


Introduction
The term dyskinesia encompasses a broad array of hyperkinetic movement disorders with varying etiopathologies and presentations.Reference Vijayakumar and Jankovic 1 , Reference Vijayakumar and Jankovic 2 Management of iatrogenic dyskinesia dominates the literature, as this includes the 2 most commonly encountered etiologies: levodopa-induced dyskinesia among patients with Parkinson’s disease and tardive dyskinesia (TD), which is related to medications that reduce dopamine neurotransmission via receptor antagonism (eg, antipsychotics, metoclopramide) or vesicular depletion (reserpine).Reference Vijayakumar and Jankovic 1 , Reference Vijayakumar and Jankovic 2 Nonetheless, clinicians should be reminded that there is a baseline rate of spontaneous dyskinesia in the general population estimated at 28.7 per 100,000 person-years, with higher rates among those with older age, female gender, and diabetes mellitus.Reference Merrill, Lyon and Matiaco 3 Using data from the pre-antipsychotic era and first episode studies, untreated schizophrenia is also associated with age-dependent risks for spontaneous dyskinesia, with estimated rates of 4% in first-episode patients, 12% for those under age 30 who are ill for several years, 25% for ages 30–50 years, and 40% for those 60 years or older.Reference Fenton 4 Not only is the dyskinesia rate 3.5 times higher in antipsychotic-naive schizophrenia patients compared to matched controls, dyskinesia is also significantly more prevalent in nonpsychotic first-degree relatives compared to controls (odds ratio 1.38, 95% CI: 1.06–1.81), suggesting a common genetic basis for dopamine dysfunction that increases risk for psychosis and movement disorders.Reference Koning, Tenback, van Os, Aleman, Kahn and van Harten 5
Irrespective of the demographic factors, when confronted with patients with drug-induced TD, clinicians have 3 viable options: drug discontinuation (when possible), switching to less potent dopamine antagonists, or use of adjunctive agents. For the cohort of patients who do not have a primary psychotic disorder discontinuing the offending agent is the most logical choice, but the long-term data show low rates of reversibility. In prospective studies where patients had dopamine modulators withdrawn (primarily antipsychotics and metoclopramide), remission rates were extremely low (2%), and response rates to drug discontinuation were in the range of 1–20%.Reference Cloud, Zutshi and Factor 6 Second generation antipsychotics generally have lower TD rates than the more potent dopamine D2 antagonist first generation antipsychotics,Reference Ryu, Yoo and Kim 7 , Reference Kinon, Kollack-Walker and Jeste 8 yet switching to a weaker D2 antagonist such as quetiapine or olanzapine may be impractical for psychiatric reasons (eg, the patient requires a higher level of D2 antagonism for optimal benefit); moreover, there are limited data to suggest reversibility of tardive syndromes upon switching to an atypical antipsychotic, with conflicting data for clozapine.Reference Vijayakumar and Jankovic 2 For patients in whom drug discontinuation does not yield substantial benefit, or who require ongoing use of dopamine blockade to treat psychiatric illness, TD management over the last 40 years necessitated choosing from an array of options, most of which demonstrated limited efficacy. However, in the past 4 years, there has been a tremendous shift away from the therapeutic nihilism surrounding TD, as the literature has been rapidly populated with papers discussing new insights into TD pathophysiology and new agents for TD management.Reference Teo, Edwards and Bhatia 9 , Reference Rana, Chaudry and Blanchet 10 These insights and pharmacological advances will be discussed here, with a focus on those agents with promising clinical data that may lead to regulatory approval.
