INTRODUCTION
Porcine neonatal coccidiosis caused by Isospora suis is found worldwide with high prevalence and has a substantial economic impact on pig breeding facilities (Lindsay et al. Reference Lindsay, Blagburn and Powe1992; Scala et al. Reference Scala, Demontis, Varcasia, Pipia, Poglayen, Ferrari and Genchi2009). Despite its veterinary importance, knowledge of the immune response towards this parasite is limited (Taylor Reference Taylor1984; Worliczek et al. Reference Worliczek, Buggelsheim, Alexandrowicz, Witter, Schmidt, Gerner, Saalmüller and Joachim2010). In order to investigate the antigen-specific immune response to I. suis in vitro, sufficient amounts of antigen with a high grade of purity are required. However, oocysts isolated from the faeces of piglets are currently the only source of antigen used for immunological studies on I. suis because there is no in vitro cultivation system available that allows propagation of this parasite in the amounts necessary for such studies. The purification of parasite material (oocysts of I. suis) from piglet faeces is a challenge because of the high fat content in diarrhoeic faeces which negatively affects the commonly used purification protocols for oocysts by flotation. Therefore, a method for isolating clean oocysts from faecal samples of piglets to retrieve antigen of high purity is needed.
Additionally, during the development of the protocol for oocyst isolation and purification, the question arose whether observed responses in immunological assays were specific for I. suis since contaminating bacterial components may have been present in antigen preparations. Pakandl et al. (Reference Pakandl, Hlaskova, Poplstein, Chroma, Vodicka, Salat and Mucksova2008) measured lipopolysaccharide (LPS) concentrations in antigen preparations from Eimeria oocysts isolated from rabbit faeces and argued that a lack of LPS demonstrates specificity of observed responses towards the parasite. However, this strategy seems to be insufficient for I. suis antigen, since possible contamination with microorganisms other than gram-negative bacteria has to be expected (Schulze, Reference Schulze1978).
Therefore, a procedure that enables high-grade purification of I. suis oocysts was developed, consisting of multiple purification steps including fluorescence-activated cell sorting (FACS) to achieve this aim. Furthermore, the feasibility of this procedure was demonstrated by applying purified I. suis oocysts as antigen in Interferon-γ (IFN-γ) ELISPOT assays to identify recall responses in lymphocytes isolated from I. suis-infected pigs.
MATERIALS AND METHODS
Infection model
Oocysts were produced in piglets (Landrace×Large White×Pietrain) that were infected orally with 1000 sporulated oocysts of I. suis (field strain Wien I) on the third day after birth. The piglets were reared conventionally with the sow in a farrowing crate on straw at the animal husbandry facilities of the Institute of Parasitology, University of Veterinary Medicine Vienna, Austria, under standardized conditions. Between days 7 and 21 after birth, individual faecal samples were taken daily to provide an oocyst count and an evaluation of faecal consistency. Piglets were re-infected with 1000 sporulated oocysts of I. suis after 5 months. Three weeks later the animals were stunned with a captive bolt pistol followed by exsanguination. Spleens were removed aseptically and kept on ice until further processing. All procedures were approved by the Austrian Federal Ministry of Science and Research and the Ethics Committee of the University of Veterinary Medicine Vienna according to the Austrian Animal Protection law.
Retrieval of sporulated oocysts
Oocysts per gram faeces (opg) were counted using a modified McMaster method (Worliczek et al. Reference Worliczek, Mundt, Ruttkowski and Joachim2009). I. suis oocysts were obtained from faeces of infected animals between days 6 and 14 post-infection. The criterion for enrichment of oocysts from faecal samples was a minimum of 10 000 opg in loose faeces to obtain oocysts with as little debris as possible. Faeces from different individuals matching these criteria were pooled. Oocysts were concentrated using a protocol modified according to Ruttkowski et al. (Reference Ruttkowski, Joachim and Daugschies2001). Faecal samples were suspended thoroughly in tap water and sedimented by centrifugation at 1700 g for 5 min. The pellet was resuspended in 25% Percoll® (GE Healthcare Biosciences AB, Uppsala, Sweden) and centrifuged again to remove the majority of the fat from the samples. The pellet containing the oocysts was washed twice with tap water. To induce sporulation the parasite material was suspended in 2% potassium bichromate in tap water and incubated for 2–3 days at room temperature with daily aeration. Sporulated oocysts were filtered through a mesh with 100 μm pore size to remove debris and stored at 11°C until further use.
