Introduction
Gene expression in oocytes is regulated and variable throughout oogenesis (Pan et al., Reference Pan, O'Brien, Wigglesworth, Eppig and Schultz2005). At the final transition from germinal vesicle (GV) to a metaphase II (MII) oocyte, there is a general RNA decay of approximately 30–50% (Biase et al., Reference Biase, Fonseca Merighe, Santos Biase, Martelli and Meirelles2008; Lequarre et al., Reference Lequarre, Traverso, Marchandise and Donnay2004). Nonetheless, some genes are upregulated during this final oocyte maturation process (Fair et al., Reference Fair, Carter, Park, Evans and Lonergan2007; Katz-Jaffe et al., Reference Katz-Jaffe, McCallie, Preis, Filipovits and Gardner2009). These maternal gene products are stored in the oocyte and are able to sustain the initial cell cycles of early embryo development. Independent experiments with α-amanitin, an RNA polymerase II inhibitor, have shown that embryos can be cultured in vitro until a developmental block occurs at the 2-cell stage in mouse, 4-cell stage in pig and rabbit, 4–8-cell stage in human, 8-cell stage in cow, and 8–16-cell stage in sheep and goat (reviewed by Memili & First, Reference Memili and First2000). These observations indicate that the maternal stock of RNA and protein present in the MII oocyte is sufficient to drive early cleavages and that embryo genome activation is likely to be controlled by maternal gene products.
Early embryo development is influenced by the quality of the oocyte. After fertilization or chemical activation, a good quality oocyte will most likely form a blastocyst, whereas a bad quality oocyte is unlikely to complete normal early development. In cattle, experimental models have been employed to characterize developmental oocyte competence based on follicle size (Blondin & Sirard, Reference Blondin and Sirard1995), age of the oocyte donor (Revel et al., Reference Revel, Mermillod, Peynot, Renard and Heyman1995), timing of the first cleavage (Lonergan et al., Reference Lonergan, Khatir, Piumi, Rieger, Humblot and Boland1999), and cumulus–oocyte complex morphology (Bilodeau-Goeseels & Panich, Reference Bilodeau-Goeseels and Panich2002). These models have been used to investigate the accumulated maternal RNAs that associate with successful embryo development (Lonergan et al., Reference Lonergan, Gutierrez-Adan, Pintado, Fair, Ward, Fuente and Boland2000; Robert et al., Reference Robert, Barnes, Hue and Sirard2000; Calder et al., Reference Calder, Caveney, Westhusin and Watson2001; Donnison & Pfeffer, Reference Donnison and Pfeffer2004; Fair et al., Reference Fair, Gutierrez-Adan, Murphy, Rizos, Martin, Boland and Lonergan2004a,Reference Fair, Murphy, Rizos, Moss, Martin, Boland and Lonerganb; Gutierrez-Adan et al., Reference Gutierrez-Adan, Rizos, Fair, Moreira, Pintado, de la Fuente, Boland and Lonergan2004; Mourot et al., Reference Mourot, Dufort, Gravel, Algriany, Dieleman and Sirard2006; Patel et al., Reference Patel, Bettegowda, Ireland, Coussens, Lonergan and Smith2007; Caixeta et al., Reference Caixeta, Ripamonte, Franco, Junior and Dode2009; Biase et al., Reference Biase, Martelli, Puga, Giuliatti, Santos-Biase, Fonseca Merighe and Meirelles2010; Romar et al., Reference Romar, De Santis, Papillier, Perreau, Thelie, Dell'aquila, Mermillod and Dalbies-Tran2011), and have identified approximately 123 genes that might be differentially expressed between developmentally competent and incompetent oocytes. Recently, we proposed a new experimental model that collected a fraction of the ooplasm for experimental analysis, allowing the remaining of the oocyte to be chemically activated and cultured in vitro. The developmental competence of the oocytes is then classified based on actual embryo development (Biase et al., Reference Biase, Martelli, Merighe, Santos Biase, Miranda, Smith and Meirelles2009). This experimental model has been used to demonstrate that the total amount of the polyadenylated RNA present in MII oocytes does not correlate with developmental competence (Biase et al., Reference Biase, Martelli, Merighe, Santos Biase, Miranda, Smith and Meirelles2009).
Results from previous studies suggest that an adequate balance of mRNA abundance for each gene in the oocyte at the moment of fertilization may determine the quality of embryonic development pre-hatching. Even though several studies have characterized RNA accumulation during oogenesis, little information is known of the role these mRNA molecules play in early stages of embryo development. We hypothesized that mRNA species present in in vitro matured oocytes are important factors in enabling embryo development from eight cells up to the blastocyst stage in cattle. To test our hypothesis we compared a representative part of the transcriptome of oocytes matured in vitro that developed into proper blastocysts with those oocytes that could not sustain embryo development after embryonic genome activation. Our aim was to identify genes in the MII oocyte that were associated with embryo development beyond the maternal–embryonic genome transition.