Pathophysiology of TD
Data from the early 1990s supported the concept that TD was a manifestation of D2 receptor upregulation and supersensitivity related to chronic reduction in dopaminergic neurotransmission, primarily from postsynaptic receptor blockade.Reference Teo, Edwards and Bhatia 9 Dopamine D2 receptors are expressed on striatal medium spiny neurons and function in an inhibitory manner to reduce the velocity and amplitude of movements through activation of the so-called the indirect basal ganglia pathway.Reference Aquino and Lang 11 The development of increased D2 receptor sensitivity would thus be expected to induce hyperkinesia. This model was supported by in vivo animal data demonstrating the development of D2 receptor upregulation and supersensitivity after exposure to D2 antagonists, and by the clinical observation that withdrawal of D2 antagonists in humans resulted in TD exacerbationReference Teo, Edwards and Bhatia 9 ; however, animal data also demonstrate that these phenomena occur very quickly after drug exposure, contrary to the clinical course of TD, and are rapidly reversible after withdrawal of the dopamine antagonist, implying that the persistent forms of TD in humans may have differing mechanisms than that associated with withdrawal dyskinesia.Reference Casey 12 , Reference Mahmoudi, Levesque and Blanchet 13 Moreover, while increased striatal D2 receptor binding can be seen in patients exposed to chronic D2 blockade, this effect is not necessarily correlated with the presence of dyskinesia in imaging or postmortem studies.Reference Teo, Edwards and Bhatia 9 Although the D2 upregulation/supersensitivity hypothesis for TD appears lacking in humans, primates exposed to clozapine or haloperidol experienced significant D3 receptor upregulation in the haloperidol cohort, with the extent of D3 binding in the nigrostriatal regions correlating with TD intensity.Reference Mahmoudi, Levesque and Blanchet 13 There is also support for the D3 hypothesis from genetic studies associating certain D3 receptor polymorphisms with increased TD risk.Reference Segman, Goltser and Heresco-Levy 14 Given the large overlap in sequence homology and ligand affinity between D2 and D3 receptors, selective D3 agents have only recently been developed for in vivo human neuroimaging, so future imaging studies may shed light on the viability of this concept.Reference Le Foll, Wilson, Graff, Boileau and Di Ciano 15
Genetic markers have also implicated numerous pathways involved in striatal dopaminergic signaling, including serotonin and dopamine receptor variations,Reference Segman, Goltser and Heresco-Levy 14 and polymorphisms in the gamma-aminobutyric acid (GABA) transporterReference Son, Lee and Yoon 16 and GABAA receptor.Reference Inada, Koga and Ishiguro 17 As will be discussed below, modulation of vesicular monoamine transporter type 2 (VMAT2) is a promising treatment modality, and several polymorphisms in the VMAT2 gene have been associated with increased TD risk.Reference Zai, Tiwari and Mazzoco 18
Other hypotheses have been advanced over the years related to in vivo animal data, and clinical and genetic human studies. The involvement of free radicals and other oxidative mechanisms was suggested 2 decades ago on the basis of animal and a small number of human studies.Reference Lohr and Browning 19 These hypotheses fell into disfavor due to the inconsistent results from vitamin E trials,Reference Lohr, Kuczenski and Niculescu 20 but they have not been completely abandoned, as some genetic studies point to increased risk among those with polymorphisms in the free radical scavenger enzyme superoxide dismutase and related anti-oxidative enzymesReference Teo, Edwards and Bhatia 9 , Reference Cho and Lee 21 and to markers related to systemic inflammation.Reference An, Tan and Shi 22 Recent animal studies have indicated that peroxisome proliferator-activated receptor (PPAR) agonists exhibit neuroprotective properties, leading to exploratory studies of the PPAR-gamma agonist pioglitazone and PPAR-alpha agonist fenofibrate in rat models of TD, with positive results.Reference Grover, Kumar, Singh, Vikram and Budhiraja 23 The observations that phenylketonuria was associated with TD risk and that administration of large phenylalanine doses worsened dyskinetic symptoms in patients with TD led to the hypothesis that failure to clear central nervous system phenylalanine might underlie TD pathophysiology. Branched chain amino acids compete with phenylalanine for transport across the blood–brain barrier, and their use was associated with TD improvement in a number of studies; however, most of these were open label and were performed by investigators at 1 institution.Reference Richardson, Small, Read, Chao and Clelland 24 No new studies have appeared in the past decade.