Purification of oocysts
For the establishment and evaluation of the whole purification procedure 8 different batches of I. suis oocysts were used. Oocysts were further purified with a density gradient in 15 ml tubes containing 5 ml of 2 m sucrose (ρ=1·25 g/l) overlaid with 5 ml of 1·25 m sucrose (ρ=1·15 g/l). The latter was overlaid with a suspension of sporulated oocysts in water (2·5 ml) with 2% potassium dichromate. The gradients were centrifuged for 7 min, at 1500 g and room temperature. Both interfaces were screened microscopically for oocysts. If oocysts were present they were collected and washed once (5 min, 1700 g, room temperature) with tap water to dilute residual sucrose solution to a density of less than 1·02 g/l to allow oocysts to sediment. After a second washing step with tap water, the pellet was finally transferred to a sterile 15 ml tube and suspended in 5 ml of sterile deionized water.
Disinfection of oocyst suspensions
To disinfect the oocyst suspension, 100 μl of a fresh NaOCl solution (12% v/v, Carl Roth GmbH, Karlsruhe, Germany) was added to a final concentration of 0·24% active chloride. The suspension was incubated for 10 min at 4°C and then washed 4 times with sterile PBS. Prior to the final washing step the suspension was filtered through a sterile CellTrics® 50 μm filter (Partec GmbH, Görlitz, Germany) to remove any coagulated debris.
To determine the efficacy of the NaOCl treatment (i. e. inactivation of living bacterial and fungal contaminants), 1 ml of oocyst suspension (containing 1000 oocysts/ml) was tested in 10 ml of BBL™ fluid thioglycollate medium (BD Diagnostics, Heidelberg, Germany) as well as in 10 ml of tryptic soy broth (Merck, Darmstadt, Germany). Aliquots of 100 μl were plated onto Difco™ Columbia agar (BD Diagnostics, Heidelberg, Germany) supplemented with 7% (v/v) sheep blood after 24 h, 48 h, 1 and 2 weeks of incubation at 37°C. Plates were incubated under aerobic, anaerobic, and microaerophilic conditions (7% CO2) for 24 to 72 h at 37°C. The viability of oocysts was tested by excystation (Hermosilla et al. Reference Hermosilla, Zahner and Taubert2006) using porcine instead of bovine bile and microscopic control for motility of sporozoites during excystation.
