


Introduction
Insects account for a large proportion of all known organisms, and as such they inevitably play a pivotal role in many ecosystems. Southwest China is one of the biodiversity hotspots of the world, and the insect populations in the region have been largely understudied. Cockroaches (Blattaria) are abundant in southwest China, yet little attention has been given to the phylogeography of the group. Understanding the geographical pattern and population structure of molecular variance within a species of Blattaria is very important to infer its origin, given that variance truly exists in the wild (de Bruyn et al., Reference de Bruyn, Grail and Carvalho2011). The double-striped cockroach, Blattella bisignata, is a wild species, which has the ability to fly over short distance, and mainly lives in leaf litter, grass or shrubs in forested areas. As the double-striped cockroach is native to southeast Asia (the epicenter for Blattella diversity), which remained uncovered by ice sheets during the Pleistocene (Williams et al., Reference Williams, Dunkerley, de Deckker, Kershaw and Chappel1998), we expect geographic structuring detectable at the genetic level. Currently, there is no information on the phylogeography of B. bisignata in southern China.
Mitochondrial DNA (mtDNA) is a popular marker for population genetic, phylogeographic and phylogenetic studies. However, mtDNA is inherited maternally and is non-recombining, which may reveal an incomplete population history when sex bias exists in dispersal (Hare, Reference Hare2001). Microsatellites, on the other hand, are extremely popular because of their high mutant rates and strong power to reveal geographic structure, historical demography and parentage, which have been used in studies of the population genetic structure of the German cockroach (Crissman et al., Reference Crissman, Booth, Santangelo, Mukha, Vargo and Schal2010; Booth et al., Reference Booth, Santangelo, Vargo, Mukha and Schal2011). Selection pressure on microsatellite loci has been previously reported (Schlotterer, Reference Schlotterer2000), thus it is important to verify that loci are not under selection before proceeding with analysis. The difference in microsatellite variations between sex chromosomes and autosomes is a representative example of such differential selection (Bachtrog et al., Reference Bachtrog, Weiss, Zangerl, Brem and Schlotterer1999). Furthermore, ‘null alleles’ sometimes fail to amplify during PCR, which can generate problems during analysis. However, all types of markers have some disadvantages, and the versatility of microsatellites in addressing many ecological questions compensates for their limitations (Selkoe & Toonen, Reference Selkoe and Toonen2006). Currently, EPICs (Exon-primed intron-crossing) are widely used in a variety of organisms, which need a certain number of genomic resources. When comparing EPICs with ANMs (anonymous nuclear markers), it has been demonstrated that the variability of ANMs is significantly higher than that of EPICs (Lee & Edwards, Reference Lee and Edwards2008). Furthermore, EPICs are more likely to undergo purifying selection because they are located near gene regions (Thomson et al., Reference Thomson, Wang and Johnson2010), whereas ANMs locate at intergenic regions.
With the massive influx of genomic resources for model organisms and the transcriptome underway for nonmodel organisms (Amaral et al., Reference Amaral, Silva, Moeller, Beheregaray and Coelho2010), single nucleotide polymorphisms (SNPs) have recently been chosen as markers for a growing number of studies (Knowles & Carstens, Reference Knowles and Carstens2007; Raychoudhury et al., Reference Raychoudhury, Grillenberger, Gadau, Bijlsma, van de Zande, Werren and Beukeboom2010). Given that there is little genomic information available for B. bisignata, we develop in our study 11 anonymous single-copy nuclear polymorphic sequence (SCNP) markers, with the aim of resolving the species’ dispersal pattern in southern China. These markers are seldom screened in insects, but recently were reported for Melanoplus oregonensis (Carstens & Knowles, Reference Carstens and Knowles2006) and in Neonympha mitchellii mitchellii (Hamm, Reference Hamm2012).