Among the more novel findings from genetic association studies are loci associated with development, cellular signaling, and neuroplasticity.Reference Teo, Edwards and Bhatia 9 , Reference Aquino and Lang 11 That TD might be best viewed as a disorder of synaptic plasticity has emerged as a leading unifying hypothesis that brings together basic science and genetic findings with the clinical observation that TD shows limited reversibility after withdrawal of offending agents.Reference Aquino and Lang 11 While striatal D2 receptor hypersensitivity might be the initial manifestation of D2 antagonist exposure, ongoing D2 blockade creates secondary effects on the plasticity of glutamatergic synapses of striatal interneurons. Aberrant glutamatergic signals to cortical structures that also have impaired plasticity results in a situation wherein withdrawal of dopamine antagonists fails to generate the expected symptomatic reversal.Reference Teo, Edwards and Bhatia 9 Not only does this model suggest that certain glutamate-based strategies might be effective for TD,Reference Teo, Edwards and Bhatia 9 but it also explains why signal interruption via surgical pallidotomy or bilateral deep brain stimulation (DBS) of the internal part of the globus pallidus (GPi) have been reported as beneficial in those with intractable TD.Reference Pouclet-Courtemanche, Rouaud and Thobois 25
Imaging studies and peripheral markers in schizophrenia patients with TD reflect this underlying cellular dysfunction. Reduced basal ganglia and thalamic volume is seen among those with TD, with the greatest reductions found in the caudate nucleus.Reference Sarró, Pomarol-Clotet and Canales-Rodríguez 26 S100B is a calcium binding protein expressed by astrocytes and involved in numerous cell regulatory processes. Peripheral S100B levels are increased after central nervous system cellular insults, and data in schizophrenia patients not only reveal higher S100B levels among those with TD, but also that serum S100B levels positively correlate with abnormal movement rating scale scores.Reference Zhang, Xiu and Chen da 27
Treatment
After an extensive review of the literature, the American Academy of Neurology (AAN) found few evidence-based therapies for TD in 2013 and concluded that the following agents are either not recommended, or have insufficient data to support (or refute) their use: acetazolamide, bromocriptine, baclofen, buspirone, diltiazem, galantamine, eicosapentaenoic acid, levetiracetam, vitamin E, vitamin B6, thiamine, selegiline, melatonin, nifedipine, yi-gan san, biperiden discontinuation, botulinum toxin type A, electroconvulsive therapy, α-methyldopa, reserpine, and pallidal DBS.Reference Bhidayasiri, Fahn and Weiner 28 The AAN review also noted that data are insufficient to support or refute TD improvement by withdrawing causative agents or switching from typical to atypical antipsychotics.Reference Bhidayasiri, Fahn and Weiner 28 Vitamin E in particular had proponents based on early small case series, but these findings failed to replicate in larger controlled studies.Reference Vijayakumar and Jankovic 2 , Reference Soares and McGrath 29 Evidence for branched chain amino acid preparations also remains inconclusive due to the paucity of controlled studies.Reference Vijayakumar and Jankovic 2
Among the small number of evidence-based options are clonazepam, ginkgo biloba, amantadine, and tetrabenazine. In a double-blind, crossover, 12-week, placebo-controlled study, clonazepam (mean dose 3.5 mg/d) was associated with 35% improvement in dyskinesia symptoms (n=19), although tolerance developed after 5–8 months of use in the 5 subjects whose treatment continued up to 9 months.Reference Thaker, Nguyen, Strauss, Jacobson, Kaup and Tamminga 30 The investigators noted that a 2-week washout was sufficient to restore the antidyskinetic effect of clonazepam. Of the 4 published studies of amantadine use, 2 employed double-blind, placebo-controlled, crossover