Fluorescence-activated cell sorting (FACS)
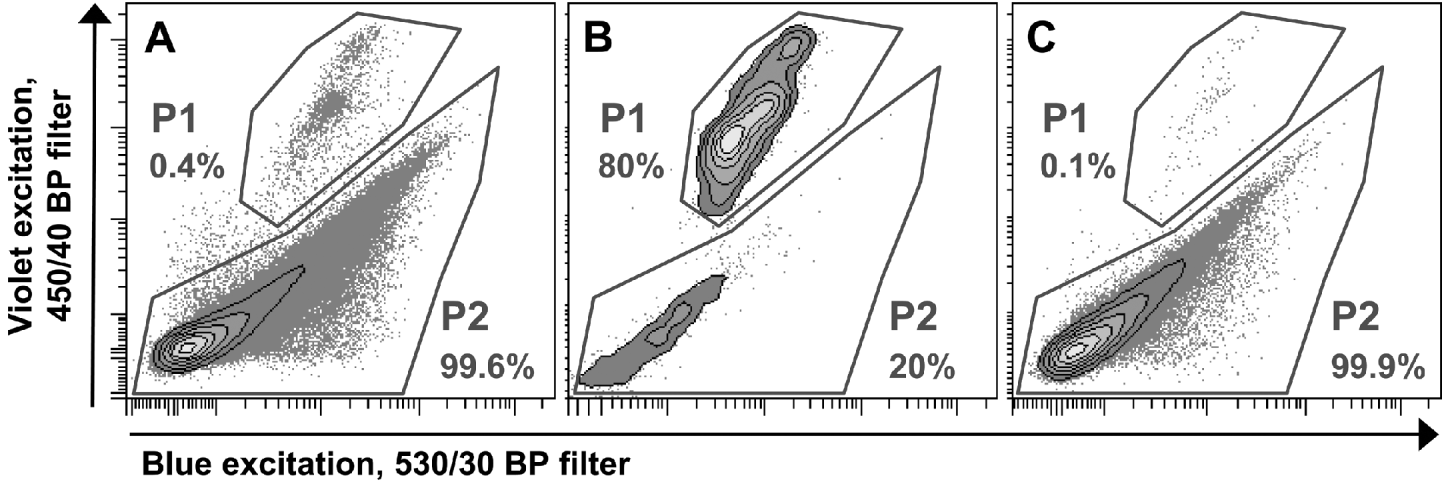
To enrich oocysts in the suspensions, FACS was performed on a FACSAria™ with FACSDiva™ Software Version 5.0.2 (BD Biosciences, San Jose, CA, USA). For this purpose oocysts were suspended in FACSFlow™ Sheath Fluid (BD Biosciences, San Jose, CA, USA). Light-scatter properties of the analysed samples were determined with a standard blue laser (488 nm). In addition, autofluorescent molecules of I. suis oocysts were excited with a violet laser at 407 nm and the blue laser, and oocyst-autofluorescence was analysed by using bandpass filters with 450/40 nm and 530/30 nm, respectively. This autofluorescence was sufficient to discriminate between oocysts and bacteria or debris, and therefore enabled sorting with an electronic gate set on the oocyst fraction (P1, Fig. 1A). The sorting process was also used to prepare a so-called mock control which is commonly used to test for the influence of by-products within an antigen preparation. For this purpose a second electronic gate containing all particles except oocysts (P2, Fig. 1A) was created and particles located in this gate were sorted into a separate tube. Collected objects from both electronic gates were suspended in PBS after sorting. To quantify the purity of the sorting process both samples with sorted objects from P1 (oocysts, Fig. 1B) and P2 (debris, bacteria, in the following called mock control, Fig. 1C) were analysed by flow cytometry with the same threshold rate for 1 min. The number of counted events in P2 of the sorted oocyst (i.e. contaminating debris) was used to calculate the appropriate dilution of the mock control with PBS to achieve an equal concentration of debris/bacteria (particles in P2) in both samples.
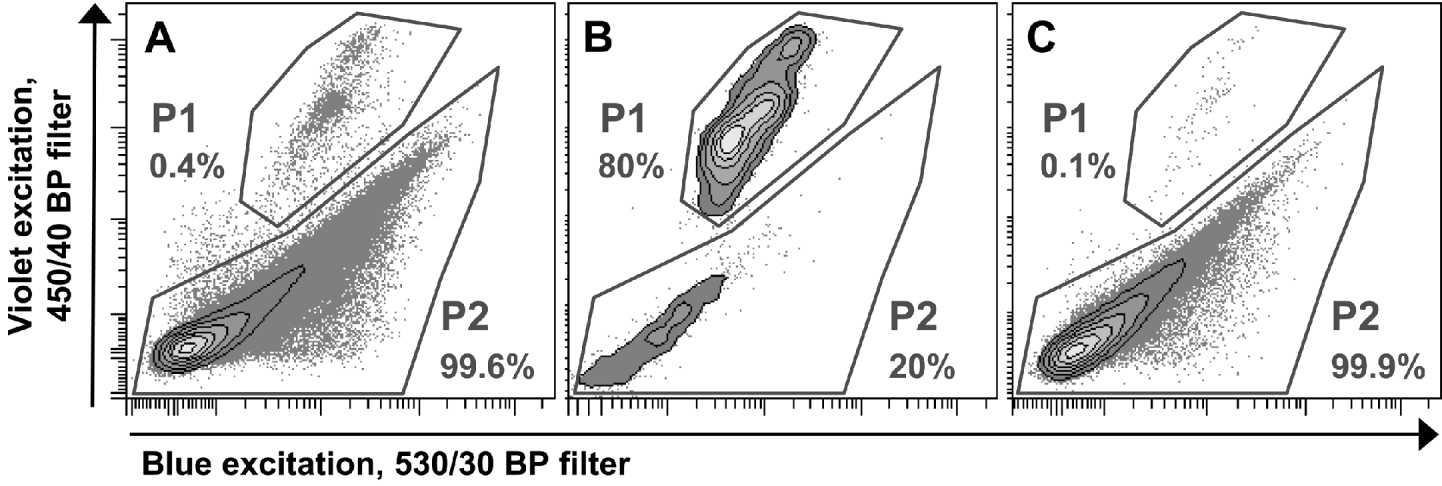
Fig. 1. Flow cytometric analysis of a single batch of Isospora suis oocyst suspensions and electronic gates for FACS sort. P1 represents oocysts, P2 represents bacteria or debris. Percentages are proportions of events in the gates in relation to all events. (A) Before sort. (B) Sorted oocysts from P1 in (A). (C) Sorted mock control from P2 in (A).
