


Introduction
Ewing's sarcoma is an aggressive malignancy involving small, round, blue cells, which commonly presents as a childhood tumour of the long bones or pelvic bones.Reference Cotterill, Ahrens, Paulussen, Jurgens, Voute and Gadner1 Primary Ewing's sarcoma of the head and neck is rare in otolaryngological practice, representing only 1–4 per cent of all Ewing's sarcoma cases.Reference Allam, El-Husseiny, Khafaqa, Kandil, Gray and Ezzat2 Such tumours usually arise in the mandible or skull base.Reference Siegal, Oliver, Reinus, Gilula, Foulkes and Kissane3
Extraskeletal Ewing's sarcoma is an uncommon variant of Ewing's sarcoma which arises in soft tissue rather than bone. The lower extremities and trunk, especially the paravertebral region, are the typical primary sites.Reference Ali, MacKenzie, Reid, O'Neill and Ganly4, Reference Kinsella, Triche, Dickman, Costa, Tepper and Glaubiger5 Extraskeletal Ewing's sarcoma is rare, with only a few reported cases in the head and neck region.Reference Cho, Park, Cho, Ryoo, Yang and Youn6–Reference Mills10
We present a case of Ewings's sarcoma arising in the masseter muscle of a young woman.
Case report
A 23-year-old woman presented with a four-year history of swelling in the right parotid area.
On palpation, tenderness was elicited over the right masseter muscle, and a firm, mobile, approximately 3 × 3 cm mass was noted in the parotid area. No trismus was present. No other abnormalities of the head or neck were found.
From the physical examination, the mass was thought to originate within the parotid gland. A fine needle aspiration biopsy (FNAB) was performed. This revealed neoplastic cells, but a definitive classification could not be made.
Additional tissue sampling was recommended, and a core biopsy was performed in the clinic without ultrasound guidance. Surprisingly, this revealed only normal parotid tissue.
A subsequent computed tomography (CT) scan revealed that the neoplasm was arising within the masseter muscle (Figure 1). The mass extended to the mandible, with destruction of the mandibular cortex and lifting of the periosteum.
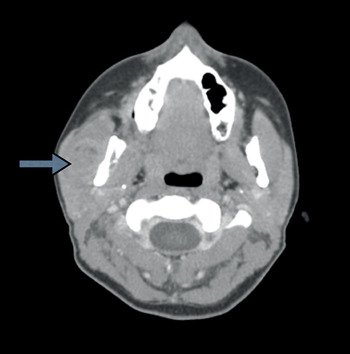
Fig. 1 Axial computed tomography scan showing a heterogeneous mass centred within the right masseter muscle, measuring approximately 2.6 × 1.7 cm (arrow).
Magnetic resonance imaging showed a poorly defined, enhancing mass centred within the right masseter muscle.
A second, ultrasound-guided core biopsy of the mass was obtained. Examination of tissue sections revealed a cellular neoplasm composed of small, round, blue cells with uniform nuclei with only mild atypia, and moderate amounts of clear cytoplasm containing intracellular glycogen, as demonstrated by periodic acid phosphatase staining with and without diastase digestion (Figure 2).
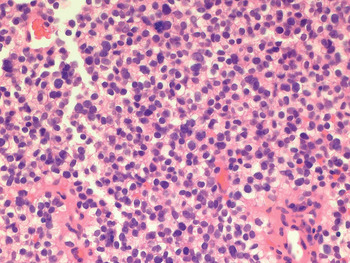
Fig. 2 Photomicrograph prepared with haematoxylin and eosin staining, showing small, round, blue, neoplastic cells. (×400)
Immunohistochemical analysis identified strong cytoplasmic membranous staining of the lesional cells with antibodies against cluster of differentiation 99 glycoprotein, in a manner typical of Ewing's sarcoma (Figure 3). Interphase fluorescent in situ hybridisation was performed using a break-apart probe for the EWSR1 (Ewing sarcoma breakpoint region 1) gene. A split signal was identified in 78.5 per cent of cells examined, consistent with a diagnosis of Ewing's sarcoma.

Fig. 3 Photomicrograph prepared with antibodies against cluster of differentiation 99 glycoprotein, showing cytoplasmic membranous staining. (×400)
Treatment was discussed by the multi-disciplinary sarcoma tumour team in London, Ontario. Induction chemotherapy was initiated, which reduced the size of the mass to 1.2 × 0.7 cm (Figure 4).
