Introduction
Molecular markers became inevitable in many fields of biological research recently and their application drastically changed many ways of living organisms’ studies at molecular, cellular, organism and population levels (Freeland, Reference Freeland, Freeland, Petersen and Kirk2011). The use of molecular genetic tools is essential for (a) precise detection and correct identification of microbes which display levels of cryptic diversity not resolved by traditional methods; (b) phylogenetic reconstructions and associated taxonomic implications; (c) determination of roles played by particular genes and gene products in biological systems. In parasitology, benefits of studies at the molecular level are augmented by the fact that parasitic microorganisms developing inside the host cell usually demonstrate reduction of cellular structures becoming as cryptic as possible (Bass et al., Reference Bass, Stentiford, Littlewood and Hartikainen2015). Moreover, genotyping of parasites provide insights into the population structure of their hosts (Timi et al., Reference Timi, Luque and Poulin2010).
Microsporidia are widespread obligate intracellular parasites of animals causing infections of different groups of their specific animal hosts and in some species switching is possible between host orders (e.g. lepidopteran vs hymenopteran insects) or classes (e.g. invertebrate vs vertebrate animals) (Tokarev et al., Reference Tokarev, Simakova, Timofeev, Malysh, Sokolova and Issi2016). Genotyping of Microsporidia is highly in demand for novel species description, molecular phylogenetic and taxonomic studies, differentiation of closely related or similarly looking forms and ecological observations, as well as diagnostics of infections in medicine, veterinary and agriculture (Deng et al., Reference Deng, Li, Zhong, Gong, Cao, Song, Wang, Huang, Liu, Hu, Fu, He, Wang, Zhang, Wu and Peng2017). Among ca 1300 species described, for as many as 150 there are nucleotide data for small subunit ribosomal RNA (SSU rRNA) gene. Dozens of species have been subjected to multi-locus sequence typing and for about 20 taxa whole genome data have been acquired with differing level of completeness. Polymorphism levels of the protein-coding genes vary greatly influencing their availability for different purposes of molecular genetic studies.
Hexokinase II (HK) is among core glycolytic enzymes of Microsporidia involved in regulation of host cell metabolic processes. Since 2012 HK has become one of the most attractive enzymes in Microsporidia research. Cuomo and coauthors used the heterologous expression in yeast to demonstrate the ability of N-terminal signals of six microsporidian HKs for direct secretion of the reporter protein. In addition, it was shown that HK of Nematocida parisii is highly expressed at the early stages of parasite intracellular development, unlike other glycolytic enzymes (Cuomo et al., Reference Cuomo, Desjardins, Bakowski, Goldberg, Ma, Becnel, Didier, Fan, Heiman, Levin, Young, Zeng and Troemel2012). Later, analysis of HK of the microsporidian Paranosema (Antonospora) locustae demonstrated secretion of the full-size enzyme into infected cells and its accumulation in the host nuclei (Senderskiy et al., Reference Senderskiy, Timofeev, Seliverstova, Pavlova and Dolgikh2014; Timofeev et al., Reference Timofeev, Senderskiy, Tsarev, Tokarev and Dolgikh2017). Biotinylation of proteins of microsporidia of the genus Nematocida in the cytoplasmic and nuclear compartment of nematode Caenorhabditis elegans ells confirmed secretion and nuclear localization of parasite HK (Reinke et al., Reference Reinke, Balla, Bennett and Troemel2017). Apparently, the nuclear localization of HK secreted by microsporidia is a universal property of this important parasitic enzyme, while secretion into the host cell is thought to be an important advantage acquired by Microsporidia during their evolutionary adaptation to intracellular parasitism (Cuomo et al., Reference Cuomo, Desjardins, Bakowski, Goldberg, Ma, Becnel, Didier, Fan, Heiman, Levin, Young, Zeng and Troemel2012). In the present paper, we tested the suitability of HK as a marker for phylogenetics, discrimination of closely related species and detection of microsporidial infections in insect hosts.
Materials and Methods
Biological material
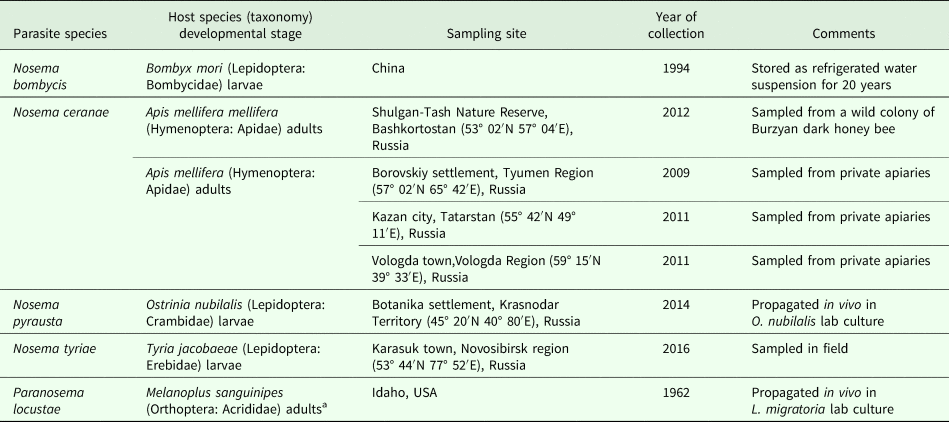
Spores of Nosema bombycis, Nosema ceranae and Nosema tyriae were available from Microsporidia collection of Prof. Irma V. Issi affiliated with All-Russian Institute of Plant Protection (St. Petersburg, Pushkin) either as water suspensions or infected insect cadavers. Paranosema locustae and Nosema pyrausta were available as live cultures of parasites propagated in vivo in their type hosts (Table 1). Partial spore purification was performed by tissue homogenization with a plastic pestle in an Eppendorf tube and several rounds of centrifugation at 4000–5000 g for 5 min followed by resuspending in distilled water. For RT-polymerase chain reaction (PCR) studies, fourth instar larvae of Locusta migratoria were fed with a mixture of wheat seedlings and P. locustae spores to provide an average dose of 5 × 106 spores per larva. Control larvae were fed with wheat seedlings without the addition of spores. Three days post infection (dpi), larvae were dissected and adipose tissue samples were frozen at −80 °C for mRNA extraction.
