Introduction
Crop collections established and maintained by genebanks provide for the present and future utilization of crop genetic resources. In the early stages of collection development, the focus was mainly on acquisition per se, and less on optimizing collection composition. Many genebank collections were started from working collections that had been used to support specific purposes, including breeding, crop improvement and taxonomic studies. In many cases, genebanks expanded their collections thereafter by including obsolete varieties, research lines or samples obtained from collecting missions to natural distribution areas of crops and their wild relatives.
Prior to the 1960s, early founded genebanks such as the N.I. Vavilov Institute at St Petersburg and the genebank of the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) at Gatersleben, Germany followed a systematic approach in building up their collections in order to gather the broadest possible genetic diversity of a given crop and sometimes of its entire gene pool (Hammer, Reference Hammer1993; Loskutov, Reference Loskutov1999). Particularly since the 1970s, when many national genebanks were established, crop collections were often developed with any germplasm that curators had access to, without a clearly defined conservation goal or mandate. Furthermore, it became common practice to request and/or exchange material among colleague genebanks. This has produced collections of considerable size, but often with unbalanced composition and/or high levels of duplication (Plucknett et al., Reference Plucknett, Smith, Williams and Anishetty1987; FAO, 1998), usually within a national context lacking coordination at the regional or global level.
Today, genebanks are increasingly concerned with acquiring knowledge about their collections and with the improvement of the composition thereof, rather than with the continuing expansion of collections (Engels and Visser, Reference Engels and Visser2003). So far, the literature on the improvement of collection composition has focused on the identification of duplicates within crop collections (van Hintum et al., Reference van Hintum, Sackville Hamilton, Engels, van Treuren, Engels, Rao, Brown and Jackson2002; van Treuren and van Hintum, Reference van Treuren and van Hintum2003), to considerations on how to rationalize collections (Sackville Hamilton et al., Reference Sackville Hamilton, Engels, van Hintum, Engels and Visser2003) and to considerations of splitting or lumping accessions (Sackville Hamilton et al., Reference Sackville Hamilton, Engels, van Hintum, Koo and Smale2002).
However, considering this history and realizing that existing collections can often be significantly improved, certainly from a regional and global perspective, conceptual strategic views to optimize crop collections are badly needed. Consequently, the present paper aims at developing such a view and identifying future research directions to develop tools that can facilitate the optimization of collection composition.
Rationale of crop collections
The basis of any discussion about approaches to optimize collection composition is the genebanks' broader mission. It is generally agreed that the main goal of genebanks, either individually or collectively, is to constitute collections that represent as wide as possible genetic diversity of a crop gene pool with a minimum of redundancy (Frankel and Brown, Reference Frankel, Brown, Holden and Williams1984), albeit sometimes within a given geographical or political area. As this goal is rather general, it immediately raises several issues and questions concerning the actual composition and diversity of existing collections. In this context, a collection is being defined as the total set of accessions belonging to a given crop or gene pool being maintained for the long term (i.e. frequently defined as the base collection) at a given genebank.
Collection composition
The first question concerning collection composition is what is actually considered to be ‘the crop gene pool’. In general, this is constituted by the domesticated and cultivated material, including landraces and bred material, together with their wild relatives. In the context of this paper, the question is how to determine the boundaries of a crop gene pool. Following the gene pool concept of Harlan and de Wet (Reference Harlan and de Wet1971), pre- and post-zygotic compatibility between taxa may be used as a guideline to determine these boundaries. This seminal concept was strongly based on the common practice of phylogenetic researchers and plant breeders, and it provided a very useful framework. However, technological advances have greatly enhanced the crossability of different species, which has resulted in an increased interest by plant breeders in species that are evolutionary more distant from the cultivated crop but do possess desired phenotypic characters or specific genes. These developments blur the gene pool concept and ask for new definitions. Nevertheless, in the context of this paper, we conform to the traditional gene pool concept as most genebank collections are still based on this concept.