Oocysts were counted in a McMaster chamber diluted in sodium chloride/sucrose solution as described above. The oocyst suspension was then diluted with PBS to an oocyst concentration of 3×104 oocysts/ml. The mock control was diluted identically to the oocyst suspension in this second dilution step.
Production of oocyst homogenate
Oocyst suspensions and controls were subjected to 3 freeze-thaw cycles in liquid nitrogen and subsequently treated with a Sonoplus™ ultrasound homogenisator (Bandelin Electronic, Berlin, Germany) at 65% power for 3 min on ice. Homogenization of the oocysts was controlled microscopically. The antigen preparations were then stored at −20°C.
Isolation of lymphocytes
Splenocytes were isolated from homogenized spleen tissue as described previously (Worliczek et al. Reference Worliczek, Buggelsheim, Alexandrowicz, Witter, Schmidt, Gerner, Saalmüller and Joachim2010). Lymphocytes were treated with GEY's Solution (Fang et al. Reference Fang, Sun, Cai, Dodge, Lotke and Williams2000) for 5 min at room temperature to lyse any contaminating erythrocytes. Subsequently, the cells were washed with PBS twice and filtered through a sterile CellTrics® 50 μm filter (Partec GmbH, Görlitz, Germany). Cells were finally suspended in cell culture medium consisting of RPMI 1640 (PAA Laboratories, Pasching, Austria) supplemented with 10% (v/v) heat-inactivated FCS, 100 IU/ml penicillin and 0·1 mg/ml streptomycin (PAA Laboratories, Pasching, Austria) and counted under Trypan blue exclusion staining.
Stimulation of cells
Splenocytes were stimulated with Isospora-homogenate or mock control in the same dilution. For the ELISPOT-assays, cells (4×105/well) were stimulated with 3 dilutions of antigen and mock control, corresponding to 150×10−5, 37·5×10−5, and 9·4×10−5 sporulated oocysts per lymphocyte, respectively. Cell culture medium was used to dilute antigen and controls.
IFN-γ ELISPOT
For the measurement of IFN-γ production by splenocytes, 96-well Multiscreen® IP plates (Millipore, USA) were coated with mouse anti-swine IFN-γ capture mAb (clone A151D5B8, Invitrogen, USA; 10 mg/ml in PBS) at 4°C overnight and blocked with cell culture medium for 2 h at 37°C. Cells in a concentration of 4×105 cells per well, and respective dilutions of antigen and mock controls (see above) were added. Each dilution was tested in duplicate. Additionally, control cultures were stimulated either with cell culture medium only (negative control) or with Concanavalin A (ConA, positive control) at a final concentration of 3 μg/ml. The plates were incubated for 48 h at 37°C, 5% CO2 and subsequently washed with buffer (PBS with 0·01% Tween 20 and 0·1% BSA). Bound IFN-γ was detected by incubation with biotinylated mouse anti-swine IFN-γ mAb (clone A151D13C5, Invitrogen, USA; 2·5 mg/ml in washing buffer) at room temperature for 1 h followed by incubation with streptavidin-bound alkaline phosphatase (dilution 1:2000 in washing buffer, Invitrogen, USA). The binding of alkaline phosphatase was visualized with SIGMAFAST® BCIP/NBT substrate (Sigma-Aldrich, Vienna, Austria) according to the manufacturer's instructions. Plates were then thoroughly washed with tap water and dried overnight at room temperature. Spots were detected with an AID ELISPOT reader (AID GmbH, Strassberg, Germany) (Pintaric et al. Reference Pintaric, Gerner and Saalmüller2008). Results of the ELISPOT assays were converted to spots per 1×106 lymphocytes for graphical presentation.