designs. The first, an 18-week trial, reported 15% improvement in dyskinesia ratings on an amantadine dose of 300 mg/d,Reference Angus, Sugars, Boltezar, Koskewich and Schneider 31 while the second study randomized patients to amantadine 100 mg/d or placebo for 2 weeks each (with a 4-day washout between treatment arms) and found a 22% reduction in abnormal involuntary movement scale (AIMS) scores.Reference Pappa, Tsouli, Apostolou, Mavreas and Konitsiotis 32 Extract of ginkgo biloba (EGb) is an antioxidant that possesses free radical scavenging properties. A standardized extract (EGb-761) was examined in a 12-week, double-blind, placebo-controlled trial in which inpatients with schizophrenia and TD were randomly assigned to EGb-761 240 mg/d (n=78) or placebo (n=79). EGb-761 was well tolerated, with 96.8% of subjects completing the study. There was a significantly greater decrease in endpoint AIMS total score in the patients treated with EGb-761 compared to the placebo cohort (p<0.0001), with ≥30% reduction in AIMS noted in 51.3% of EGb-761 but only 5.1% of the placebo group.Reference Zhang, Tan, Zhang, Chan, Wu and Zhou 33
By the 1960s, it was known that TD was the result of increased dopamine signaling, and this led to the search for agents that could modulate dopamine neurotransmission without directly antagonizing postsynaptic receptors. Reserpine’s effect on presynaptic vesicle monoamine content would not be elucidated until the 1960s; however, by the mid-1950s it was known that reserpine had antipsychotic properties and was useful for movement disorders such as Huntington’s disease, but with significant tolerability issues related to orthostasis.Reference Chandler 34 , Reference Lazarte, Petersen and Baars 35 Tetrabenazine (TBZ) was developed in the 1950s as an antipsychotic based on in vivo models that predicted reserpine-like effects, but with markedly reduced orthostasis risk.Reference Quinn, Shore and Brodie 36 The first TD study with TBZ was published in 1972 with the authors rationalizing the choice of TBZ due to its lower risk for hypotension than reserpine.Reference Kazamatsuri, Chien and Cole 37 Though TBZ has been available in Canada, Great Britain, and Europe for decades, it was not approved in the US until August 15, 2008, with an indication for the management of chorea in patients with Huntington’s disease.
While the mechanism that differentiated reserpine’s and TBZ’s clinical properties was not understood, by the mid-1980s it became clear that integral membrane transporters were necessary to package neurotransmitters into synaptic vesicles of presynaptic neurons (Figure 1).Reference Scherman and Weber 38 This discovery led to the characterization of multiple vesicular transporters, including those with specificity for acetylcholine,Reference Roghani, Feldman and Kohan 39 or for monoamines such as dopamine, serotonin, norepinephrine, epinephrine, and histamine.Reference Erickson, Eiden, Schafer and Weihe 40 The vesicular monoamine transporter (VMAT) was found to exist in 2 isoforms (VMAT1 and VMAT2) that vary in distribution: VMAT1 is expressed mainly in the peripheral nervous system, while VMAT2 is expressed mainly in monoaminergic cells of the CNS.Reference Erickson, Schafer, Bonner, Eiden and Weihe 41 TBZ’s improved tolerability profile was related to the fact that it was a specific and reversible VMAT2 inhibitor, while reserpine was an irreversible and nonselective antagonist of both VMAT isoforms. TBZ and reserpine also have different binding sites on VMAT2 (Figure 2).

Figure 1 Location and function of VMAT. ADP: adenosine biphosphate, ATP: adenosine triphosphate, VMAT: vesicular monoamine transporter.

Figure 2 VMAT2 structure.Reference Jankovic and Clarence-Smith 46 Adapted from: Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Review of Neurotherapeutics 2011; 11(11):1509-2, reprinted by permission of the publisher Taylor & Francis Ltd. (http://www.tandfonline.com).