The second question related to collection composition is what is actually meant by ‘as wide as possible genetic diversity’. Clearly, expansion of a collection with accessions possessing novel variation that belong to the crop gene pool increases the genetic diversity within that collection. This strategy has been followed, certainly during the initial stages of collection development, but due to various constraints, including storage, regeneration and handling capacities, the number of accessions is ultimately limited for any given genebank.
We briefly outline two options for dealing with this limitation. The first option is to stop further expansion and consider collection composition as final. However, since the total genetic diversity of a crop gene pool is generally not quantified, it remains unclear how representative the collection is with respect to the total genetic diversity. Therefore, this option may conflict with the general aim of genebanks to represent as widely as possible the genetic diversity of a crop gene pool. The second option is to treat the composition of a crop collection of certain limited size as flexible and dynamic. Given the unknown extent to which a collection represents the total genetic diversity of a crop gene pool, curators always need to consider acquisition of newly available materials, such as from unexplored parts of the distribution range of a crop and/or its wild relatives, if these would broaden the genetic diversity within the collection. Assuming that the limits of the collection size of a given crop gene pool have been reached, adding new material to the collection would imply the removal of existing accessions. Through this second option, a stepwise increase in the genetic diversity of the collection will gradually improve the representation of the total genetic diversity of a crop gene pool.
The third question regarding collection composition is what is actually meant by ‘minimum redundancy’. Redundancies may include genetically identical (duplicates) as well as genetically very similar accessions. For many genebank accessions, the presence of intra-accession variation highly complicates the identification of ‘redundant’ germplasm. This is particularly true for highly outcrossing organisms that usually show a large degree of overlap in genetic variation among accessions (van Treuren et al., Reference van Treuren, Bas, Goossens, Jansen and van Soest2005; van Hintum et al., Reference van Hintum, van Treuren, van de Wiel, Visser and Vosman2007). In case accessions are not identical, the question is how large the differences should be in order to consider them non-redundant, requiring analytical procedures that are by no means straightforward (van Treuren and van Hintum, Reference van Treuren and van Hintum2003). An option to avoid problems with the definition of redundancy is to follow the core collection approach (van Hintum et al., Reference van Hintum, Brown, Spillane and Hodgkin2000) by selecting a subset of accessions that collectively contribute most to the genetic diversity within that collection ( Jansen and van Hintum, Reference Jansen and van Hintum2007). The accessions that do not form part of the selected subset may then be considered as redundant and removed from the collection for the purpose of optimization.
Collection diversity
Concerning collection diversity, the question is what is actually meant by ‘the total genetic diversity’ of a crop gene pool. In the widest sense, this is the total variation occurring in the entire genome among all individuals constituting the crop gene pool. Clearly, this genetic diversity is unknown and capturing the entire diversity for most taxa would be an elusive and absurd goal. The question then is on which part of the genetic diversity we should focus. For most organisms, the largest part of the genome consists of genetic diversity considered to be selectively neutral. Much of this neutral diversity is formed by variation in DNA that seems to have no particular biological function, and therefore may not be relevant from the perspective of collection representation. However, neutral variation may also occur in coding regions of the DNA that may become relevant in the future, because of changing consumer demands or environmental conditions (Endler, Reference Endler1986). In addition, variation may exist in coding regions of the genome that are currently under selective pressure, either by natural or artificial causes. The user community of genetic resources is mainly interested in the genetic diversity in a usually limited number of phenotypic characters that currently are of adaptive significance (short-term objective, specific genetic diversity), whereas conservationists try to also maintain genetic diversity that may have relevance for the future (long-term objective, broad genetic diversity). Therefore, the target genetic diversity in composing a crop collection may vary, depending on disciplinary bias. If the aim is to satisfy both perspectives, as is true for many genebanks, variation in (subsets of the) coding regions of the genome, either under current selective pressure or not, may be the target diversity to focus on in composing crop collections.
Optimizing crop collections
The strategy that we are proposing for optimizing the composition of germplasm collections consists of three steps: (1) defining the population structure of the crop gene pool in terms of an hierarchy, describing subsets that we call ‘end-groups’; (2) distributing the total number of accessions that a collection ideally may contain (hereafter referred to as ‘the capacity of the collection’) among the end-groups; and (3) optimizing the diversity within the end-groups.