Table 1. Isolates of Microsporidia used in the present study
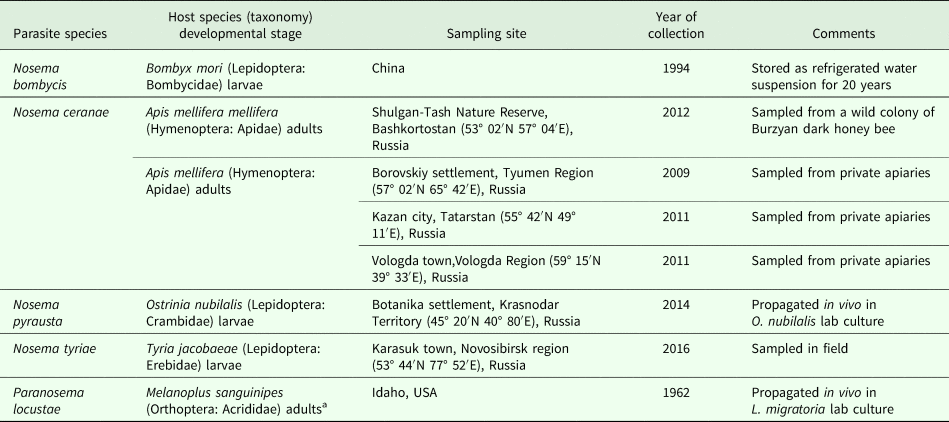
a See Henry (Reference Henry2017) for details.
Nucleic acid extraction and reverse transcription
For extraction of nucleic acids the samples of purified spores were crushed in liquid nitrogen by pestle and mortar and processed routinely using either phenol-chloroform method for genomic DNA (Sambrook et al., Reference Sambrook, Fritsch and Maniatis1989) or ‘Trizol reagent’ (Thermo Fisher Scientific, USA) and the Deoxyribonuclease I (Thermo Fisher Scientific, USA) for total RNA extraction according to the manufacturer instruction. The concentration of DNA and RNA in the samples was measured using spectrophotometry. The cDNA synthesis was performed with RevertAid Reverse Transcriptase and Oligo(dT)18 Primer (Thermo Fisher Scientific, USA) using 2 µg of total RNA as a matrix to the final volume of 20 µL.
DNA amplification, purification, cloning and sequencing
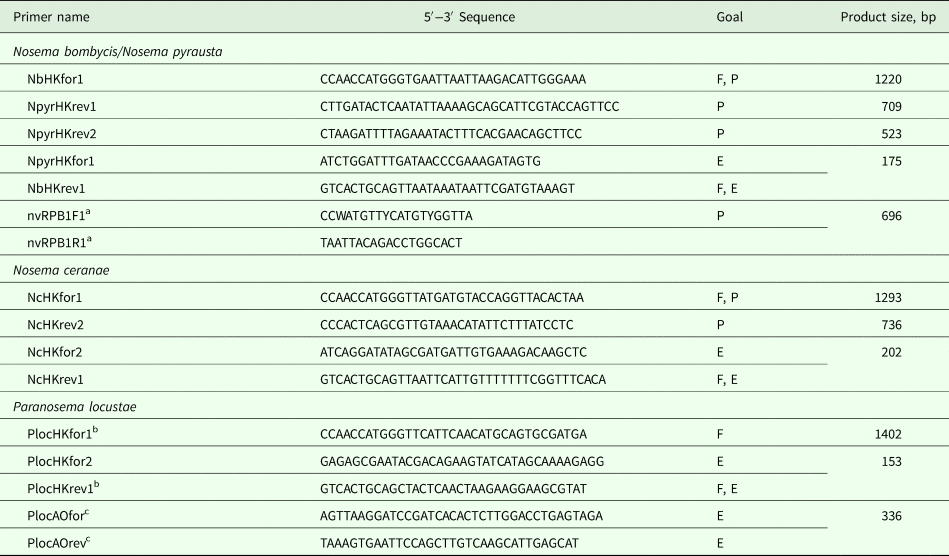
For PCR amplification of the sequences studied, 500 ng of genomic DNA or cDNA were used for the single reaction in 20 µL of the mixture. The PCR was performed according to a standard protocol using DreamTaq polymerase (Thermo Fisher Scientific) as a ready-to-use mixture (https://www.thermofisher.com/order/catalog/product/EP0701). Species-specific primers used are listed in Table 2. PCR products were separated by agarose gel electrophoresis and purified using silicium dioxide (Vogelstein and Gillespie, Reference Vogelstein and Gillespie1979). Amplicons sized over 500 bp were sequenced directly in both directions using ABI Prism Genetic Analyzer 3500. Amplicons sized below 200 bp and RNA polymerase (RPB1) amplicons for which direct sequencing showed the presence of double peaks were cloned in pAL-TA vector (Evrogen, Russia). The resulting plasmids, purified with phenol-chloroform, were sequenced using standard M13f(−20) primer.