Material and methods
Oocyte collection, cytoplasm biopsy and embryo culture
The chemicals used for in vitro culture were purchased from Sigma-Aldrich Co. (Oakville, Ontario, Canada) unless otherwise stated. Oocyte collection and biopsy were conducted as described elsewhere (Biase et al., Reference Biase, Martelli, Merighe, Santos Biase, Miranda, Smith and Meirelles2009). Briefly, bovine cumulus–oocyte complexes (COCs) were aspirated from antral follicles of 3–8 mm in diameter. Those COCs that contained compact layers of cumulus cells and homogeneous oocyte cytoplasm were matured in vitro (TCM199 medium, Gibco, Burlington, Ontario, Canada) for 20 h in 5% CO2 and a humidified atmosphere at 38°C. Cumulus cells were removed from oocytes by manual pipetting in hyaluronidase solution (2 mg/ml), and 120 oocytes containing the GV were selected for micro-manipulation and cytoplasm collection. Oocytes were placed in cytochalasin B (5 μg/ml) for 10 min before micro-manipulation. A fraction of the cytoplasm (approximately 10–20%) was aspirated from the oocyte on the opposite site of the polar body. The cytoplasm fraction was placed in 2 μl of phosphate-buffered saline that contained polyvinylalcohol (0.1%) and RNase inhibitor (1 U/μl), transferred to 30 μl of Trizol Reagent (Invitrogen, Carlsbad, California, USA) and frozen at –80°C until RNA extraction. The corresponding oocytes were placed individually in a Terasaki plate (Nunc, Inter-Med, Denmark) containing 10 μl of synthetic oviduct fluid (SOF) medium (Tervit et al., Reference Tervit, Whittingham and Rowson1972). Then, 26 h after the beginning of in vitro maturation, oocytes were chemically activated individually using 5 μM of ionomycin in TCM199 medium containing bovine serum albumin (BSA, 0.03 mg/ml) for 5 min following incubation in SOF medium containing 6-dimethylaminopurine (6-DMAP, 2 mM) for 3 h at 38°C in 5% CO2 and a humidified atmosphere. Embryos were individually cultured in vitro (Carolan et al., Reference Carolan, Lonergan, Khatir and Mermillod1996) on a Terasaki plate (Nunc, Inter-Med, Denmark) that contained 15 μl of SOF medium supplemented with 2% fetal calf serum (FCS) and BSA 0.005 μg/μl per well. The experiment was performed twice. For each replicate, a control group of 25 oocytes was matured in vitro, was not micro-manipulated and was chemically activated; the embryos were cultured in vitro in groups using the same conditions as the experimental oocytes and embryos but in 100 μl of the supplemented SOF medium under mineral oil.
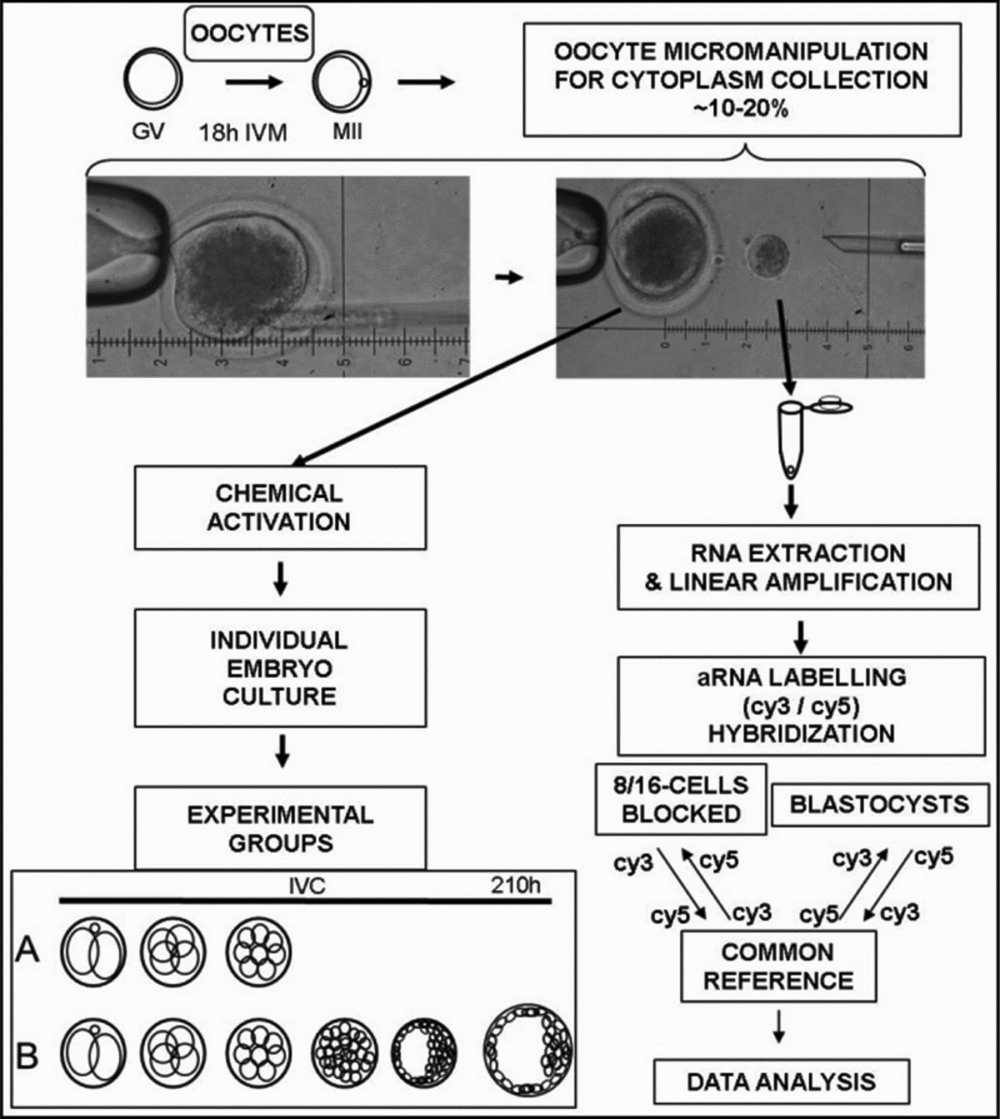