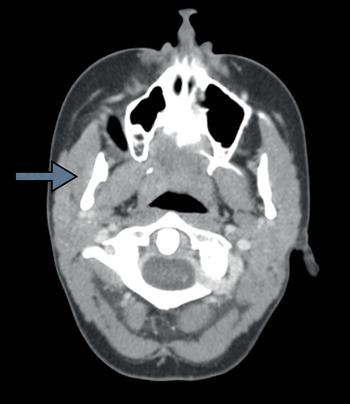
Fig. 4 Axial computed tomography scan taken after induction chemotherapy, showing a poorly defined mass in the right masseter muscle measuring 1.2 × 0.7 cm (arrow). This represents a significant reduction in size.
Following chemotherapy, surgical removal of the mass was undertaken. This involved wide resection of the masseter muscle and underlying mandibular ramus, with complete mobilisation and preservation of the facial nerve, via a transcervical parotidectomy approach (Figure 5). The composite mandibular defect was reconstructed with an osseus fibular free flap (Figure 6).

Fig. 5 Intra-operative photograph showing the free flap in situ, positioned medial to the branches of the facial nerve (indicated by arrows). Forceps indicate the de-epithelialised portion of the fibular flap, used to restore the facial contour.

Fig. 6 Appearance four months after surgery.
Histological examination of the surgical specimen revealed no residual malignant cells, negative margins, and lymph nodes without metastatic disease. Due to this lack of residual disease, the patient did not undergo radiation therapy.
Ten months post-operatively, the patient was doing well, and was considered to be in complete remission.
Discussion
Ewing's sarcoma was first described as an osteolytic lesion by James Ewing in 1921.Reference Ewing11 It is considered to be a highly malignant bone tumour of neuroectodermal origin, which usually arises in the long bones or the pelvis.Reference Chao, Chang and Sheen7
The extraskeletal form was first recognised as a distinct small cell tumour by Tefft et al. in 1969.Reference Tefft, Vawter and Mitus12 Extraskeletal Ewing's sarcoma usually presents as a rapidly growing tumour of the lower limb or paravertebral area.Reference Siegal, Oliver, Reinus, Gilula, Foulkes and Kissane3, Reference Ali, MacKenzie, Reid, O'Neill and Ganly4 A review of extraskeletal Ewing's sarcoma undertaken by O'Keefe et al. found that half of all reported cases were located in the extremities, and the remainder in the chest, pelvis or abdomen.Reference O'Keefe, Lorigan and Wallace13 Approximately 9 per cent of extraskeletal Ewing's sarcoma cases occur in the head and neck region.Reference Mills10
Although imaging is non-specific, extraskeletal Ewing's sarcoma tends to be well circumscribed and of generally low attenuation on CT, compared with muscle. It is commonly hypoechoic on ultrasound, a finding thought to signify cyst formation or necrosis. Vascularity is variable.Reference Siegal, Oliver, Reinus, Gilula, Foulkes and Kissane3 In our patient, the mass was poorly defined, but did exhibit low attenuation respective to muscle and significant vascularity.
Histologically, extraskeletal Ewing's sarcoma is described as a poorly differentiated neuroectodermal tumour with small, uniform, round, blue cells thought to arise from neural crest cells.Reference Heare, Hensley and Dell'Orfano14 However, extraskeletal Ewing's sarcoma lacks any specific morphological features, resulting in a high misdiagnosis rate when examined by light and electron microscopy alone.Reference Zagar, Triche and Kinsella15
Immunohistochemistry and genomics are essential in making an accurate diagnosis. Ewing's sarcoma is associated with a tumour-specific chimeric gene, usually derived from a characteristic translocation, t(11;22)(q24;q12), which results in the fusion of the EWS and FLI-1 genes.Reference Heare, Hensley and Dell'Orfano14, Reference Zagar, Triche and Kinsella15 This translocation is present in 85–95 per cent of Ewing's sarcoma cases, and was identified in our patient.Reference Heare, Hensley and Dell'Orfano14, Reference Zagar, Triche and Kinsella15 Immunohistochemical staining for cluster of differentiation 99 glycoprotein (a cell surface glycoprotein) is also important in making the diagnosis.Reference Zagar, Triche and Kinsella15
While there is not yet strong consensus on the best treatment of extraskeletal Ewing's sarcoma, aggressive multi-modal treatment appears to offer the best prognosis.
The high risk of recurrence and metastatic disease necessitates systemic therapy, so induction chemotherapy is commonly used. Several chemotherapeutic drugs have been identified as effective, including vincristine, doxorubicin and cyclophosphamide.Reference Ali, MacKenzie, Reid, O'Neill and Ganly4 Venkitaraman et al. compared outcomes using the vincristine, adriamycin, cyclophosphamide (VAC), vincristine, cyclophosphamide, doxorubicin, actinomycin D (VACA) and vincristine, adriamycin, cyclophosphamide/ifosfamide, etoposide (VAC/IE) regimes, in a small group of 15 patients with extraskeletal Ewing's sarcoma.Reference Venkitaraman, George, Ramanan and Sagar16 The cohort treated with VAC/IE is a chemotherapeutic regimen in which vincristine, adriamycin and cyclophosphamide is alternated with ifosfamide and etoposide had substantially better outcomes, with all five patients achieving a complete response and remaining disease-free at a median follow up of 12 months. This is a standard chemotherapeutic approach for Ewing's sarcoma, and appears to work well for its extraskeletal subtype.