Table 2. Primers designed for specific amplification of hexokinase (HK), largest subunit RNA polymerase II (RPB1) and alternative oxidase (AO) genes
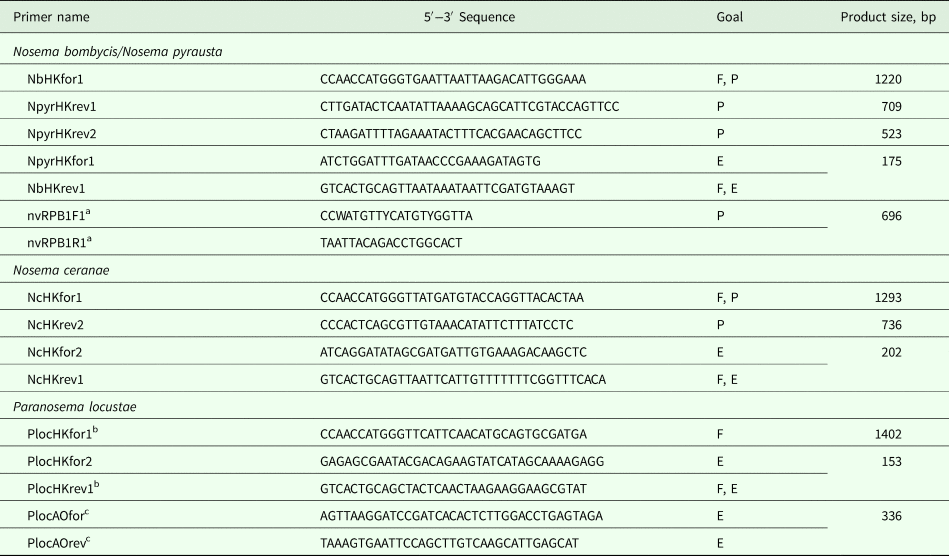
The primers designed either for full-length gene amplification (F), genetic polymorphism (P) or gene expression (E) studies. Primer pairs are matched by an identical letter (F, P, E) within each group.
a Primers from Grushevaya et al. (Reference Grushevaya, Ignatieva, Malysh, Senderskiy, Zubarev and Kononchuk2018).
b Primers from Senderskiy et al. (Reference Senderskiy, Timofeev, Seliverstova, Pavlova and Dolgikh2014).
c Primers from Dolgikh et al. (Reference Dolgikh, Senderskiy, Pavlova, Naumov and Beznoussenko2011).
Bioinformatics
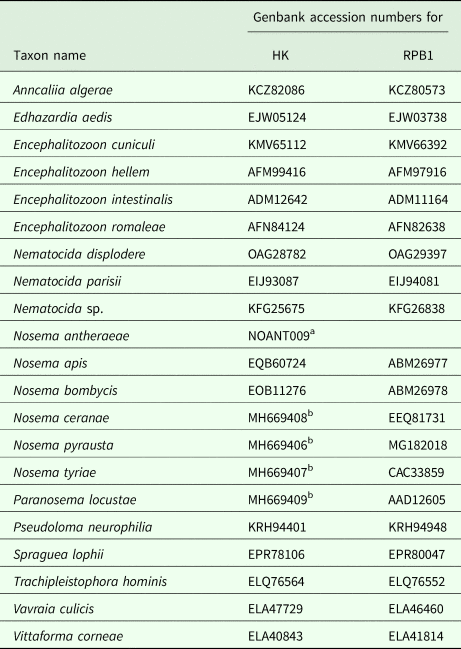
Newly obtained sequences were corrected manually in BioEdit and compared with Genbank entries using BLAST utility. Genbank (http://www.ncbi.nlm.nih.gov.nuccore/) and whole genome sequencing project database for Nosema antheraeae (http://silkpathdb.swu.edu.cn/datasets) were used to extract nucleotide sequence data for HK and largest subunit RNA polymerase II (RPB1) genes (Table 3). The nucleotide sequences were aligned in BioEdit (Hall, Reference Hall1999), nucleotide and amino acid sequence similarity and pairwise genetic distances were calculated using built-in utilities. Primers were designed manually basing upon Genbank-accessible as well as newly obtained sequences aligned in BioEdit. For phylogenetic reconstructions, amino acid sequence alignments of HK (603 aa long) and RPB1 (1674 aa long) of 21 taxa representing five major clades of Microsporidia Tree of Life were prepared (including indels). Maximum Likelihood approach using JTT model (Jones et al., Reference Jones, Taylor and Thornton1992) with gamma distributed rates among sites (+G) and a proportion of invariable sites (+I) was applied with 100 bootstrap replicates in raxmlGUI v.1.5 (Silvestro and Michalak, Reference Silvestro and Michalak2012).