Embryo development was observed in two stages; cleavage was evaluated at 90 h post activation (hpa), when 10 μl of the medium was replaced with 10 μl of fresh medium, and blastocyst formation was evaluated 190 hpa. The oocytes were classified according to embryo development: 8–16-cell stage embryos that had arrested development before the blastocyst stage (group A); and embryos that formed a blastocyst (group B). Only the oocyte samples that produced embryos in groups A and B were used for microarray experiment, as demonstrated in Fig. 1.

Figure 1 Experimental design. Oocytes were matured in vitro and 10–20% of the cytoplast was collected through micro-manipulation. Oocytes were then chemically activated in individual micro-wells in Terasaki plates. Presumptive zygotes were cultured individually in vitro and embryonic development was categorized into two groups: cleavage arrest after the 8-cell stage but before blastocoel formation (A) or blastocyst formation (B). The cytoplast was frozen and subjected to a microarray analysis to compare the gene expression patterns between oocytes from groups A and B.
RNA extraction, amplification, labeling and microarray hybridization
Twenty-five cytoplasm fractions from oocytes from group A and 55 cytoplasm fractions from group B were used for the experiment. Each cytoplasm biopsy fraction was kept in a tube with Trizol® Reagent. Five of these tubes from the same oocyte group were pooled to make one biological replicate. Therefore there were respectively five and 11 biological replicates from group A and group B oocytes. Total RNA was extracted with Trizol® Reagent according to the manufacturer's instructions with the addition of 0.1 μg of linear acrylamide (Ambion®, Austin, Texas, USA) at the first step. Antisense oocyte RNA was amplified and labelled using a TargetAmp™ 2-Round Aminoallyl-aRNA Amplification kit 1.0 (Epicentre® Biotechnology, Madison, Wisconsin, USA). One nanogram of reference RNA was also amplified using the same procedure. Three micrograms of aRNA were subjected to integration of either Cy3-ester or Cy5-ester (Amersham, Piscataway, New Jersey USA). The aRNA from each oocyte pool was combined with reference aRNA and co-hybridized on a 13,257-element bovine oligoarray at 42°C for 40 h (GEO:GPL2853), following successive stringency washes for non-hybridized aRNA removal, essentially as described previously (Everts et al., Reference Everts, Chavatte-Palmer, Razzak, Hue, Green, Oliveira, Vignon, Rodriguez-Zas, Tian, Yang, Renard and Lewin2008; Loor et al., Reference Loor, Everts, Bionaz, Dann, Morin, Oliveira, Rodriguez-Zas, Drackley and Lewin2007). Microarray images were obtained with an Axon 4000B scanner (Molecular Devices, Sunnyvale, CA) and processed with GenePix 6.0 software (Molecular Devices, Sunnyvale, CA). Microarray data are available to the community via the NCBI GEO database (GSE29191), following MIAME standards.