Investigation of TBZ’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ, which have high affinity for VMAT2 and are the pharmacologically active agents.Reference Kilbourn, Lee, Vander Borght, Jewett and Frey 42 , Reference Kilbourn, Lee, Heeg and Jewett 43 The α-DH-TBZ isomer is metabolized via cytochrome P450 (CYP) 2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.Reference Mehanna, Hunter, Davidson, Jimenez-Shahed and Jankovic 44 , Reference Muller 45 Due to the short half-life of DH-TBZ and the existence of 2D6 polymorphisms, use of TBZ for Huntington’s disease carries recommendations for thrice daily (TID) dosing, and for CYP 2D6 genotyping to screen for poor metabolizer status when exceeding 50 mg/d.Reference Jankovic and Clarence-Smith 46 To obviate these issues, 2 different pharmacological strategies were explored to moderate TBZ’s metabolism, to permit once-daily dosing, and also to improve tolerability.
Deutetrabenazine
The use of the stable isotope deuterium to replace selected hydrogen atoms in a molecule can result in a compound with similar pharmacodynamic properties but different kinetics, as the carbon-deuterium covalent bond requires 8 times more energy to break than a carbon-hydrogen bond. 47 A deuterated form of TBZ deutetrabenazine (Deut-TBZ) was synthesized (Figure 3) with such purpose in mind. While the active metabolites of Deut-TBZ retain the VMAT2 affinity of the nondeuterated DTBZ forms, the substitution of deuterium for hydrogen at specific positions markedly slows the breakdown of metabolites, resulting in a pharmacokinetic profile with longer metabolite duration of action, greater active drug exposure (Figure 4), and less impact of 2D6 genotype on drug exposure, eliminating the need for genotyping. 47 , Reference Frank, Testa and Stamler 48 Deut-TBZ was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study in which 90 patients were randomized 1:1 to receive Deut-TBZ or placebo twice daily.Reference Frank, Testa and Stamler 48 The study involved an 8-week titration period and 4-week maintenance period followed by a 1-week washout. The maximum daily Deut-TBZ dose was 48 mg, but was reduced to 36 mg in those receiving a strong CYP 2D6 inhibitor (bupropion, fluoxetine, or paroxetine). No dose modification was needed based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for Deut-TBZ compared to 14.4% for placebo.Reference Frank, Testa and Stamler 48 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the Deut-TBZ arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of Deut-TBZ subjects and at a rate ≥2 times that of placebo was somnolence: 11.1% for Deut-TBZ vs. 4.4% for placebo.

Figure 3 Structures of tetrabenazine and deutetrabenazine.
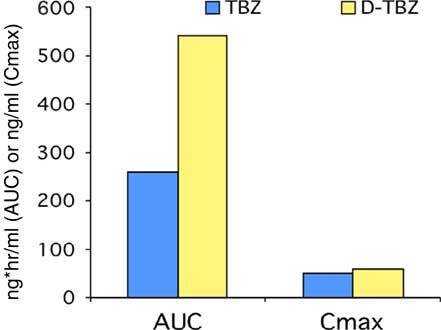
Figure 4 Kinetics of tetrabenazine and deutetrabenazine. 47
A subsequent TD study was performed in a similar design with 117 subjects randomized in 1:1 manner to Deut-TBZ or placebo.Reference Anderson, Factor and Hauser 49 The population demographics were as follows: mean age 54.9±9.8 years, 59% female, 79% Caucasian, 80.5% of whom were receiving ongoing dopamine antagonists, mean TD duration of 75.0±81.9 months. The mean baseline AIMS score for items 1–7 was 9.6±3.9, with 85.8% of subjects having AIMS≥6. Study treatment retention was high, with 6 drop-outs in the Deut-TBZ arm vs 7 in the placebo arm. There was a mean 3.0 point decrease in AIMS for Deut-TBZ compared to 1.4 for placebo (p=0.019). Among those with baseline AIMS≥6, there was a 3.4 point decrease in AIMS for Deut-TBZ compared to 1.9 for placebo (p=0.027). There were no adverse effects that occurred in ≥5% of Deut-TBZ subjects and at a rate ≥2 times the rate in placebo.