Previous phylogeographic and population genetic studies of Blattaria have relied mainly on mitochondrial sequences (Chinn & Gemmell, Reference Chinn and Gemmell2004; Maekawa et al., Reference Maekawa, Kon, Matsumoto, Kitade and Araya2007), microsatellites (Booth et al., Reference Booth, Bogdanowicz, Prodohl, Harrison, Schal and Vargo2007) and rDNA-RFLP (Mukha et al., Reference Mukha, Kagramanova, Lazebnaya, Lazebnnyi, Vargo and Schal2007). Considering all of the points addressed above, our findings may prove valuable for further population studies.
Materials and methods
Collection of samples
Collection sites and the number of individuals used in our study are shown in fig. 1. All the individuals were collected from five different locations in southern China and stored in 100% ethanol. The details of sampling sites are in table S1.

Fig. 1. Sampling sites: 1, Jinghong (JH); 2, Baise (BS); 3, Mayanghe (MYH); 4, Anqing (AQ); 5, Zhenjing (ZJ). M01, Hengduan Mountain Range; M02, Phoenix Mountain.
Genomic library construction and cloning
We constructed a genomic library from a fresh B. bisignata specimen sampled from Baohua Mountain (Zhenjiang, Jiangsu Province, China). Tissue was obtained from the dorsal anterior thorax, and total genomic DNA was extracted following standard phenol-chloroform procedures (Sambrook et al., Reference Sambrook, Fritsch and Maniatis1989). The extract was concentrated to 3.2μgμl–1 (assayed using a NanoDrop® ND-3300 Fluorospectrometer (PeqLab, Erlangen, Germany)) using a vacufuge (Eppendorf, Cambridge, UK). About 15μg of DNA was fragmented by restriction digest using AfaI (RsaI) for over six hours. The sheared DNA was visualised on a 1.5% agarose gel, and the target products ranging from 0.75 to 1.5kb were excised with sterile blades and purified using a QIAGEN Gel Extraction Kit (Qiagen, Hiden, Germany) (eluted in 50μl elution buffer). This size-selected DNA was vacufuged from the initial concentration of 15ngμl–1 to a final concentration of 45ngμl–1 in order to reach the proper molar ratio (>10:1) for insertion into the vector for optimal blunt-ended ligation. Approximately 150ng of DNA was ligated into 25ng of PCR Zero Blunt vector (Invitrogen, NY, USA), then transformed into chemically-competent One Shot TOP10 E. coli cells (Invitrogen). These were then plated on LB agar plates containing kanamycin (50μgml–1) and grown overnight in an incubator at 37 °C following the method described by Noonan & Yoder (Reference Noonan and Yoder2009).
Finally, we picked 96 colonies individually into 1.5ml SOB-kan (50μgml–1) liquid medium using autoclaved pipette tips, and grown overnight at 37 °C with continuous gentle shaking. Each PCR reaction was 16.5μl, containing 15μl TransTaq PCR SuperMix (Beijing TransGen Biotech Co., Beijing, China), 0.5μl of each primer (M13F (–20) & R provided with the Invitrogen kit), and a 0.5μl template directly pipetted from each individual overnight culture. These 96 PCR products were subsequently visualised on a 1% agarose gel to confirm the product size. We chose 80 products to sequence, ranging in size from 800 to 1500bp, using the primers used in PCR on an ABI-PRISM™ 3730 Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA).
Amplification of loci
Fifty sequences were excluded, either because they aligned with other sequences via local BLAST (n = 16), were extremely AT rich (n = 9), returned positive BLAST hits against coding sequences, SINEs (short interspersed repetitive element) or other retrotransposons (n = 11, B. germanica × 5, Bombyx mori × 2, Nasonia × 1, Reticulitermes flavipes × 1, Aedes aegypti × 2) or did not sequence properly (n = 14). The remaining sequences were then translated into amino acid sequences. We chose 30 sequences, using the NCBI ORF finder (http://www.ncbl.nlh.gov/projects/gorf/), that were apparently from noncoding regions of the B. bisignata genome, after which we designed species-specific polymerase chain reaction (PCR) primers for the sequences. Primer design was carried out using Primer Premier 5.0 (Clarke & Gorley, Reference Clarke and Gorley2001). These primers were tested on a small panel (two individuals from B. bisignata, and one each from B. germanica and B. lituricolliss) and a distantly related cockroach, Eupolyphaga sinensis Walker (Eupolyphaga). A 25μl volume containing 2.5μl 10 × LA PCR Buffer, 0.4mM of each dNTP, 1.25 units of LA Taq polymerase (TaKaRa Biotech, Dalian, China), 2.5mM MgCl2, 0.5μl each of primers and approximately 10ng of DNA was used per PCR with the following protocol: initial denature 94 °C for 5min, 30 cycles at 94 °C for 30s, optimal X°C for 30s (see table 1 for locus-specific annealing temperatures), 72 °C for 30s and a final extension at 72 °C for 10min. PCR products with single amplified bands were first purified and then subcloned in one individual per locus. Finally, 4–5 clones were sequenced in both the forward and reverse directions to make sure each locus was a single copy.