Microarray data analysis
Microarray data were imported and analyzed using R programming language (Ihaka & Gentleman, Reference Ihaka and Gentleman1996) and the Bioconductor (Gentleman et al., Reference Gentleman, Carey, Bates, Bolstad, Dettling, Dudoit, Ellis, Gautier, Ge, Gentry, Hornik, Hothorn, Huber, Iacus, Irizarry, Leisch, Li, Maechler, Rossini, Sawitzki, Smith, Smyth, Tierney, Yang and Zhang2004) package Limma (Smyth, Reference Smyth2005). For each microarray, foreground data (median) were subjected to background subtraction (Ritchie et al., Reference Ritchie, Silver, Oshlack, Holmes, Diyagama, Holloway and Smyth2007), and filtering of the spots for which signal intensity was lower than 300 arbitrary units. The remaining spots were subjected to within-array normalization using print-tip LOESS quantile normalization between arrays (Smyth, Reference Smyth2003). Differential gene expression was assessed using moderated t-statistics (Smyth, Reference Smyth2004; Jeanmougin et al., Reference Jeanmougin, de Reynies, Marisa, Paccard, Nuel and Guedj2010), considering correlation between the duplicate spots (Smyth et al., Reference Smyth, Michaud and Scott2005). Probes with a false discovery rate (FDR; Benjamini & Hochberg, Reference Benjamini and Hochberg1995) corrected P (H0)-value less than 0.05 and B greater than 0.5 were assumed to be differentially expressed between the two groups tested.
Functional annotation of the genes for the corresponding probes was obtained from Gene Ontology (Ashburner et al., Reference Ashburner, Ball, Blake, Botstein, Butler, Cherry, Davis, Dolinski, Dwight, Eppig, Harris, Hill, Issel-Tarver, Kasarskis, Lewis, Matese, Richardson, Ringwald, Rubin and Sherlock2000) and Gene Enrichment Analyses were performed using Babelomics v4.1 web tools (Medina et al., Reference Medina, Carbonell, Pulido, Madeira, Goetz, Conesa, Tarraga, Pascual-Montano, Nogales-Cadenas, Santoyo, Garcia, Marba, Montaner and Dopazo2010).
Validation of microarray result with RT-qPCR
The following genes were selected to validate the microarray result: IGF2R, DDR1, DUSP6, HERC2, NDUFB6, NDUFS4, PFKFB3, SFRS14, UQCRH. Primers were designed for these genes based on reference sequences from the GenBank database (Table 1). Reverse transcription (RT) was performed using 100 ng of amplified RNA and SuperScript® III Reverse Transcriptase (200 U, Invitrogen, Carlsbad, CA), using random hexamers (500 ng), dNTP mix (0.5 mM), RNAseOUT (40 U, Invitrogen, Carlsbad, CA), following manufacturer's instructions for incubation temperature and time, in a final volume of 20 μl. Real-time polymerase chain reactions (PCRs) were set up with one-quarter of the RT reaction volume, using SYBR® GREEN PCR Master Mix (Warrington, UK), following the manufacturer's instructions. PCRs were run on a 7900 HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA), following preset cycling parameters, with an annealing temperature of 60°C.
Table 1 Primers used for qPCR
Relative expression values were calculated using the comparative cycle threshold (Ct) approach (Schmittgen & Livak, Reference Schmittgen and Livak2008). Only samples with Ct values bellow 35 were subjected to analysis. The expression values from the samples were quantified relative to ACTB and GAPDH control genes using the methods presented elsewhere (Hellemans et al., Reference Hellemans, Mortier, De Paepe, Speleman and Vandesompele2007). The significance of the difference of the relative abundance values from two oocyte groups was assessed using a Wilcoxon test (Yuan et al., Reference Yuan, Reed, Chen and Stewart2006), performed in R (Ihaka & Gentleman, Reference Ihaka and Gentleman1996). Fold changes were calculated for the group of good quality oocytes relative to the bad quality oocytes (calibrator group). Differences with a P (H0)-value less than 0.05 were considered to be significant and the gene was inferred to be differentially expressed, unless otherwise stated.
Results
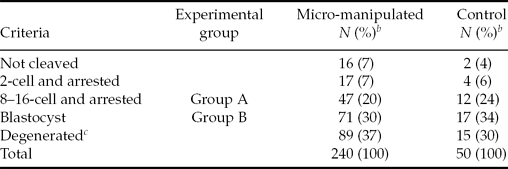
The number of oocytes that developed to blastocyst in experimental oocytes and their control non-manipulated counterparts was similar (29.6% vs. 34.0%, P > 0.05; Table 2), indicating that the developmental competence of the oocytes was not significantly reduced due to micro-manipulation and ooplasm removal.
Table 2 Oocyte classification and distribution according to the embryo in vitro culture outcomea
aData from two replicates.
bPercentages were calculated according to the total of oocytes activated and placed for in vitro embryo culture.
cDegenerated: oocytes were not classified due to degeneration of the zygote or blastomeres.