Valbenazine
Each of TBZ’s active metabolites α-and -DH-TBZ possess multiple chiral centers, yielding a total of 8 possible isomers (Figure 5), each of which has different VMAT2 activity.Reference Muller 45 , Reference Yao, Wei and Wu 50 Characterization of isomers with greatest VMAT2 affinity (Table 1) led to the development of valbenazine, a prodrug that is metabolized into the most active DH-TBZ isomers.Reference Muller 45 Importantly, valbenazine was designed to be metabolized slowly, and thereby minimize high peak plasma concentrations, decrease peak-to-trough ratios, and reduce inter-subject variability. The TMax for the active metabolites is 4–10 hours with a half-life of approximately 20 hours, allowing once-daily dosing.Reference O’Brien, Jimenez and Hauser 51 Due to the limited range of metabolites (2), it was also designed to limit off-target receptor binding that was theoretically possible with certain DH-TBZ isomers.Reference Muller 45 Data from a randomized, 6-week, double-blind, placebo-controlled, dose-titration study in subjects with TD was published in 2015 (n=100).Reference O’Brien, Jimenez and Hauser 51 The population demographics were as follows: mean age 56.2±10.3 years, 57% male, 79% Caucasian, 73% of whom were receiving ongoing antipsychotic treatment, mean TD duration of 7 years. The mean baseline AIMS score for items 1–7 was 8.0±4.0. The valbenazine starting dose was 25 mg, and this could be escalated in 25 mg increments every 2 weeks to a maximum of 75 mg. At study endpoint, 75% of subjects were on the maximum dose, and there were no study drop-outs due to adverse events in the valbenazine cohort. There was a mean 3.6 point decrease in AIMS for valbenazine compared to 1.1 for placebo (p=0.0005), with lower baseline severity and mood diagnosis (vs schizophrenia spectrum) moderating factors that improved treatment response.Reference Josiassen, Remington and Burke 52 The following adverse events occurred in ≥5% of valbenazine subjects and at a rate ≥2 times that of placebo: fatigue: 9.8% for valbenazine vs 4.1% for placebo; headache: 9.8% for valbenazine vs 4.1% for placebo; decreased appetite: 7.8% for valbenazine vs 0% for placebo.

Figure 5 Dihydrotetrabenazine.
Table 1 Dihydrotetrabenazine (DH-TBZ) isomer affinity for VMAT2 (Ki±SEM nM)Reference Yao, Wei and Wu 50

A subsequent 6-week study using similar design was presented in 2016 using dosing data derived from the prior trial.Reference Marder, Knesevich and Hauser 53 In this design 234 subjects with TD were randomized in a 1:1:1 manner to placebo, valbenazine 40 mg once daily or valbenazine 80 mg once daily. Completion rates were high (87.6%), with only 2 drop-outs due to adverse events in each of the placebo and 40 mg arms, and 3 in the 80 mg group. There were no adverse events in either valbenazine arm that exceed 5% in frequency and were ≥2 times that of placebo. Subject demographics were similar to the prior trial but with higher baseline AIMS scores: mean 10.4±3.6.Reference Marder, Knesevich and Hauser 53 The prior trial demonstrated a least squares mean 2.5 point difference in AIMS score between valbenazine and placebo, while for this trial the least squares mean change from baseline to week 6 was 3.1 points between valbenazine 80 mg (–3.2) and placebo (–0.1) (p<0.001). The effect size (Cohen’s d) was large, at 0.90.