Structuring the crop gene pool
The proposed optimization strategy largely builds on the concepts of core collection (van Hintum et al., Reference van Hintum, Brown, Spillane and Hodgkin2000) and core selection (van Hintum, Reference van Hintum1999). These concepts are based on structuring collections and aim at the identification of subsets of accessions that collectively maximize variation while minimizing the number of accessions. They are basically user-oriented as the user can directly influence the final result through changing weighting factors that reflect the (subjective) importance of specific parts or end-groups within the collection. However, instead of focusing on structuring an existing collection, our proposed optimization strategy aims at structuring the entire crop gene pool.
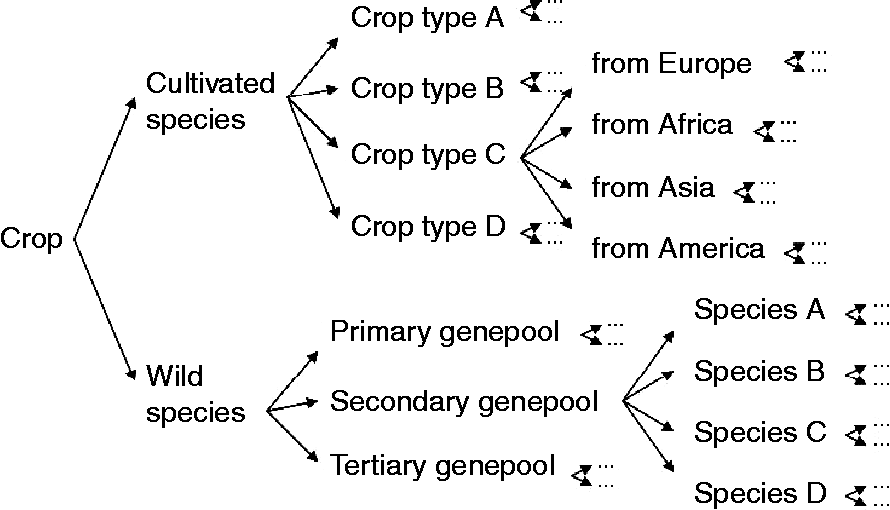
For every crop, the total gene pool can be subdivided into smaller subunits (Fig. 1), with a relatively large proportion of genetic diversity being distributed between units when compared with within units. The first division may distinguish between cultivated material and the crop-related wild gene pool. Often, the cultivated material can be further subdivided into distinct crop types within a botanical species. For example, butterhead, crisp, cutting, cos, stalk, oilseed and Latin are recognized crop types in lettuce, while each crop type may comprise series of varieties, landraces and research/breeding material, which in turn can be further divided into subgroups with genetically distinguishable material. The wild relatives can usually be grouped into primary, secondary and tertiary gene pools according to the aforementioned gene pool concept of Harlan and de Wet (Reference Harlan and de Wet1971). Each species may be represented by a series of sampled populations representing the natural distribution of the species. The population structure depicted in Fig. 1 merely presents an example of how a gene pool could be structured, but, obviously, this may vary depending on the curator or the crop. For example, in the original gene pool concept of Harlan and de Wet, the cultivated material is not treated separately from the wild germplasm as the cultivated material is by definition included in the primary gene pool. However, it should always be possible to draw a tree representing the structure of the genetic diversity within the crop, given sufficient pre-existing knowledge. The large body of literature on taxonomic classifications, domestication and breeding history that is available for many crops facilitates this structuring.