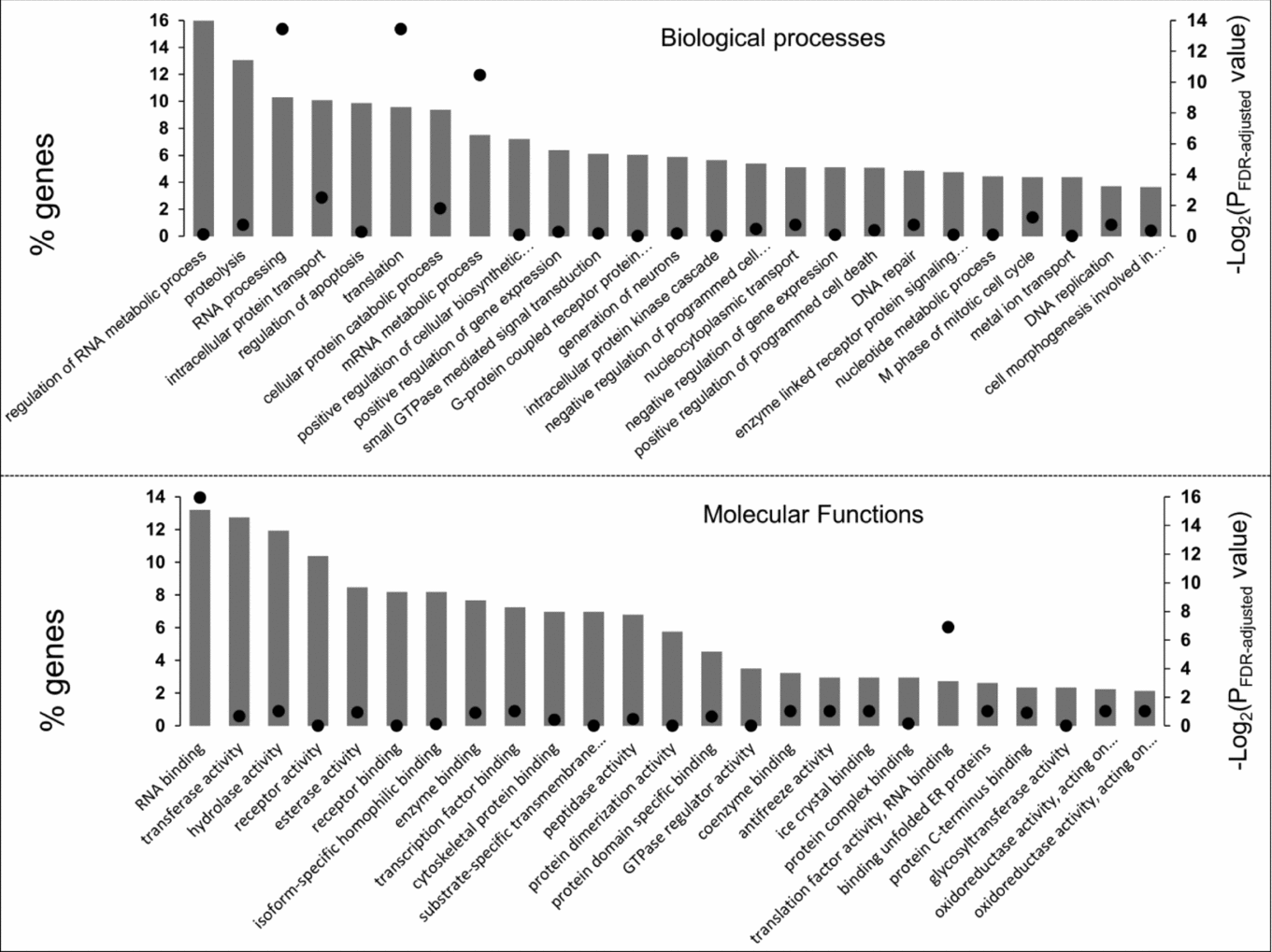
Using multiple samples from in vitro matured bovine oocytes, we evaluated the relative expression of 4414 probes out of the 13,257 oligonucleotides present on the microarray slide. These probes are equivalent to 4320 annotated genes, from which a subset of 2301 genes have an annotation for biological processes and 2134 genes for molecular function. The 25 categories with the highest number of genes are presented in Fig. 2.
Figure 2 Functional categorization of the genes expressed in ooplasm after in vitro maturation. For each chart, vertical bars represent the 25 categories with most number of genes (left axis) while the dots are P-values from the functional enrichment analysis (right axis).
Expressed genes were significantly enriched in the functional categories ‘RNA processing’, ‘translation’ and ‘mRNA metabolic process’ compared with the microarray gene set. Amongst molecular function classes, we observed ‘RNA binding’ (N = 282) as the category with highest number of expressed genes. Along with ‘translation factor activity, RNA binding’ (N = 58), the ‘RNA binding’ category was also enriched in the set of expressed genes.
