetic seawater mixture. Geochemical modelling using PhreeqC showed that Mg2+ cations preferentially form MgCO30 rather than MgSO40 in our experiments, predicting 11.45 mmol/kg of MgCO30 and 1.0 mmol/kg of MgSO40 in natural seawater (Table S2). Also, the experimental threshold for ikaite precipitation occurs after the concentration of both Mg2+ and MgCO30 has exceeded the concentration of Ca2+ (Fig. 5).
Fig. 5. Ion speciation calculated by PhreeqC for the experiments in series 1 and 2 as a function of Mg concentration in solution. The dashed lines show the corresponding zone from the experiments as a function of Mg concentration in solution.
It has been shown that when calcite and monohydrocalcite (MHC) precipitate from highly supersaturated solutions, the first phase to nucleate is metastable amorphous calcium carbonate (ACC) which is transformed to vaterite and then to the more stable phase calcite (Bots et al., Reference Bots, Benning, Rodriguez-Blanco, Roncal-Herrero and Shaw2012). If the solution has high Mg/(Mg + Ca) ratio the ACC will be transformed to MHC (Rodriguez-Blanco et al., Reference Rodriguez-Blanco, Shaw, Bots, Roncal-Herrero and Benning2014). We only detected an amorphous phase in one of our experiments (exp. 33) but this might be because the XRD analyses were not done immediately after precipitation. However, in a recent experimental study (Rodriguez-Ruiz et al., Reference Rodriguez-Ruiz, Veesler, Gómez-Morales, Delgado-López, Grauby, Hammadi, Candoni and García-Ruiz2014), calcium carbonate was precipitated in confined nano- and pico-volumes at 20°C and the first phase to crystallize was ikaite and no ACC was detected. This could mean that ikaite preferentially crystallizes directly from a supersaturated solution. One reason could be that in a confined environment the volume of ikaite occupies less space than the equivalent volume of CaCO3 (calcite) and 6H2O (Lennie, Reference Lennie2005). However, this is not applicable to our study. Purgstaller et al. (Reference Purgstaller, Dietzel, Baldermann and Mavromatis2017) showed from in situ monitored experiments that the first precipitates (ikaite or ACC) transformed into more stable anhydrous CaCO3 polymorphs depending on the Mg/Ca ratio in the reactive solution. We cannot rule out the possibility that in the experiments in which calcite was found, that ikaite precipitated first and thereafter transformed to calcite.
Our other observation, that pH controls ikaite precipitation, is consistent with previous experimental work by Hu et al. (Reference Hu, Wolf-Gladrow, Dieckmann, Völker and Nehrke2014). These authors investigated controlling factors of ikaite precipitation in sea ice and found pH to be the controlling factor, which could be attributed to higher CO32– concentration at higher pH. They showed that this results in increased supersaturation of ikaite and that ikaite precipitated earlier. However, they did not test the effect of Mg in their study.
In our experiments, we simulated general conditions in Ikka Fjord and make no claim to have captured the full complexity of the Ikka Fjord system where other factors such as trace-element concentrations (cf. Meyer, Reference Meyer1984) and microbial life (Trampe et al., Reference Trampe, Larsen, Glaring, Stougaard and Kühl2016) might also stabilize and preserve the tufa columns. Instead, we conclude that high pH and Mg2+ as a calcite growth inhibitor are important factors that can allow ikaite formation. Here, we consider how these factors might contribute to ikaite formation in other environments. We consider ikaite formation in sea ice, marine sediments and a lacustrine environment (Mono Lake, California, USA).
The mechanism of ikaite formation in sea ice is not fully understood but might relate to elevated salinity and pH as sea ice starts to form (Geilfus et al., Reference Geilfus, Carnat, Dieckmann, Halden, Nehrke, Papakyriakou and Delille2013). Hu et al. (Reference Hu, Wolf-Gladrow, Dieckmann, Völker and Nehrke2014) showed experimentally that pH and PO43– were controlling factors of ikaite precipitation in mixed CaCl2 and NaHCO3 solutions; vaterite precipitated in experiments without PO43– but ikaite precipitated both with and without PO43– in seawater. We speculate that Mg in the seawater could have inhibited calcite nucleation in their experiments allowing ikaite to form. In nature, elevated pH, salinity, Mg2+ concentration and low temperature as sea ice starts to form might favour ikaite precipitation.
As with Ikka Fjord ikaite formation in sediments tends to be associated with cold alkaline conditions, arising because of organotrophic or methanotrophic sulfate reduction (Suess et al., Reference Suess, Balzer, Hesse, Muller, Ungerer and Wefer1982; Teichert and Luppold, Reference Teichert and Luppold2013). Ikaite crystals precipitate in marine sediments from interstitial solutions when anoxic organic matter or methane releases CO2 during microbial decomposition (Suess et al., Reference Suess, Balzer, Hesse, Muller, Ungerer and Wefer1982; Schubert et al., Reference Schubert, Nürnberg, Scheele, Pauer and Kriews1997). Suess et al. (Reference Suess, Balzer, Hesse, Muller, Ungerer and Wefer1982) suggested that Ca2+ was supplied from interstitial seawater and that ikaite could be favoured by the presence of Mg2+ in the seawater. This is in accordance with our study; however, marine sediments can be enriched in other elements (e.g. Fe, Mn) that could also act as calcite growth inhibitors (Meyer, Reference Meyer1984) allowing ikaite to form.
In Mono Lake, ikaite precipitates from a mixture of spring water and alkaline lake water. In contrast to Ikka Fjord, precipitation of ikaite is a seasonal phenomenon; ikaite only precipitates during cold winters. No precipitation occurs during warmer periods. The fact that calcite does not precipitate instead of ikaite in warmer periods suggests that its growth is inhibited by an inhibitor that does not affect ikaite (Bischoff et al., Reference Bischoff, Stine, Rosenbauer, Fitzpatrick and Stafford1993b; Council and Bennett, Reference Council and Bennett1993). The spring waters have low concentrations of Mg but the lake water, which dominates the mixing zone where ikaite crystals form, (Council and Bennett, Reference Council and Bennett1993) has a Mg2+/Ca2+ ratio of ~10 (Bischoff et al., Reference Bischoff, Fitzpatrick and Rosenbauer1993a,b). This implies that the concentration of Mg exceeds that of Ca, implying that Mg could inhibit calcite formation in Mono Lake. However, the lake water also contains 60 mg/L of PO4, a perhaps more probable inhibitor of calcite formation (Bischoff et al., Reference Bischoff, Fitzpatrick and Rosenbauer1993a).
Conclusions
We conclude that Mg concentration and pH are important controls of ikaite precipitation in Ikka Fjord and potentially elsewhere. Ikaite precipitation is favoured by high pH and Mg concentration, with ikaite precipitation in a mixture with natural seawater requiring a pH > 9.3. We further conclude that one reason that ikaite precipitation occurs is that Mg2+ inhibits calcite precipitation. Our contribution towards the understanding of the chemical controls on ikaite formation has implications for the understanding of ikaite occurrence in nature and its possible role in part of the carbon cycle, especially in cold marine environments.
Acknowledgements
We thank M. Ahlbom, V. Brüchert, J. Ek, P. Hjelmqvist, J. Langhof, C.C.N. Lee, H. Skogby, M. Söderman and J. West for assistance in lab and the Bolin Centre for financial support.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2018.110