RESULTS
Retrieval of oocysts
Oocysts were sporulated to obtain antigen from sporozoites as well as from oocyst walls. The majority of oocysts was sporulated after 3 days. The percentage of sporulated oocysts varied between samples taken from different days post-infection and different litters with values of 40–90%. Despite the use of 25% Percoll®, contamination with fat compounds and also the occurrence of debris, fibres, inorganic matter and bacteria was observed after the isolation of oocysts from faeces (Fig. 2A). Inorganic matter and the majority of fibres could be removed by a filtration step through a mesh with 100 μm pore size (Fig. 2B). The recovery rate after filtration was 86±14%.

Fig. 2. Oocyst suspensions of Isospora suis during purification procedure. (A) After isolation and sporulation in 2% potassium dichromate. (B) After filtration through mesh with 100 μm pore size. (C) After density-gradient centrifugation. (D) After FACS sort. Pictures were obtained by using differential interference contrast. For pictures (B)–(C) oocysts were suspended in the same volume as in (A) after treatment. Oocysts in (D) were suspended in 2 ml of PBS after FACS.
Density gradient
As a second cleaning step oocysts were centrifuged on a density gradient made of tap water and sucrose. The majority of oocysts was found at the interface between water and 1·25 m sucrose; a significant cleaning effect could be observed (Fig. 2C). Only a small proportion of oocysts was recovered from the interface between 1·25 m and 2 m sucrose. This proportion was not used for further processing since this interface was heavily contaminated with debris. The recovery rate from the upper interface was 66±18%.
Sterility of oocyst suspensions
All tested oocyst suspensions were bacteriologically sterile after treatment with NaOCl. The oocysts themselves were still viable, showing distinct sporozoite motility during and after excystation.
Enrichment and production of mock control by FACS
Oocysts could easily be identified by their autofluorescence which was excited by a violet laser at 407 nm. They were discriminated from other particles by comparing their fluorescence in the 450/40 nm band-pass filter with their fluorescence after blue excitation in the 530/30 band-pass filter (Fig. 1A). The use of these 2 filters showed the best separation of oocysts from other particles and allowed specific electronic gating. Oocysts had a mean proportion of 0·6±0·4% on all events. Due to low oocyst recovery rates during FACS, only samples with at least 70% sporulation rate were used for sorting, although it was possible to enrich sporulated oocysts only. The FACS procedure enriched oocysts significantly (Fig. 1B) to a mean proportion of 70·8±27·7% of all events (calculated by all sorted batches of oocysts) and to a high grade of purity (Fig. 2D). To ensure successful sorting, filtration of the suspension through the 50 μm filter and also the suspension of oocysts in FACSFlow™ Sheath Fluid (BD Biosciences, San Jose, CA, USA), which contains detergents, was crucial. Without FACSFlow™, threshold rates during sorting were insufficient. Nevertheless, some oocyst batches could not be sorted as the threshold rate broke down continuously.
The proportion of oocysts in the recovered mock control was marginal (Fig. 1C) and in the final dilution of the mock control for immunological assays no oocysts were found. The mean recovery rate of oocysts by FACS was 21±6%. The overall recovery rate in relation to sporulated oocysts isolated from the faeces was 14±4%.