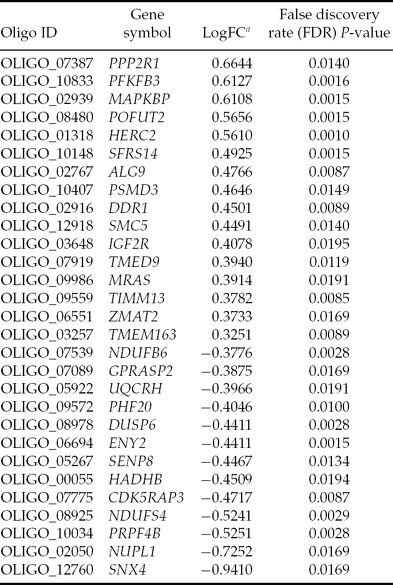
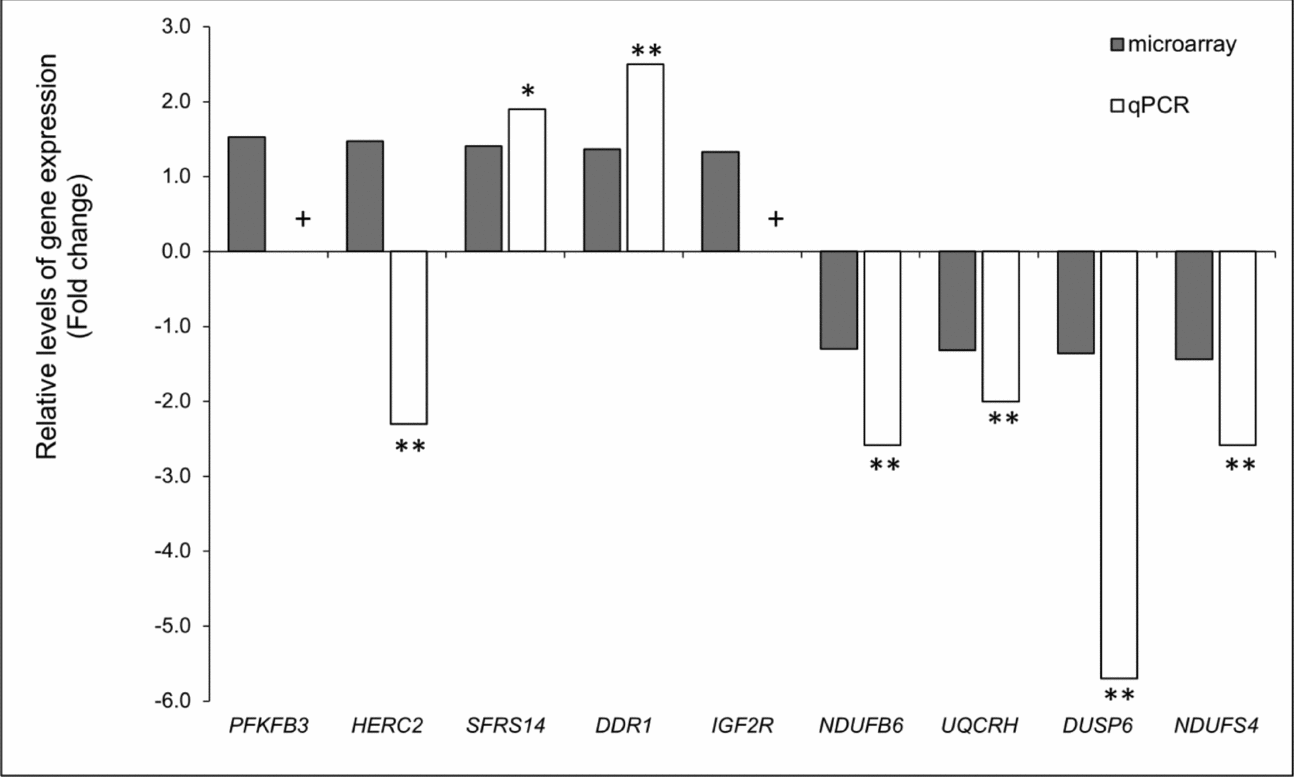
Twenty-nine genes were differentially expressed between good and bad quality oocytes (Table 3). Sixteen genes were upregulated in oocytes that sustained early embryo development to blastocyst stage. There was no enrichment of functional or molecular categories in the list of differentially expressed genes (DEG) as compared with either the microarray set of genes or the expressed genes in in vitro matured oocytes. Validation of the microarray result was performed by analyzing nine genes and qPCR assays with the same samples used for microarray hybridizations. With the exception of HERC2, nine out of the nine genes examined were in agreement with the microarray results (Fig. 3). No fold change was calculated for the genes PFKFB3 and IGF2R because no fluorescence signal was detected from amplification or the Ct values for some samples were higher than 37, therefore above our threshold for data analysis. Thus, these two genes were validated because specific amplification was detected in all samples of good quality oocytes.
Table 3 Differentially expressed genes in good versus bad quality oocytes
aLog(fold change) = Log2(expression in good quality oocytes) – Log2(expression in bad quality oocytes).
Figure 3 Validation of microarray experiment. **P < 0.05, *P < 0.10; + No fold change was calculated because fluorescence from no sample (PFKFB3) or only one sample (IGF2R) was detected from bad quality oocytes.