Table 3. Accession numbers for amino acid sequences of hexokinase-II (HK) largest subunit RNA polymerase II (RPB1) used in phylogenetic reconstructions
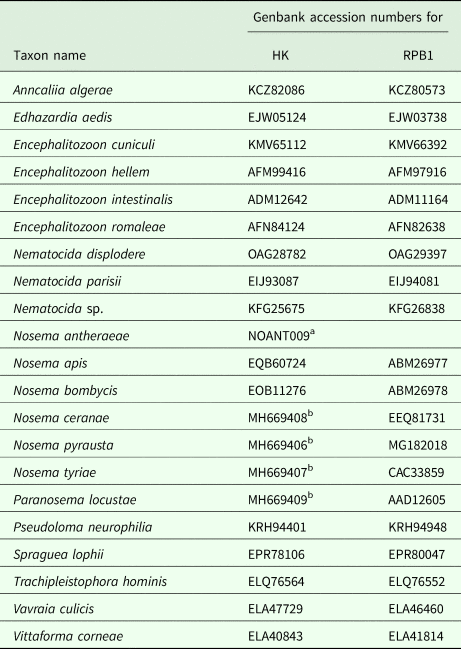
a Contig number from genome sequencing project at http://silkpathdb.swu.edu.cn/datasets.
b Genbank accession numbers for nucleotide sequences were available only for these entries at the time of the manuscript submission.
Results
Intra- and interspecific polymorphism of HK gene in Microsporidia
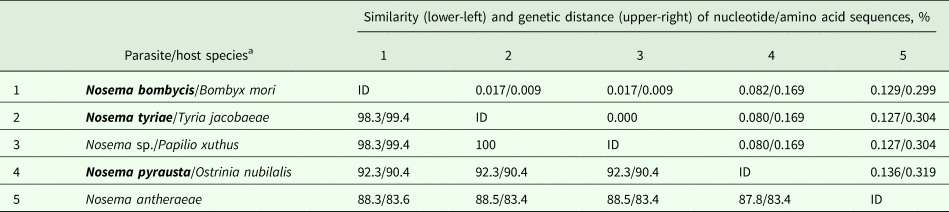
The use of primers flanking full-length gene of N. bombycis (Table 1) allowed amplification of this locus in N. bombycis, N. tyriae and N. pyrausta, that is expectable given that these three taxa are closely related and their SSU rRNA gene sequences are 99.7–99.8% similar (Tokarev et al., Reference Tokarev, Malysh, Kononchuk, Seliverstova, Frolov and Issi2015). The nucleotide sequence of N. bombycis was 100% identical to that of N. bombycis from B. mori (sampled in China) available at Genbank (# EOB11276). Surprisingly, protein BLAST search showed 100% identity of N. tyriae-derived HK sequence to an entry XP_013169030 (coded by a reference sequence XM_013313576) annotated as Papilio xuthus (Lepidoptera: Papilionidae) HK II. Closer investigation showed the presence of only four nucleotides (per ~1300 bp) and one amino acid substitutions (per ~430 aa) as compared with N. bombycis. It is, therefore, obvious that the sequence was incorrectly annotated as an insect genome locus while it is, in fact, a microsporidian gene. Its similarity to N. bombycis was 98.3% (nucleotide) and 99.4% (amino acid). As for N. pyrausta, it was similar to N. bombycis by 92.3% (nucleotide) and 90.4% (amino acid), respectively. Another member of true Nosema for which full genome (and therefore HK sequence) was available, N. antheraeae, showed slightly lower similarity to these taxa: ~83% for nucleotide and ~88% for amino acid sequences (Table 2). Thereafter, even short portions of HK gene sequence alignments contained a reliable number of SNPs to differentiate between these closely related species (Fig. 1A).

Fig. 1. Alignments of nucleotide sequences of hexokinase (HK) and largest subunit RNA polymerase II (RPB1) genes. Dots show the identical positions. (A) A portion of alignment of HK of Nosema bombycis and allied species showing SNPs in closely related taxa (see Table 3). P.x. – Papilio xuthus. (B) An alignment of HK of Nosema ceranae showing SNPs as compared between isolate BLDR01 used for genome sequencing project (Cornman et al., Reference Cornman, Chen, Schatz, Street, Zhao, Desany, Egholm, Hutchison, Pettis, Lipkin and Evans2009) and isolates sequenced in the present study from a wild-hive honey bee (Shulgan-Tash) and from private apiaries in Russia (Tyumen, Kazan, Vologda). Paragraph signs and ‘~’ mark the ‘hidden’ columns devoid of polymorphic sites. (C) A portion of directly sequenced RPB1 sequence of Nosema pyrausta (above) aligned against the respective chromatogram and molecular clones (1, 3–5). Red contour arrows indicate ambiguous sites in the upper sequence, double peaks in the chromatogram and respective SNPs in molecular clones of this amplicon.