Evaluation of in vitro application
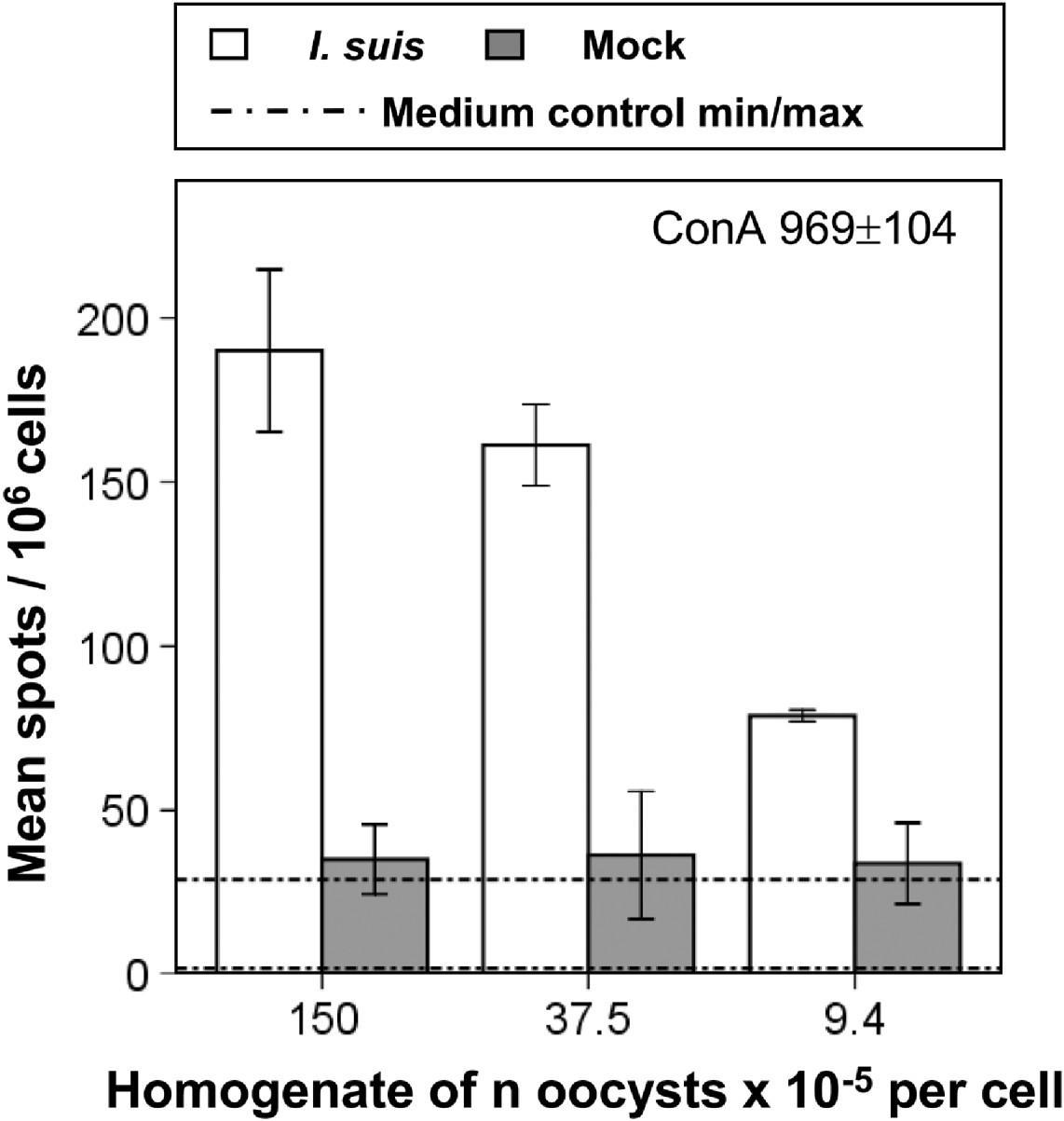
To test purified oocysts and mock controls for their suitability in immunological re-stimulation assays, homogenates of both were used for stimulation of splenocytes from previously I. suis-infected and challenged pigs. As a readout system IFN-γ ELISPOTs were performed to detect the number of cells producing this key effector molecule of the cellular immune response. The ELISPOT assays were sensitive enough to detect the relatively low frequency of IFN-γ secreting cells after 48 h of incubation (Fig. 3). Moreover, there was a clear dose-dependent response of IFN-γ producing cells when the oocyst homogenate was used for stimulation. In Fig. 3 the results of 1 animal are shown which are representative for the pattern of IFN-γ production among 6 investigated animals (further data not shown). In contrast, frequency of cells producing IFN-γ after mock-treatment was only slightly higher than in cells cultivated solely in medium, and showed no distinct dose-dependence. Stimulation with ConA induced a 5-fold higher frequency of IFN-γ-producing cells compared to I. suis-stimulated cells. In summary, by using IFN-γ ELISPOTs an I. suis-specific recall response was detected.

Fig. 3. Results of IFN-γ ELISPOT of splenocytes after in vitro re-stimulation with homogenates of Isospora suis oocysts and mock control. The result for stimulation with ConA as positive control is given in numbers. Representative experiment out of 6 experiments. Error bars extend to +/−1 standard deviation.
DISCUSSION
In the present study a protocol to isolate, purify and enrich oocysts of I. suis to obtain material suitable for immunological research, but also for other parasitological in vitro studies was established.