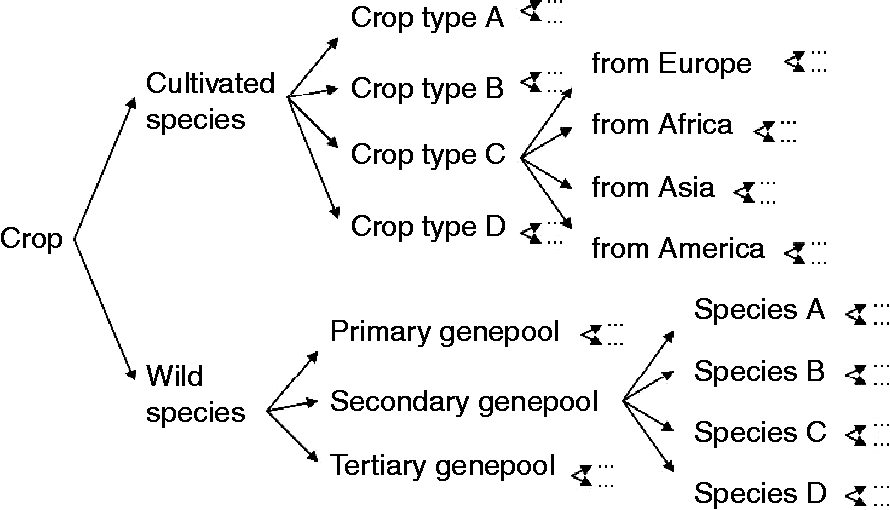
Fig. 1 Example of the first subdivisions within the population structure (diversity tree) of a crop gene pool.
End-groups constitute the lowest hierarchical levels in the gene pool structure presented in Fig. 1. For example, an end-group may consist of ‘north-western European populations of the wild species Lactuca serriola (prickly lettuce)’ or ‘yellow-leaved butterhead lettuce from The Netherlands’. The number of end-groups will vary among gene pools, is to some extent arbitrary and will depend on the degree of structure in the gene pool, i.e. the number of species, the number of crop types and the global distribution of the crop and its wild relatives. Information from plant breeders may be used to define the end-groups for cultivated material, and the botanical literature may be surveyed to define the end-groups for the crop-related wild gene pool. In practice, groups denoted as ‘unknown’ can be included at different levels of the hierarchy to cover accessions with insufficient data or species for which their relationship to other species is unclear.
Distributing capacity over end-groups
The different groups within the hierarchy (Fig. 1) will rarely be of equal importance. For example, past breeding efforts may have varied considerably among different crop types, resulting in different levels of variation. Wild species of the tertiary gene pool will generally be less important to potential users, such as traditional plant breeders, than would be species of the primary gene pool. Therefore, the question is how to ideally distribute the capacity of a collection over end-groups. This issue can be addressed by assigning relative weights to the subgroups in each group within the diversity tree, based on the estimated importance of the subgroup. For example, if the first division is in cultivated vs. wild material, it may be decided that the relative importance to the collection of the cultivated material is four times higher than that of the wild material, resulting in relative weights of 4 and 1, respectively. Similar decisions need to be made at each branching point. This should be done in consultation with experts and stakeholders to ensure an optimal balance between the different groups.
Several factors may be considered in assigning relative weights to groups, such as considerations on genetic relationships, importance to anticipated users and practical aspects, such as complexity of maintenance of the species. Which aspects to consider and how to weight them will depend on the curator, the purpose of the germplasm collection and available knowledge about the groups. For example, the relative weight of individual species might logically decline with increasing evolutionary distance from the cultivated crop (Lebeda et al., Reference Lebeda, Doležalová and Astley2004a) because of decreasing crossability and the higher probability that favourable new alleles can easily be found at the species level instead of only in a few specific populations of a given species. The advantage of small numbers of populations included from evolutionarily distant species makes it less important to define the exact boundaries of the crop gene pool. The populations of a species to include in a collection can be a random selection. However, if available, knowledge about habitat variation and information about the natural distribution area of the species should be used to maximize the diversity captured (Lebeda et al., Reference Lebeda, Doležalová, Feráková and Astley2004b).
When relative weights have been assigned to each of the groups at each branching point within the diversity tree, the desired capacity for a collection will determine how many accessions should ideally be included in each end-group. If the capacity of the collection in the aforementioned example is 2000 accessions, the collection should ideally consist of 1600 cultivated and 400 wild accessions. This process of distributing the capacity of groups according to the weight of the subgroups continues until a group is no longer subdivided (so-called end-groups) or until the remaining capacity has decreased to zero.