In N. ceranae, intraspecific diversity of nucleotide and amino acid sequences was found at the level of 99.7% (3 SNPs per 1242 bp) and 99.8% (1 amino acid substitution per 413 aa), respectively, between BLR01 and Russian isolate from Bashkortostan (Shulgan-Tash). The three SNPs were located at positions 105 (T/A), 296 (A/G) and 603 (C/T) so we designed additional reverse primer NcHKfor2 to flank about 700 bp (Table 2) region containing these SNPs. Sequencing the respective region of HK gene in N. ceranae from Vologda, Kazan and Tyumen showed its higher similarity to BLR01 isolate as they possessed the same SNPs as in BLR01 at positions mentioned above (being therefore less similar to the Bashkortostan isolate) and were different only by 1 SNP at position 603 (G/T) per 700 bp (Fig. 1B) providing 99.9% nucleotide sequence similarity and 100% amino acid sequence similarity (Table 4).
Table 4. Pairwise similarity and genetic distance of hexokinase sequences from microsporidia of the genus Nosema obtained from Genbank and from this study (in bold).
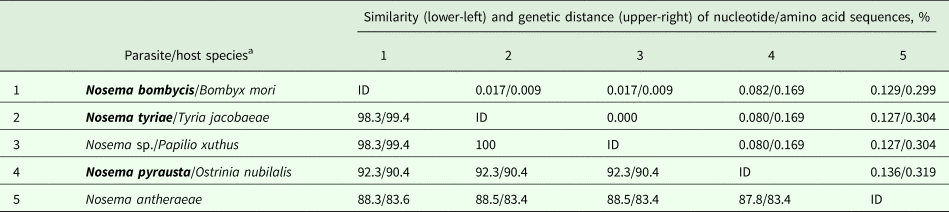
ID, identical entry.
a See Table 2 for Genbank accession numbers.
The alignment length was 1157 bp and 385 aa.
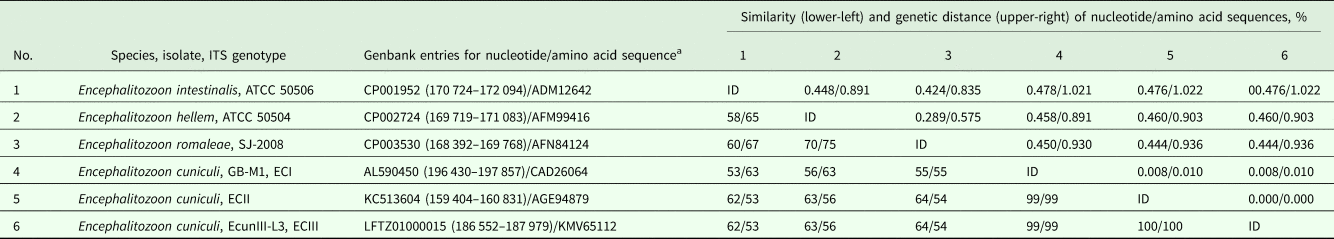
To test whether HK is useful to estimate inter- and intraspecific genetic variation in other groups of microsporidia, a set of Genbank-accessible sequences of Encephalitozoon species and genotypes was analysed. As a result, the between-species similarity of both nucleotide and amino acid sequences reached ~50–75%, which is remarkably lower as compared with the closely related group of true Nosema (see above) and reflects higher dissimilarity of SSU rRNA gene sequences of congeneric species within Encephalitozoon group. To compare, the between-species similarity of nucleotide (~4500 bp) and amino acid (~1500 bp) RPB1 gene sequences was ~80–86 and 90–94%, respectively. The Encephalitozoon cuniculi isolate belonging to the ECI genotype showed 99% sequence similarity to genotypes ECII & ECIII, which were identical to each other (Table 5). Similarly, RPB1 nucleotide sequences of ECII (coding for amino acid sequence under accession #AGE95912) and ECIII (# KMV66392) were 100% identical to each other, being 99.5% similar to ECI (#CAD26175) (data not shown).
Table 5. Similarity of nucleotide and amino acid sequences of hexokinase from microsporidia of the genus Encephalitozoon obtained from Genbank.
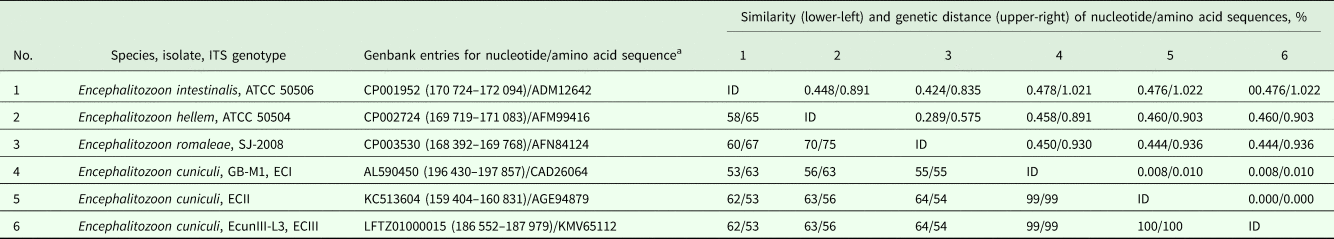
ID, identical entry.
a Nucleotide positions within a chromosome/whole genome shotgun sequence are given in parenthesis.