Discussion
In this study, we obtained an overall view of the global gene expression pattern of bovine in vitro matured oocytes using a small fraction of their cytoplasm. After the removal of the cytoplasm fraction, these oocytes were activated subsequently to examine their ability to undergo early embryonic development in vitro. Then, by comparing the transcriptome of oocytes that sustained embryo development to blastocyst stage with those that arrested development before blastula formation, we were able to associate specific gene products in the oocyte to its ability to undergo normal development.
Extraction of a portion of the cytoplasm did not affect the capability of competent oocytes to sustain early embryo development, as shown by the similarity between the blastocyst development ratio among micro-manipulated oocytes compared with the control group. Embryo culture from micro-manipulated oocytes yielded a similar blastocyst rate as observed previously in both individual embryo culture (Carolan et al., Reference Carolan, Lonergan, Khatir and Mermillod1996) and culture following an oocyte biopsy in microdrops that contained groups of 20 embryos (Biase et al., Reference Biase, Martelli, Merighe, Santos Biase, Miranda, Smith and Meirelles2009).
To avoid removing the metaphase spindle, ooplasm samples were aspirated from the opposite site of the polar body. The mammalian oocyte is a polarized cell (Gardner, Reference Gardner2001) and mRNA localization has been demonstrated for different higher eukaryotic organisms (Palacios & St Johnston, Reference Palacios and Johnston2001). Therefore, it is possible that our sampling was not a full representation of the mRNAs accumulated during oogenesis. Cytoplasm stratification remains a controversial subject in mammalian eggs. The comparison of sister blastomeres from 2-cell murine embryo suggested that transcripts of both blastomeres is very similar (Tang et al., Reference Tang, Barbacioru, Nordman, Bao, Lee, Wang, Tuch, Heard, Lao and Surani2011), but some genes have different mRNA quantities between the sister blastomeres (Roberts et al., Reference Roberts, Katayama, Magnuson, Falduto and Torres2011), a result that suggested that there might be non-uniform distribution of maternal mRNA in the cytoplasm. Nonetheless, as all biopsies were recovered consistently away from animal pole (region containing the metaphase plate), it is likely that the sampled region and its molecular composition were also relatively consistent throughout. Previous studies have identified between approximately 9000–11,000 genes expressed in bovine in vitro matured oocytes (Misirlioglu et al., Reference Misirlioglu, Page, Sagirkaya, Kaya, Parrish, First and Memili2006; Fair et al., Reference Fair, Carter, Park, Evans and Lonergan2007; Kues et al., Reference Kues, Sudheer, Herrmann, Carnwath, Havlicek, Besenfelder, Lehrach, Adjaye and Niemann2008), however we quantified the expression for 4320 in the ooplasm fragments, suggesting that the difference may be due to the restricted sampling region, i.e. vegetal pole, chosen for these studies.
The functional characterization of the genes expressed in in vitro matured oocytes revealed that a representative portion is dedicated to regulation of transcription and RNA processing and protein synthesis and degradation, corroborating previous findings (Cui et al., Reference Cui, Li, Yin, Kong, Kang and Kim2007; Fair et al., Reference Fair, Carter, Park, Evans and Lonergan2007). The RNA and protein synthesis and processing are certainly key functions that mediate activation of the embryonic genome. The presence of genes that function in DNA repair, replication, and the cell cycle in the MII oocyte is also compatible with the need to sustain three cell cycles without major gene transcription. During the initial three cleavages, blastomeres are not susceptible to apoptosis due to different inhibition mechanisms (Brad et al., Reference Brad, Hendricks and Hansen2007; Carambula et al., Reference Carambula, Oliveira and Hansen2009), however we observed expression of genes related to negative (n = 124) and positive (n = 117) regulation of cell death in our sample of bovine MII oocytes and supporting findings that oocytes accumulate pro- and anti-apoptotic regulators (Dalbies-Tran & Mermillod, Reference Dalbies-Tran and Mermillod2003; Fair et al., Reference Fair, Carter, Park, Evans and Lonergan2007; Fear & Hansen, Reference Fear and Hansen2011).
Comparative analysis between oocytes identified 29 DEG between embryos that reached the blastocyst stage and embryos that arrested at the 8–16-cell stage. Of these, 16 genes were more abundant in good quality oocytes and 13 were more abundant in bad quality oocytes when compared with their counterparts. The DEG found in our study have not been previously associated with oocyte quality in cattle, however we found that four genes have been described in studies that compared oocytes from women at different ages (49–51). The rationale of this model is that there is a significant negative correlation between in vitro blastocyst developmental ratio and age of women (Janny & Menezo, Reference Janny and Menezo1996). Transcripts for DDR1 were relatively more abundant in GV stage oocytes from younger women (27–35 years) when compared with oocytes from older subjects (37–39 years) (Grondahl et al., Reference Grondahl, Yding Andersen, Bogstad, Nielsen, Meinertz and Borup2010). The levels of mRNA for DUSP6 and NUFS4 were more abundant in in vitro matured oocytes from older women (>40 years) compared with oocytes from younger patients (<32 years) (Steuerwald et al., Reference Steuerwald, Bermudez, Wells, Munne and Cohen2007). In our study, the expression of PPP2R1B was more abundant in good quality oocytes, but was also more abundant in oocytes from older women (Steuerwald et al., Reference Steuerwald, Bermudez, Wells, Munne and Cohen2007). We find it worth noting that genes belonging to the same family of PSMD3, TIMM13 and NDUFS4 were differentially expressed in matured eggs from young and old mice (Hamatani et al., Reference Hamatani, Falco, Carter, Akutsu, Stagg, Sharov, Dudekula, VanBuren and Ko2004).