Induction chemotherapy is usually followed by surgical resection and/or irradiation. When the entire family of Ewing's sarcoma is considered, there is some evidence that chemotherapy followed by resection and post-operative irradiation leads to better outcomes than chemotherapy and irradiation alone. Bacci et al. reported a local control rate of 88 per cent for the former approach and 80 per cent for the latter.Reference Bacci, Longhi, Ferrari, Mercuri, Barbieri and Bertoni17 However, there are no published trials specifically comparing radiation therapy with surgery for local control of extraskeletal Ewing's sarcoma.Reference Zagar, Triche and Kinsella15 Currently, most protocols recommend surgical resection when possible, followed by radiation for positive margins or residual disease.Reference Zagar, Triche and Kinsella15 This was the approach taken with our patient.
Regarding radiation therapy, there is limited evidence that irradiation at greater than 50 Gy may lead to better outcomes. Venkitaraman et al. examined nine patients diagnosed with extraskeletal Ewing's sarcoma and treated with chemotherapy and irradiation.Reference Venkitaraman, George, Ramanan and Sagar16 Six received 40 Gy and three received more than 50 Gy. Fifty per cent of the 40 Gy cohort relapsed, while all three patients who received more than 50 Gy remained disease-free after a mean follow up of 12 months. In contrast, other authors advocate the use of lower irradiation doses (30–36 Gy) in order to minimise the dose to normal tissues and to reduce the risk of side effects, including second malignancies.Reference Zagar, Triche and Kinsella15
• Extraskeletal Ewing's sarcoma is a poorly differentiated neuroectodermal tumour with small, uniform, round, blue cells thought to arise from neural crest cells
• Ewing's sarcoma is rare in the head and neck region, and has not previously been reported to arise in the masseter muscle
• Immunohistochemistry and genomics are essential in diagnosing this tumour
• Advances in multi-disciplinary management have significantly improved prognosis
• Aggressive treatment, including multi-agent chemotherapy, surgical resection and irradiation, appears to offer the best prognosis
Ewing's sarcoma has historically been considered to have a poor prognosis. Verrill et al. examined Ewing's sarcoma and primitive neuroectodermal tumours in adults, and found a 38 per cent overall five-year survival rate and a 27 per cent probability of progression-free survival.Reference Verrill, Judson, Harmer, Fisher, Thomas and Wiltshaw18 Paulino et al. assessed only non-metastatic Ewing's sarcoma, and found a five-year survival rate of 55.5 per cent.Reference Paulino, Nguyen and Mai19 The prognosis of Ewing's sarcoma depends on the tumour site and volume at presentation, the patient's age and response to primary treatment, and the presence of metastases at diagnosis.Reference Cotterill, Ahrens, Paulussen, Jurgens, Voute and Gadner1, Reference Verrill, Judson, Harmer, Fisher, Thomas and Wiltshaw18 When only the head and neck region is considered, some authors have reported better outcomes.Reference Allam, El-Husseiny, Khafaqa, Kandil, Gray and Ezzat2 In a study of 24 cases of Ewing's sarcoma of the head and neck encountered between 1975 and 1996, the five-year overall survival rate for the entire group was 53 per cent while the disease-free survival rate was 30 per cent.Reference Allam, El-Husseiny, Khafaqa, Kandil, Gray and Ezzat2
Extraskeletal Ewing's sarcoma generally has a worse prognosis. Venkitaraman et al. assessed 19 patients with extraskeletal Ewing's sarcoma between 1992 and 2003, and found three- and five-year disease-free survival rates of 38 and 19 per cent, respectively, and overall three- and five-year survival rates of 47 and 24 per cent, respectively.Reference Venkitaraman, George, Ramanan and Sagar16 After exclusion of patients with metastases on presentation, overall survival rates at three and five years became 60 and 30 per cent, respectively.
Conclusion
The presented case highlights the diagnosis and management of extraskeletal Ewing's sarcoma of the head and neck. Genomics and immunohistochemistry are essential diagnostic tools. Aggressive multi-modal treatment, including multi-agent chemotherapy, surgical resection and irradiation, appears to offer the best prognosis. Contemporary reconstructive techniques can restore function and form in cases with challenging defects.
Acknowledgements
The authors thank Drs D Logan, J A Hammond and D Ho for their input into the manuscript. We also extend special thanks to Dr Ho for supplying and interpreting the imaging.