The alignment length was 1308 bp and 436 aa.
HK gene-based phylogenetics of Microsporidia
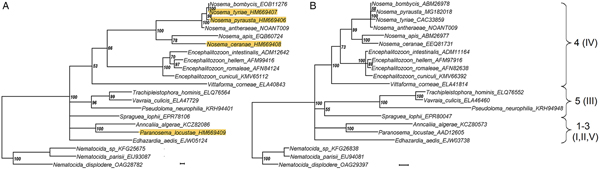
The phylogenetic reconstructions were compared based upon either HK or RPB1 gene sequences of 21 species of Microsporidia for which HK sequences were available from genome projects or sequenced within the present study. The bootstrap support was higher in case of a RPB1-based tree, as expected given the higher level of conservatism of this locus (see above), yet the tree topology was essentially the same (Fig. 2), is largely consistent with previously published studies. In particular, two major groups known as Clade 4 (previously clade IV) and Clade 5 (previously Clade III) were found in the crown of the tree in sister position to each other. Within Clade 4, Vittaforma corneae was in a basal position to the rest of the clade including two branches of Nosema and Encephalitozoon lineages in sister position to each other. Within Clade 5, Vavraia culicis and Trachipleistophora hominis were preceded by Pseudoloma neutrophila and Spraguea lophii was in a basal position to the whole group (though with bootstrap support below 50%). This topology fairly reproduced both SSU rRNA gene (Vossbrinck et al., Reference Vossbrinck, Debrunner-Vossbrinck, Weiss, Weiss and Becnel2014) and whole genome-based reconstructions (Ndikumana et al., Reference Ndikumana, Pelin, Williot, Sanders, Kent and Corradi2017). The base of the tree was not completely resolved to result in a polytomy of the node giving rise to Clades 4 and 5, as well as to members of other clades, namely Edhazardia aedis (Clade 1), Paranosema locustae (Clade 2) and Anncaliia algerae (Clade 3) and this was similar to the study of Ndikumana et al. (Reference Ndikumana, Pelin, Williot, Sanders, Kent and Corradi2017) where position of A. algerae and E. aedis was poorly resolved in a genome-based tree.
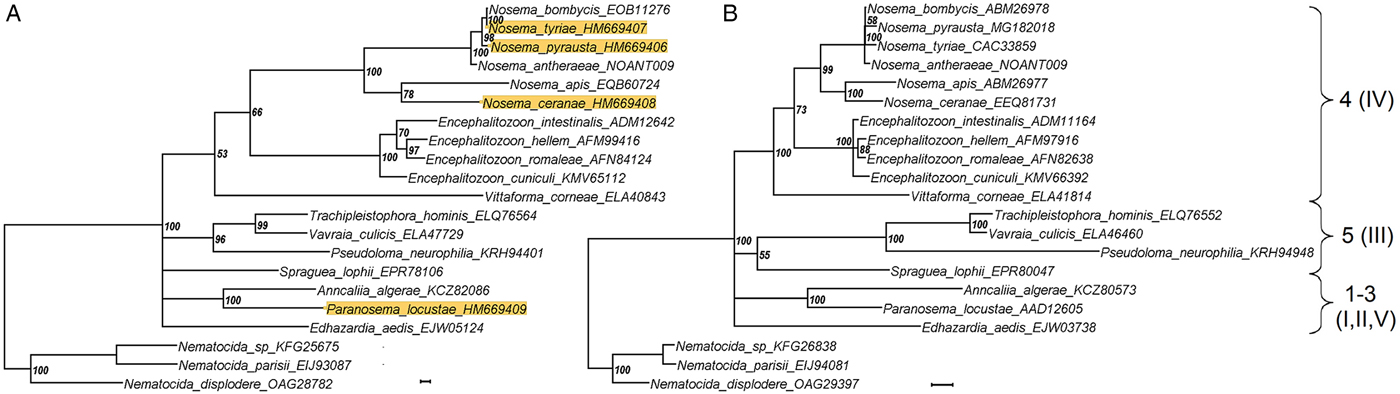
Fig. 2. A phylogenetic reconstruction based upon 21 amino acid sequences of hexokinase (A) and largest subunit RNA polymerase II gene obtained from public databases and from the present study (background highlighted). The taxon names are annotated with accession numbers as in Table 3. Numbers of major clades as classified by Vossbrinck and Debrunner-Vossbrinck (Reference Vossbrinck and Debrunner-Vossbrinck2005), I–V, and Vossbrinck et al. (Reference Vossbrinck, Debrunner-Vossbrinck, Weiss, Weiss and Becnel2014), 1–5, are shown to the right. Scale bar = 0.01 expected changes per site.