The desired capacity of the entire collection will obviously depend on the objectives set for that collection, resource availability and the available options to reduce or expand the current collection size. It is realized by the authors that several aspects related to the decision-making on the capacity of the entire collection as well as the relative weights assigned to the various end-groups are to some extent subjective, and that more research is needed to further increase the representativeness of the total genetic diversity in a crop gene pool in as few as possible accessions across end-groups to ensure an efficient and rational long-term conservation of such genetic diversity.
Optimizing diversity within end-groups
Comparison of the distribution of capacity among the end-groups and the actual number of accessions within the end-groups may result in three possible scenarios. First, the actual number of accessions available for an end-group may be lower than the target value based on the capacity distribution. Such end-groups are under-represented and can be considered ‘collection gaps’ when even a single accession is unavailable. In this case, optimization should focus on targeted acquisition of material for that specific end-group. Second, the actual number of accessions within an end-group may resemble the target value based on the capacity distribution. In such cases, no optimization step is needed. However, it should be noted that groups that are adequately represented in terms of accession numbers may still display a qualitatively poor within-group composition. This issue is not further addressed here, but is of considerable importance to genebanks when novel germplasm becomes available and replacement of existing accessions has to be considered. Third, the actual number of accessions within an end-group may exceed the target value based on the capacity distribution. The germplasm in such end-groups can be considered to be over-represented. Then, optimization would involve the selection and maintenance of those accessions that collectively contain the highest level of genetic diversity in that end-group, and the excess can be considered as redundant.
Apart from the above-mentioned optimization steps, removal of obvious duplicates and the addition of new and genetically distinct material should always be considered. In the case of novel material belonging to an end-group that has already reached its size limit, the existing accession that contributes least to the genetic diversity of the group, e.g. in terms of phenotypic or molecular diversity, can be replaced by the candidate accession. Replaced accessions may be given a reduced priority, such as the ‘archive status’ introduced by the Centre for Genetic Resources, The Netherlands (CGN). This status is used for material that is still stored but is no longer an integral part of the active nor of the base collection (Engels and Visser, Reference Engels and Visser2003). Compared with discarding samples, the advantage of the archive status is that the material is still present and its status can be reconsidered, if necessary, as long as its viability lasts. When additional data about the candidates for acquisition in the collection are not yet available, decisions might be postponed or the candidates could be added to the collection on a provisional basis until such data become available.
Illustration of the proposed strategy
To illustrate our strategy, an analysis was performed using potato. First, the potato gene pool, including both cultivated and wild germplasm, was structured for the purpose of the core selector (van Hintum, Reference van Hintum1999), resulting in 350 end-groups, such as ‘potato → wild tuber-bearing species → endosperm balance number (EBN, indicating the sexual compatibility group; Johnston et al., Reference Johnston, den Nijs, Peloquin and Hanneman1980; Hanneman, Reference Hanneman1994) larger than 1 → clade 4 (Spooner and Castillo, Reference Spooner and Castillo1997) → series Cuneolata → Solanum infundibuliforme → Argentina → Province Jujuy → Department Yavi’.
Second, relative weights were assigned to each subgroup in all of the groups within the structured gene pool. In our example, six regions were distinguished in Argentina, and the Yavi Department from Jujuy Province was given a relative weight of 0.125, indicating that one-eighth of the samples in this group of Argentinean germplasm should ideally consist of accessions from the Yavi Department.
Third, for the purpose of this illustration, the total optimal collection size was considered identical to the current size of CGN's potato collection, and this figure was used to distribute the number of accessions over all groups within the structured gene pool. Subsequently, these numbers were compared with the actual numbers within each group in CGN's current collection.