In cases where oocysts are used as infection material for their natural hosts the purity of the oocyst suspension is not crucial; faeces with a sufficiently high oocyst content can be used for the recovery of parasite material regardless of the faecal consistency. Moreover, faeces with normal consistency often contain a high number of oocysts that can be enriched (Mundt et al. Reference Mundt, Joachim, Becka and Daugschies2006). However, in the present study only faeces from swine with diarrhoea were used to minimize contamination with fibres, inorganic matter or straw to obtain the cleanest material possible. Heavy contamination with smaller particles cannot be removed by density-gradient centrifugation and in our experience disturbs subsequent FACS, resulting in the breakdown of threshold rates during the sorting process. Some oocyst batches were not suitable for FACS most probably due to this problem.
The treatment with NaOCl turned out to be a feasible method to produce infectious parasite material in the absence of viable bacterial contaminants. Such oocysts can be used, for instance, in experiments where sterile material is necessary, such as infection of gnotobiotic piglets or cell cultures.
For antigen-specific immunological assays, bacteria killed by the NaOCl treatment would either have to be removed completely from the antigen preparation to exclude stimulation by bacterial components, or a sufficient control has to be included. Such a control can be produced in an elegant manner by FACS as shown above, where the frequency of contaminating particles within purified oocysts was used to calculate and adjust the same frequency of these particles in a corresponding mock control.
It was previously shown that coccidian oocysts can be isolated by FACS using fluorochrome labelling (Ferrari et al. Reference Ferrari, Vesey, Davis, Gauci and Veal2000; Everson et al. Reference Everson, Ware, Dubey and Lindquist2002) or utilizing the autofluorescence of oocysts in flow cytometry (Fuller and McDougald, Reference Fuller and McDougald1989; Everson et al. Reference Everson, Ware, Dubey and Lindquist2002). Since I. suis oocysts are also known to display autofluorescence (Daugschies et al. Reference Daugschies, Bialek, Joachim and Mundt2001), this was applied in an approach using FACS of this material for enrichment and for the retrieval of mock controls. With a recovery rate of only 14% after the whole purification procedure including FACS (with the highest losses being encountered during this step), the loss of oocysts during the whole process is considerable but seems acceptable when taking into account that a highly purified population of oocysts is obtained. Moreover, an appropriate mock control for in vitro stimulation assays of lymphocytes is also acquired with the developed procedure. Faeces from different animals and litters have to be pooled to obtain enough oocysts for antigen-specific stimulation, and different batches of oocysts can vary considerably in their purity when purified and enriched without FACS. Considering these differences in the grade of contamination in different batches of oocysts, the method presented for production of matched mock controls provides a solution for this issue. The excystation of sporulated oocysts and the use of subsequently purified sporozoites as antigen source (used e. g. for Eimeria spp. by Schmatz et al. Reference Schmatz, Crane and Murray1984; Breed et al. Reference Breed, Schetters, Verhoeven, Boot-Groenink, Dorrestein and Vermeulen1999; Hermosilla et al. Reference Hermosilla, Barbisch, Heise, Kowalik and Zahner2002) might solve the problem of bacteria attached to oocyst walls. Because of limited amounts of parasite material and insufficient excystation rates, this method is not applicable for I. suis at the moment. Future refinement of excystation methods and in vitro cultivation of I. suis might enable the production of antigen without any contamination by faecal material.
After in vitro re-stimulation mock-treated cells showed a very weak response that was only slightly stronger than the medium control but distinctly lower than the response to the Isospora antigen, allowing a clear differentiation between the Isospora-specific response and response to other antigens which may have contaminated the antigen preparation. Especially in cases of a low frequency of reactive cells – as it was shown for I. suis-specific lymphocytes in this study – and therefore a possibly weak response in the applied readout systems, an appropriate control for the specificity of the stimulation is needed and can easily be provided by the method described in this paper.
In summary, material (antigen and controls) provided by the preparation of I. suis oocysts by centrifugation, sterilization and FACS is suitable for sensitive immunological readout systems and therefore may also be applied to other coccidian species where faecal material is used for this purpose. Moreover, the detected Isospora-specific IFN-γ recall response encourages further functional studies on the cellular immune response to I. suis.
ACKNOWLEDGEMENTS
The authors thank Ewald Denner for the bacteriological examination, and the staff of the Institute of Parasitology, University of Veterinary Medicine Vienna for collecting faecal samples.