Table 1. Primer sequences (5′ − 3′) for 11 anonymous nuclear loci and optimal PCR annealing temperatures (°C) cross several cockroach species.

B.b., Blattella bisignata; B.g., Blattella germanica; B.l., Blattella lituricollis; J.t., Jacobsonina tortuosa; R.f., Rhabdoblatta formosana; P.a., Periplaneta americana; n/a, not applicable.
Data analysis
Sequence assembly was conducted using SeqMan (DNASTAR, Inc., Madison, WI, USA), and alignments were produced by Clustal W (Thompson et al., Reference Thompson, Higgins and Gibson1994). All alignments were checked visually, and sequences of different individuals for each locus were cut to equal length. The analyses of DNA sequence polymorphisms, such as number of variable sites (S), nucleotide diversity (π), and haplotype diversity (Hd), were calculated with DnaSP Version 5.00 (Librado & Rozas, Reference Librado and Rozas2009). We also calculated the minimum number of recombination events (Hudson & Kaplan, Reference Hudson and Kaplan1985), Tajima's D (Tajima, Reference Tajima1989) and LD (linkage disequilibrium) level for each marker with DnaSP Version 5.00. In the LD analysis, the chi-square test was used to estimate the significance of associations between all parsimony-informative polymorphic sites, following the Bonferroni procedure for multiple comparisons. In this study, the Dunn-Šidák correction was used instead of the the Bonferroni correction, because the Dunn-Šidák correction gives a stronger bound than the Bonferroni correction (Abdi, Reference Abdi and Salkind2007). Maximum parsimony (MP) and neighbor-joining (NJ) phylogenetic analyses were used to identify major clades and evaluate the relationships among the haplotypes, using B. germanica as the outgroup. The model selected by the Akaike information criterion (AIC) in Modeltest 3.7 (Posada & Crandall, Reference Posada and Crandall1998) was used in the maximum parsimony analyses PAUP* 4.0b10 (Swofford, Reference Swofford2003). The NJ tree was constructed in MEGA 5 (Tamura et al., Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011) with both bootstrap and interior branch support tests. The confidence level of the nodes in the NJ and MP trees were estimated using 1000 bootstrap pseudoreplicates. To test for hierarchical population genetic structure in B. bisignata, we performed analysis of molecular variance (AMOVA) in ARLEQUIN 3.11 (Excoffier L. Zoological Institute, University of Berne, Switzerland) with 1000 permutations to examine partitioning of genetic diversity within and among populations. F ST values were calculated to evaluate genetic partitioning among populations. Mismatch distribution analyses were performed using ARLEQUIN 3.11 to detect recent demographic signatures of population expansions (Rogers, Reference Rogers1995).