Fourth, a similar analysis was performed using combined data from the potato collections maintained at CGN, the IPK at Gatersleben (Germany) and the Scottish Crop Research Institute (SCRI) at Invergowrie (Scotland). These collections are considered to be the major collections within the European Union (EU), and their combined dataset is hereafter referred to as the ‘EU collection’. Prior to all analyses, obvious duplicates within and between the collections, defined as accessions originating from the same original collection, were removed from the dataset. The collection sizes after this correction, i.e. 2792 and 6016 accessions for the CGN and EU collections, respectively, were considered actual and optimal collection sizes for the purpose of our illustration.
The results of our analyses are presented in Table 1 for a single group and its subgroups, namely the eight series of species belonging to clade 4 of wild tuber-bearing species, with EBN>1. Obviously, for the purposes of this illustration, we will not focus on a detailed explanation of how this grouping was accomplished. Our illustration is just to show that at any level within a hierarchy, there will be groups of material subdivided into a number of subgroups. Table 1 shows that within CGN's actual collection, the presented group is over-represented by 285 accessions, the actual size being 1803 and the optimal size being 1518 accessions. Despite the over-representation of the group as a whole, deficiencies were observed for certain subgroups (i.e. series Conicibaccata, Longipedicellata and Demissa). In contrast to CGN's collection, the EU collection showed an under-representation of 587 accessions for the presented group. As for CGN's collection, substantial over- and under-representation can be observed for separate subgroups, indicating that despite the over- or under-representation of the group as a whole, some of its constituent subgroups may show the opposite. The added value gained from combining these three collections was low for some subgroups (series Conicibaccata, Longipedicellata and Demissa) as deficiencies were observed in CGN's collection as well as in the EU collection. However, for the series Tuberosa, the rather large over-representation of 232 accessions within the CGN collection partly compensated for large deficiencies within the other two collections, resulting in an under-representation of 197 accessions within the EU collection. A similar finding was observed for series Yungasensa. These results indicate that virtually combining collections may improve the balance in the overall composition since some groups will be over-represented in some collections and under-represented in others. This complementation can occur when different genebanks have specialized on different parts of the gene pool.
Table 1 Comparison of the actual and optimal accession numbers for a given group (clade 4 of wild tuber-bearing species with an EBN larger than 1) in the potato gene pool (see text for details). Data are presented for each of the eight series of species belonging to this group and comprise the sum of several end-groups. Analyses were performed for the data from the collection of CGN and for the combined dataset of the collections of CGN, IPK and SCRI (EU collection)
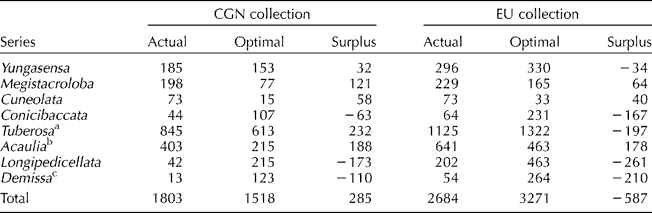
CGN, Centre for Genetic Resources, The Netherlands; EU, European Union; IPK, Leibniz Institute of Plant Genetics and Crop Plant Research.
a Only species belonging to the Brevicaule complex (van den Berg et al., Reference van den Berg, Miller, Ugarte, Kardolus, Villand, Nienhuis and Spooner1998).
b Including Solanum demissum.
c Excluding S. demissum.
This illustration was based on the personal knowledge and views of a single curator. However, for cooperating genebanks, the process of structuring of the gene pool and the assigning of relative weights to groups within the gene pool should be collaborative, involving the various collection holders. In our illustration, an optimal total collection size equal to the actual size was presented, but similar analyses may be performed to establish the optimal composition for collections to be reduced or enlarged.
An index for collection composition
Data on the distribution of the actual and optimal accession numbers may be used to quantify overall collection composition. If for each end-group the optimal and actual accession numbers were identical, one would consider the collection composition as perfect. The larger the difference between the actual and optimal numbers within the end-groups, the larger the imbalance in collection composition. One can define a parameter to estimate this imbalance, the collection composition imbalance index (CCII). Let the actual and optimal numbers within an end-group be denoted by N actend and N optend, respectively, and the actual and optimal total collection size by N acttot and N opttot, respectively, then the CCII value for n end-groups can be defined as
The potential range of CCII values is given by
and may range from 0 (no imbalance) to 1 (maximum imbalance), when N acttot = N opttot. When applied to our illustration in potato where we defined the optimal collection sizes as equal to the actual sizes, the CCII value were 0.54 and 0.53 for the CGN and EU collections, respectively, indicating that the composition imbalance is of similar magnitude in both collections.