Detection of Microsporidia in tissues of infected hosts
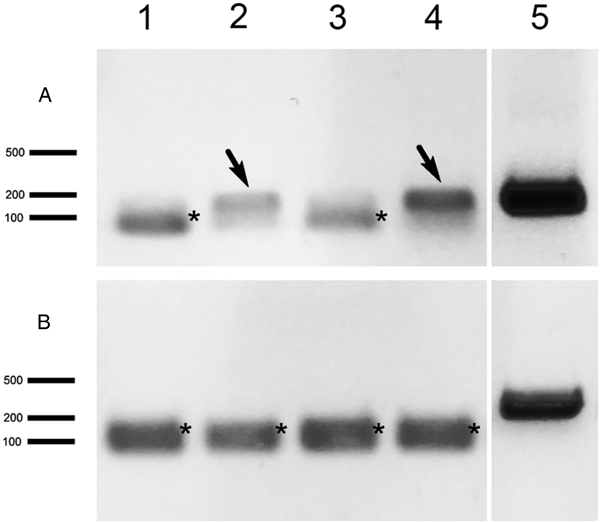
Primers specific for P. locustae, N. pyrausta and N. ceranae targeting a short HK gene region, about 150–200 bp long, were designed (Table 1) to increase the sensitivity of PCR. A series of genomic DNA sample dilutions were subjected to PCR and the positive reaction was observed in samples with a dilution factor of 100 or even higher (Fig. 3), indicating a high sensitivity of these markers. For gene expression, we collected larval adipose tissue from L. migratoria 3 dpi for RT-PCR analysis of HK, expected to be expressed early in the parasite's proliferation cycle, and alternative oxidase (AO), which is expressed only in the spores (Dolgikh et al., Reference Dolgikh, Senderskiy, Pavlova, Naumov and Beznoussenko2011) to serve as a control. HK transcripts were detected in 2 out of 4 locusts whereas no AO transcription activity was found in these samples (Fig. 4). To verify the PCR results, we performed molecular cloning of the amplicons and plasmid sequencing which showed 100% sequence similarity of the obtained products to the target locus.

Fig. 3. Electrophoretic profiles of short fragments of N. ceranae (A) and N. pyrausta (B) hexokinase amplified using genomic DNA samples diluted by 100 (1), 1000 (2) and 10 000 (3). Size of three reference bands of the molecular weight marker (M) is indicated (bp). Primer dimers are marked with asterisks.
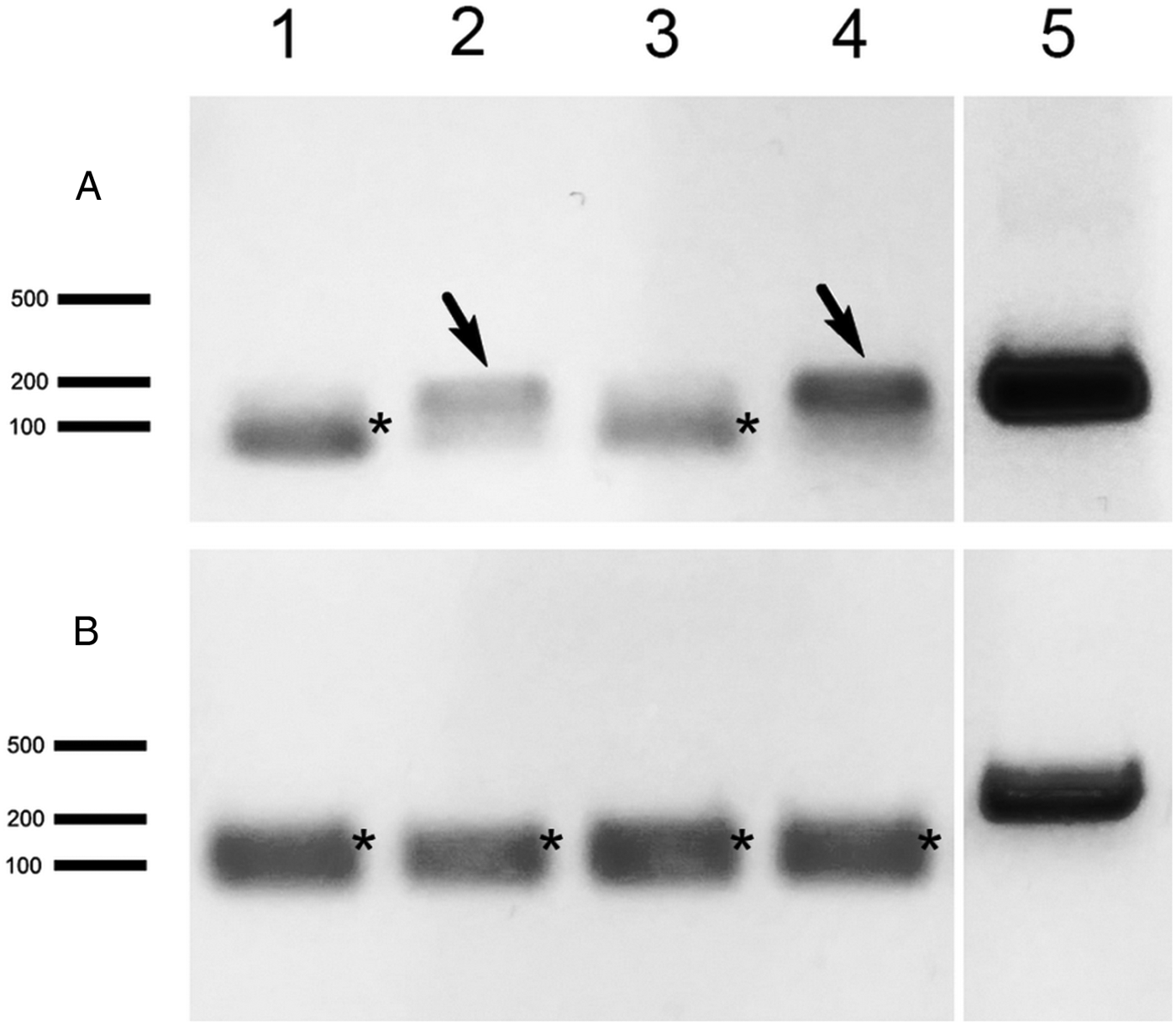
Fig. 4. RT-PCR analysis of P. locustae hexokinase (A) and alternative oxidase (B) in L. migratoria tissue samples 3 days after infection. Locust individuals are marked as 1–4, positive samples are indicated by arrows. 5 – Positive control (P. locustae genomic DNA used as a matrix). Other indications as in Fig. 3.