Conclusions
There have been significant advances in the understanding of schizophrenia pathophysiology, but knowledge of tardive dyskinesia has lagged behind. As with other complex disorders, genetic load and environmental factors impose risks. Besides exposure to dopamine modulating agents, sources of oxidative stress may be another contributing factor to TD development. Recent data indicate that the final common pathway for persistent TD may relate to aberrations in synaptic plasticity, while the model of receptor upregulation/supersensitivity applies more appropriately to withdrawal dyskinesia, with its high rates of reversibility. On the other hand, TD shows very low rates of improvement after antipsychotic discontinuation, indicating a more durable problem with persistently abnormal synaptic connections.Reference Teo, Edwards and Bhatia 9
Although the developments discussed above have suggested some potential evidence-based treatment options, such as ginkgo biloba extract,Reference Zhang, Tan, Zhang, Chan, Wu and Zhou 33 clinicians have been discouraged by positive case reports for a variety of agents, many of which have limited high-level evidence for TD treatment.Reference Bhidayasiri, Fahn and Weiner 28 However, more rigorous clinical research with the VMAT2 inhibitor tetrabenazine has amassed a considerable evidence base for TD treatment.Reference Leung and Breden 54 Importantly, characterization of TBZ’s metabolic pathway and the activity of its isomers has yielded 2 divergent strategies for optimizing response to TBZ: (1) deuterated tetrabenazine (Deut-TBZ), where the substitution of deuterium for selected hydrogen atoms increases bond strength 8-fold, delays breakdown of active metabolites, and minimizes variability in drug exposure based on 2D6 genotype; and (2) valbenazine, a prodrug for the active TBZ isomers. Both of these options appear effective in phase 3 studies, with kinetic profiles that permit once-daily dosing and obviate the need for 2D6 genotyping.
Given the widespread use of antipsychotics in the management of schizophrenia, bipolar mania, bipolar depression, and adjunctively for unipolar depression, persistent TD will remain a clinical feature of modern psychiatry. The resurgent interest in TD has led to valuable insights into pathophysiology and mechanisms for TD management. The clinical development of 2 new compounds based on the older but useful agent tetrabenazine has yielded therapies that that may be soon available for treatment of this vexing problem. With the routine use of atypical antipsychotics, many clinicians have developed the perception that persistent TD was a thing of the past. For those who work with the chronically and persistently mentally ill, or who are unfortunate to have higher functioning mood patients with this disorder, TD is an issue that clearly is not gone, and should not be forgotten.
Disclosures
Dr. Meyer reports having received speaking or advising fees from Acadia, Alkermes, Forum, Merck, Otsuka-USA, Sunovion, and Teva.
Optional CME Posttest and Certificate
CME Credit Expires: November 30, 2019
CME Posttest Study Guide
NOTE: The posttest can only be submitted online. The below posttest questions have been provided solely as a study tool to prepare for your online submission. Faxed/mailed copies of the posttest cannot be processed and will be returned to the sender. If you do not have access to a computer, contact NEI customer service at 888-535-5600.
-
1. Positive data for which of the following compounds provide compelling evidence that antioxidant treatment can improve tardive dyskinesia symptoms?
-
A. Reserpine
-
B. Ginkgo biloba extract
-
C. Vitamin E
-
D. N-Acetylcysteine
-
E. 2 and 3
-
F. B, C, and D
-
-
2. Which of the following models is the best explanation for the persistence of tardive dyskinesia symptoms in most patients after withdrawal of antipsychotic medication?
-
A. Neurotoxicity
-
B. Abnormal synaptic plasticity
-
C. Postsynaptic receptor upregulation
-
D. Postsynaptic receptor supersensitivity
-
E. All of the above
-
-
3. Which of the following agents has positive data from double-blind, placebo-controlled trials for the treatment of patients with tardive dyskinesia?
-
A. Deuterated tetrabenazine
-
B. Tritiated tetrabenazine
-
C. Valbenazine
-
D. 9-Fluororeserpine
-
E. A and C
-
F. A, B, and C
-
G. A–D
-
Optional CME Online Posttest and Certificate Instructions
There is no posttest fee nor fee for CME credits.
-
1. Read the article.
-
2. Complete the posttest and activity evaluation, available only online at www.neiglobal.com/CME (under “CNS Spectrums”).
-
3. Print your certificate, if a score of 70% or more is achieved.
Questions? call 888-535-5600, or email CustomerService@neiglobal.com