It should be noted that similar CCII values do not necessarily indicate a similar collection composition. CCII values evaluate how well accessions are distributed over different hierarchical levels within a predefined structure, but different compositions may lead to similar levels of imbalance. The main feature of the outlined concept is that the quality of the collection composition can be quantified and evaluated, and that it may support a curator in decisions concerning collection management.
Discussion
Proposed optimization strategy
The outlined optimization strategy is based on the hierarchical structuring of a crop gene pool and the assignment of relative weights to the constituting groups. For some crops, the classification of accessions into predefined groups may not always be straightforward because of blurred boundaries between groups. These may, for instance, be due to cross-breeding between groups resulting in intermediate types (van Treuren et al., Reference van Treuren, van Hintum and van de Wiel2008). Despite the fact that a predefined gene pool structure may not always be perfect, it is still functional as a practical tool when based on sufficient available knowledge. Moreover, the predefined structure should not be regarded as fixed because it can be gradually improved in response to new insights, e.g. by splitting of divers groups, by combining groups with low genetic differentiation or by introducing new groups. The assignment of relative weights to the groups within the hierarchy is to some extent a subjective process, depending on the curators' views and the availability of pre-existing knowledge. However, a range of objective data covering conservation and use aspects may be considered, including breeding history, economical relevance, red-book listings, data on species vulnerability, geographical distribution of landraces and populations of wild species (Greene and Morris, Reference Greene and Morris2001; Morris and Greene, Reference Morris and Greene2001).
The main advantage of the outlined optimization strategy is that it can be used to gradually increase the genetic diversity of a collection in a systematic way, thereby contributing to a better representation of the genetic diversity present in a crop gene pool, while controlling the overall number of accessions. The potential to replace existing accessions by novel materials makes a collection flexible and dynamic. A flexible and dynamic collection offers the advantage that the relative importance of the various elements in the crop gene pool can be altered in response to improved insights and that collection composition can be optimized accordingly. For example, the importance of a crop's wild gene pool may increase significantly when novel resistance characters are needed in response to new diseases, to an accelerated breakdown of existing resistance mechanisms (Bonnier et al., Reference Bonnier, Reinink and Groenwold1992) or to address the consequences of climate change. In addition, consumer demands may change or the importance assigned to the conservation of specific wild species may increase when a species becomes more endangered in situ and/or access to its genetic diversity decreases significantly. These additional considerations require regular communication between curators and their respective user communities to evaluate changing demands, and with the conservation community to monitor for changes in the occurrence of wild species in their natural habitats (Widrlechner, Reference Widrlechner1997; Widrlechner and Burke, Reference Widrlechner and Burke2003).
When the collection is the product of a regional or global process and countries have used their sovereign right over germplasm within their borders to include specific accessions, it might be more difficult to exchange accessions with others. In such cases, it might only be possible to add accessions rather than to replace them.
Needed tools
In order to be able to optimize collections, particularly those with over-represented end-groups, a meaningful parameter is needed to quantify genetic diversity within the end-groups. The data types normally available for optimization may include passport, morphological, evaluation and molecular data. Each of these data types has its own features with information about different aspects of genetic diversity. How these data types can be best integrated in a comprehensive approach to obtain a meaningful parameter of genetic diversity goes beyond the scope of this paper, but is an important area for further research. Other relevant aspects in future research include appropriate ways to quantify and incorporate intra-accession variation, to estimate the added value of haploid material, such as pollen instead of seeds or tissue, to deal with differences in the number of plants studied per accession, to handle incomplete datasets and to weight different data types. Weighting of data types may be necessary because the number of characters per data type may differ considerably, and because data types per se may be considered of different relative importance (e.g. disease resistance characters vs. random molecular markers). Consequently, further research may include theoretical/statistical elements as well as case studies from existing collections with ample available accession data.