Results and discussion
Cross-species amplification
Of all 30 loci, 11 loci may be ideal anonymous single-copy nuclear sequence markers because they amplified a single-copy product in three Blattella species and contained nucleotide variation, while only one locus (AL51) for E. sinensis. We also tested these 11 pairs of primers in other representative species of the cockroach families: Jacobsonina tortuosa (Blattellidae), Rhabdoblatta formosana (Epilampridae) and Periplaneta americana (Blattidae). Our study showed some loci can cross-amplify at high taxonomic levels, after testing all these loci in representative species of Blattellidae, Epilampridae and Blattidae, respectively (see table 1). As expected, cross-species amplification success decreased with increasing genetic distance. A previous study of the microsatellite loci in B. germanica (Booth et al., Reference Booth, Bogdanowicz, Prodohl, Harrison, Schal and Vargo2007) showed they were applicable to only one additional B. asahinai, but not to Parcoblatta pennsylvanica and Supella longipalpa (family Blattellidae), P. americana and P. fuliginosa (family Blattidae). High cross-species applicability of microsatellites loci in Hymenoptera (Henshaw et al., Reference Henshaw, Toth and Young2011), Diptera (Augustinos et al., Reference Augustinos, Asimakopoulou, Papadopoulos and Bourtzis2011) and Lepidoptera (Sinama et al., Reference Sinama, Dubut, Costedoat, Gilles, Junker, Malausa, Martin, Neve, Pech, Schmitt, Zimmermann and Meglecz2011) has been previously reported, so our anonymous single-copy nuclear markers were more time efficient and less costly to some extent, at least in Blattaria.
Genetic diversity
The primers were tested on B. bisignata individuals collected from southern China. Direct sequencing of PCR products was conducted twice as well as a clone strategy for some heterozygous individuals to obtain the precise sequences. A total number of 151 variable sites or SNPs were recognized in 4157bp sequenced markers with an average 3.6 SNPs per 100bp. The nucleotide variation observed in our loci is higher than that of grasshoppers (Carstens & Knowles, Reference Carstens and Knowles2006). See table 2 for the summary of data for these 11 loci. The nucleotide diversity (π) of each locus varied from 0.002 (AL57) to 0.018 (AL51). These differences likely result from the specific genomic locations of the sequences and their distances away from the neighbouring functional regions of the genes, which have been shown to influence substitution rates (Ji et al., 2011). It has been inferred that despite some conservative noncoding regions, many anonymous regions of the genome may be free from selection (Lee & Edwards, Reference Lee and Edwards2008). We detected recombination at six of the 11 loci (AL12, AL42, AL45, AL51, AL53, AL65), and no significant linkage disequilibrium (LD) was observed after correcting for multiple comparisons, indicating that these loci are unlinked.
Table 2. Summary data for 11 anonymous nuclear loci screened for variation in B.bisignata.

bp, size of alignable sequence in base pairs; N, sample size; S, number of variable sites; H, number of haplotypes; π, nucleotide diversity per site; Hd, Haplotype diversity; θw, Watterson's estimate of theta per site; indel, presence or absence of insertions/deletions; D, Tajima's D; LDD, the mean linkage disequilibrium (Hedrick, Reference Hedrick2005).
Population genetic structure and demographic analysis
The MP trees and NJ trees based on three anonymous loci (AL31, AL51, AL65) revealed relatively consistent results (figs 2–4). Both trees consisted of two main clades, and one clade contained haplotypes or individuals from JH and BS, while the other contained those from MYH, AQ and ZJ (tables S2–S4). We didn't find any shared haplotypes, which was likely a result of the lack of abundant sampling sites. AMOVA showed clear population genetic structure among the B. bisignata population (F ST varying from 0.52 to 0.87, P < 0.01). When we regarded every sample site as one population, the AMOVA based on haplotype frequencies analyses revealed that variations among populations were far greater than those within populations. We divided the populations from five sampling sites into two or three groups according to their geographic locations, and the F CT value achieved a maximum when they were divided into two groups (JH + BS and MYH + AQ + ZJ). This is consistent with results of MP and NJ trees to some extent. The pairwise F ST at all locations for three anonymous loci are shown in tables S5–S7. JH and BS have a relatively short genetic distance, and this is also true for AQ and ZJ. Not surprisingly, a long genetic distance was detected between the group (JH, BS) and group (AQ, ZJ). MYH was a debatable location for its closer geographical distance to JH and BS than AQ and ZJ, but the results showed its close relationship with the southeast populations (AQ and ZJ). Considering the locations of Phoenix Mountain and the Hengduan Mountain range (fig. 1), we infer that the mountains may have formed natural barriers to the expansion of B. bisignata. The mismatch distributions for all individuals of each loci did not consist of a distinct unimodal curve (figs S1–S3), which suggested that a massive population expansion in B. bisignata did not occur. The mild climate, the mountain barriers and some other factors may have caused a relatively stable population structure of B. bisignata in southern China. As the number of sampling sites increases in the future, we believe a more clear and convincing population genetic structure of B. bisignata will become evident.