We found two receptors that are more abundant in good quality oocytes compared with their bad quality counterparts: DDR1 and IGF2R. The protein DDR1 can interact with type I to IV collagens (Koo et al., Reference Koo, McFadden, Huang, Abdulhussein, Friese-Hamim and Vogel2006) and its activation transduces signals that function towards differentiation, extracellular matrix remodeling and cell cycle control (Vogel et al., Reference Vogel, Abdulhussein and Ford2006). Along with other receptors, the DDR1 protein may mediate cell communication between oocytes and cumulus cells during maturation, when the synthesis and deposition of collagen type IV increases on the cell surface (Sutovsky et al., Reference Sutovsky, Flechon and Pavlok1995).
The other membrane protein differentially expressed was IGF2R. Messenger RNA for IGF2R was previously detected in mature human and bovine oocytes (Lighten et al., Reference Lighten, Hardy, Winston and Moore1997; Yaseen et al., Reference Yaseen, Wrenzycki, Herrmann, Carnwath and Niemann2001; Katz-Jaffe et al., Reference Katz-Jaffe, McCallie, Preis, Filipovits and Gardner2009; Wang et al., Reference Wang, Feng, Ma, Cang, Li, Yan, Zhou, Wen, Bou and Liu2009), and IGF2R protein was present in the plasma membrane (Wang et al., Reference Wang, Feng, Ma, Cang, Li, Yan, Zhou, Wen, Bou and Liu2009), where it may respond to autocrine and paracrine stimulus from the synthesis of IGF2 from oocyte and cumulus cells (Wang et al., Reference Wang, Feng, Ma, Cang, Li, Yan, Zhou, Wen, Bou and Liu2009). Experiments in vitro have shown that oocytes and early developing embryos respond to the presence of IGF2 in culture medium (Warzych et al., Reference Warzych, Wrenzycki, Peippo and Lechniak2007; Wang et al., Reference Wang, Feng, Ma, Cang, Li, Yan, Zhou, Wen, Bou and Liu2009). The abundance of IGF2R mRNA in MII oocytes is altered by in vitro maturation. Oocytes cultured in vitro showed four-fold fewer transcripts compared with in vivo matured oocytes (Katz-Jaffe et al., Reference Katz-Jaffe, McCallie, Preis, Filipovits and Gardner2009). Our results support the fact that lower IGF2R mRNA expression in mature oocytes result in arrest of early embryonic development. Recent findings of the imprinted status of IGF2R in immature, but fully grown competent, oocytes showed one differentially methylated region (DMR) on intron 2 with a methylation profile of 31% on the groups of oocytes analyzed (O'Doherty et al., 2011). We hypothesize that the lower amount of IGF2R transcripts in incompetent oocytes may be associated with the alteration in the methylation profile of the DMR in the IGF2R gene.
The RNA and proteins accumulated in mature oocytes drive the regulation of gene expression from the embryonic genome and the efficacy of oocyte maturation and may determine the fate of the embryo after the 8–16-cell stage (Memili & First, Reference Memili and First2000). Our analysis revealed four DEG that encode for proteins related to gene expression or mRNA metabolic processes: ALG9, SFRS14, ENY2, PHF20, PRPF4B; and one zinc finger protein: ZMAT2. Another group of five genes of similar function were also differentially expressed: HERC2, MRAS, TIMM13, NUPL1 and SNX4. According to gene ontology, the last group of genes code for proteins that function in protein localization and protein transport. The correct location of proteins throughout the ooplasm is important for maintaining the homogeneity of protein segregation among the blastomeres during the early cleavages, cell compaction, and cell differentiation that forms the trophectoderm and inner cell mass (Chen et al., Reference Chen, Wang, Wu, Ma and Daley2010).
In summary, we were able to characterize the transcriptome of in vitro matured oocytes from a cytoplast fraction and correlate gene expression with the embryonic development until the blastocyst stage. Using our innovative approach, 29 genes were associated with oocyte quality, some of which had been reported previously in bovine, mouse or human experiments. Genes discovered in the present study are potential markers of mammalian oocyte quality. A better understanding of the gene expression architecture of a developmentally competent oocyte will lead us to create tools to improve fertility rates in in vitro fertilization services.
Acknowledgements
This study was financed partially by the following agencies: FAPESP, Brazil (2006/57973-7); USDA-ARS (AG 58-1265-2-020).