Based on these genetic diversity parameters, algorithms are needed to evaluate over-represented end-groups and identify those accessions that collectively contain the highest level of genetic diversity given a certain group size. Because the number of accessions within an end-group may be substantial, a large number of different combinations of accessions may require evaluation. Therefore, such algorithms should allow straightforward identification of optimal or near-optimal configurations. Algorithm development may build on recent and ongoing research in defining core collections, such as the concept of ‘genetic distance sampling’ ( Jansen and van Hintum, Reference Jansen and van Hintum2007).
In addition to algorithm development, the development of tools to visualize the effects of certain optimization decisions may be helpful to guide curators in the application of optimization procedures. Such tools could include software to display the effects of optimization methods by visualizing the genetic diversity within the collection before and after optimization.
Genetic diversity issues
An important point of attention in defining strategies to optimize collections is the crop's breeding system. As a consequence, different approaches for predominantly outbreeding, predominantly self-fertilizing and vegetatively propagated species may be required. Within some gene pools, a range of different breeding systems can be found, impacting the conservation strategy to be used (Engels and Visser, Reference Engels and Visser2003). For example, differences between accessions of highly outcrossing crops are often reflected in allele frequency differences between accessions, rather than in the differential fixation of alleles between accessions (van Treuren et al., Reference van Treuren, Bas, Goossens, Jansen and van Soest2005). Although careful curation of a given collection aims at maintaining the genetic integrity of individual accessions, allele frequencies will change during the regeneration process, and decisions about the optimization of collection composition of highly outcrossing crops may thus be influenced by the regeneration protocol being used (van Hintum et al., Reference van Hintum, van Treuren, van de Wiel, Visser and Vosman2007).
In addition to removing and adding accessions to collections, optimization may also be achieved by combining accessions with extensive overlap in genetic diversity (Sackville Hamilton et al., Reference Sackville Hamilton, Engels, van Hintum, Koo and Smale2002; van Hintum et al., Reference van Hintum, Sackville Hamilton, Engels, van Treuren, Engels, Rao, Brown and Jackson2002). The development of guidelines is needed for the bulking of accessions based on phenotypic and molecular marker data (van Treuren et al., Reference van Treuren, van Soest and van Hintum2001; Cruz et al., Reference Cruz, Nason, Luhman, Marek, Shoemaker, Brummer and Gardner2006), and considering effects of regeneration on the genetic constitution of accessions (van Hintum et al., Reference van Hintum, van Treuren, van de Wiel, Visser and Vosman2007).
Concluding remarks
Collection optimization has become of considerable interest in genetic resources management. However, methods how to best achieve optimization are scanty in the literature. With the present paper, describing a practical strategy, we invite readers to contribute to discussions on this topic. An appealing feature of the proposed strategy is that it can be applied not only to single collections, but also to collections of consortia of cooperating genebanks. The latter may be particularly relevant for ongoing efforts to share responsibilities in the framework of a European genebank integrated system (AEGIS project web site: available online at http://www.ecpgr.cgiar.org/AEGIS/AEGIS.htm), which aims at establishing a virtual European collection on the basis of identifying the most appropriate genetically unique accessions on a crop basis. Another example is provided by the current efforts of the global cacao genetic resources network (CacaoNet) to establish a global strategic cacao base collection that will represent the total available genetic diversity in the Theobroma cacao gene pool in as few as possible accessions (CacaoNet web site: available online at http://www.cacaonet.org). With increasing cooperation between genebanks, not only in Europe but possibly also on a future global scale, database interoperability is likely to become of crucial importance. In general terms, a higher level of efficiency in collection management may become within reach through the further development and application of optimization strategies outlined in this paper.
Acknowledgements
The authors would like to thank L. Frese, H.-R. Gregorius, H. Dempewolf, V.R. Rao, Dapeng Zhang, B. Visser and four anonymous reviewers for their contributions and comments on earlier versions of this paper.