Conclusions
The present study developed 11 anonymous single-copy nuclear polymorphic loci from B. bisignata using an insert-genomic library, which is critical to phylogeographic research of Blattaria. All of the markers had some level of cross-species amplification success and, furthermore, exhibited abundant variation. These anonymous markers seem to be adequate in determining the population genetic structure of B. bisignata. We have presented here the foundation of the phylogeography of B. bisignata in southern China, and a further study will be carried out with a larger number of samples from the region. These informative markers, along with other available markers, are likely to reveal a more complete picture of the evolution and dispersal patterns of cockroaches in southern China.

Fig. 2. Maximum parsimony tree of AL31 of B. bisignata based on JC substitution model. Values represent MP bootstrap values. Bootstrap values of 50% or greater are shown.

Fig. 3. Maximum parsimony tree of AL51 of B. bisignata based on Tvmef substitution model. Values represent MP bootstrap values. Bootstrap values of 50% or greater are shown.
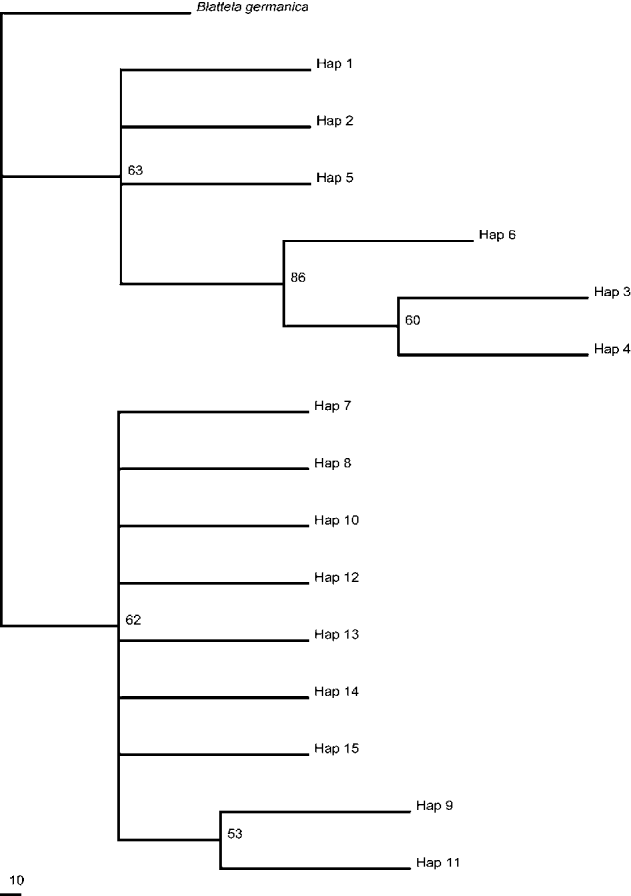
Fig. 4. Maximum parsimony tree of AL65 of B. bisignata based on F81 substitution model. Values represent MP bootstrap values. Bootstrap values of 50% or greater are shown.
Acknowledgements
We thank Dr Qi-Xin He and Mr Benjamin Blanchard (Department of Ecology and Evolutionary Biology, University of Michigan, MI, USA) for their help on some data analyses and language. We also thank Dr Bo Xiao for the sample collection and Zhuo Chen for laboratory assistance. This work was supported by grants from the Natural Science Foundation of China (No. 30970339) awarded to G.F. Jiang, and Nanjing Normal University Innovative Team Project (No. 0319PM0902).
Supplementary material
The online figures and tables can be viewed at http://journals.cambridge.org/ber.