Discussion
Multilocus sequence typing is essential in many fields of biology as precise identification of organisms is important at species, subspecies and population level (Deng et al., Reference Deng, Li, Zhong, Gong, Cao, Song, Wang, Huang, Liu, Hu, Fu, He, Wang, Zhang, Wu and Peng2017). In this respect, acquisition of sequence information for as many genomic loci as possible is essential. However, due to the weird abundance of microorganisms taxa and genetic information, whole genome sequencing of all species of interest is not always possible and is often substituted with a core set of genes to be sequenced for particular purposes. We, therefore, found it appropriate to test HK sequence information for bioinformatics studies and compare it with other molecular genetic loci such as SSU rRNA and RPB1, which are the most widely exploited genes in microsporidia studies.
HK showed clear differentiation between N.bombycis, N. pyrausta and N. tyriae that would be problematic if SSU rRNA was compared in these species due to the very low level of divergence of the latter locus (Tokarev et al., Reference Tokarev, Malysh, Kononchuk, Seliverstova, Frolov and Issi2015). Nosema antheraeae is more diverged in its SSU rRNA gene sequence from N. bombycis than N. tyriae and N. pyrausta and this is fairly reflected by differences in their HK gene sequences. As for N. ceranae, intraspecific differences could be demonstrated between the isolates from the domesticated A. mellifera and wild A. mellifera mellifera. The latter lives under protection in forest mountain zone of South Ural and is referred to as a purebred population (Nikolenko et al., Reference Nikolenko, Ilyasov, Poskryakov and Petukhov2015). The Shulgan-Tash isolate of N. ceranae is, therefore, of great interest as it shows clear genetic differentiation from globally distributed N. ceranae in domesticated and interbred European honey bee. Finally, HK sequences in the species of the genus Encephalitozoon were more polymorphic but showed the same level of divergence in intraspecific isolates of E. cuniculi when compared with RPB1. Noteworthy, direct sequencing resulted in unambiguous reading of HK gene which could not be achieved for other protein-coding genes which are polymorphic (Ironside, Reference Ironside2013). For example, multiple double peaks corresponding to SNPs within a single isolate were observed within the chromatogram of RPB1 gene of N. pyrausta and molecular cloning was necessary to determine the exact combination of SNPs within two genetic variants present (Fig. 1C).
The PCR analysis with primers specific to parasite genes is an effective tool for microsporidia detection in tissues of infected hosts. This method is more sensitive than light microscopy and provides an opportunity to detect parasite in the early stages of infection (Kaushik et al., Reference Kaushik, Saha, Das, Ramachandran, Goel, Cohen, Lajtha, Lambris, Paoletti and Rezaei2017). However, in the case of artificial infection, performed by feeding or injecting microsporidia spores to the hosts, the PCR signal can be resulted not from developing parasites but from introduced spores remaining in the host organism at the early stages of infection. For this reason, HK gene is a perfect target as it is intensively expressed in active stages of intracellular development of parasite, but not in the spores, as shown for P. locustae (Senderskiy et al., Reference Senderskiy, Timofeev, Seliverstova, Pavlova and Dolgikh2014). Thereby analysis of transcription activity of this enzyme allowed us to identify infected insects in really early and non-symptomatic stage of infection.
To conclude, the HK gene of Microsporidia examined in the present study showed a higher level of nucleotide/amino acid sequence polymorphism as compared with another protein-coding gene, RPB1, while genetic polymorphism within individual isolates was not observed. And though in an HK-based phylogenetic reconstruction the bootstrap support was lower as compared with RPB1, the overall topology of both trees was congruent, indicating HK as a good candidate for multigene phylogenetic studies of Microsporidia. Finally, its expression at the beginning of the parasite development is helpful to detect early infections. Different tasks of molecular genetic investigations of microsporidia can be thus achieved using the HK gene, and degenerate primer sets with a broad range of target species can be designed for further studies exploiting amplification of this locus, in contrast to the present work which used genus-specific primers.
Acknowledgements
Authors are thankful to Marina V. Bakalova (Shulgan-Tash State Nature Biosphere Reserve, Bashkortostan, Russia) for the gift of Burzyan honey bee sample.
Financial support
This work was supported by Russian Scientific Foundation [grant number 16-14-00005].
Conflict of interests
None.
Ethical standards
Not applicable.