


Introduction
Maternal nutrition has a profound impact on fetal development and growth and influences the future health of the offspring.Reference Gluckman, Hanson, Cooper and Thornburg1, Reference Symonds, Sebert, Hyatt and Budge2 However, the mechanisms linking altered maternal nutrition to changes in fetal growth and developmental programming are poorly understood. Previous studies in rodents and sheep implicate changes in placental growth, structure and function as critical mediators of adverse pregnancy outcomes when maternal nutrient availability is altered.Reference Jansson, Pettersson and Haafiz3–Reference Belkacemi, Nelson, Desai and Ross9 Here, we review changes in placental nutrient transport in response to altered maternal nutrition in pregnant women and in relevant animal models. The concept of maternal nutrition is defined broadly as the ability of the maternal supply line to provide nutrients and oxygen to the placenta. Our discussion will therefore also include placental responses to compromized utero-placental blood flow, maternal hypoxia and iron deficiency.
The placental barrier and factors influencing placental transfer
Fetal nutrient and oxygen availability depend on the rate of transfer across the ‘placental barrier’. In the human term placenta, there are only two cell layers separating fetal and maternal circulations; the fetal capillary endothelium and the syncytiotrophoblast (Fig. 1).Reference Kaufmann and Burton10 The syncytiotrophoblast is the transporting epithelium of the human placenta and has two polarized plasma membranes: the microvillous plasma membrane (MVM) directed toward maternal blood in the intervillous space and the basal plasma membrane (BPM) facing the fetal capillary. In the mouse and rat placenta, three trophoblast layers form the placental barrier, and accumulating evidence suggests that the maternal-facing plasma membrane of trophoblast layer II of the mouse placenta is functionally analogous to the MVM in the human placenta.Reference Kusinski, Jones, Baker, Sibley and Glazier11 In the hemochorial placenta of primates and rodents, the trophoblast is directly in contact with maternal blood. However, in the synepitheliochorial placenta of the sheep the maternal capillary endothelium and uterine epithelium remain intact and fetal binucleate cells migrate and fuse with the uterine epithelium, creating a syncytium of mixed maternal and fetal origin.Reference Burton, Kaufmann and Huppertz12, Reference Wooding and Flint13

Fig. 1 The placental barrier in the human term placenta. The figure represents a cross-section of the human placenta. The insert to the right shows a schematic illustration of the placental barrier, which at term mainly consists of the syncytiotrophoblast (ST) cell and the fetal capillary (FC) endothelial cell. Of these structures, it is primarily the two polarized syncytiotrophoblast plasma membranes, the microvillous plasma membrane (MVM) and the basal plasma membrane (BPM) that restrict the transfer of molecules like ions and amino acids. N, nucleus of syncytiotrophoblast cell; IVS, intervillous space; SA, spiral artery; VT, villous tree; UC, umbilical cord. Reproduced by permission from Elsevier Ltd.
Net maternal–fetal transfer is influenced by a multitude of factors. These include utero-placental and umbilical blood flows, available exchange area, barrier thickness, placental metabolism, concentration gradients and transporter expression/activity in the placental barrier. Placental transfer of highly permeable molecules such as oxygen is non-mediated and particularly influenced by changes in barrier thickness, concentration gradients, placental metabolism and blood flow.Reference Carter14 In contrast, the rate-limiting step for maternal–fetal transfer of many ions and nutrients, such as amino acids, is the transport across the two plasma membranes of the syncytiotrophoblast, which express a large number of transporter proteins. Thus, changes in expression or activity of placental nutrient and ion transporters in response to altered maternal nutrition may influence fetal nutrient availability and growth. Regulation of placental nutrient transporters may therefore constitute a link between maternal nutrition and developmental programming.
In this review, we will focus on changes in transporter activity determined in vitro and transplacental transport measured in vivo. Furthermore, we will discuss factors circulating in maternal and fetal blood and placental signaling pathways that have been shown to regulate key placental nutrient transporters. A detailed discussion of general mechanisms of maternal–fetal exchange, placental blood flow, metabolism, energy availability and ion gradients, all factors affecting placental transport indirectly, is beyond the scope of this paper and have been reviewed elsewhere.Reference Jansson and Powell15–Reference Atkinson, Boyd and Sibley18
Placental transport in response to maternal undernutrition: two models
There are two fundamentally different, but not mutually exclusive, models for how the placenta responds to changes in maternal nutrition (Fig. 2). In the placental nutrient sensing model,Reference Jansson, Pettersson and Haafiz3, Reference Rosario, Jansson and Kanai8, Reference Jansson and Powell19 the placenta responds to maternal nutritional cues, resulting in downregulation of placental nutrient transporters in response to maternal undernutrition or restricted utero-placental blood flow. As a result, fetal nutrient availability is decreased and intrauterine growth restriction (IUGR) develops (Fig. 2). Placental nutrient sensing therefore represents a mechanism by which fetal growth is matched to the ability of the maternal supply line to allocate resources to the fetus. In this model, changes in placental growth and nutrient transport directly contribute to or cause altered fetal growth. On the other hand, predominantly based on elegant mouse studies it has been proposed that placental function is primarily controlled by fetal demand.Reference Sibley, Brownbill, Dilworth and Glazier20–Reference Angiolini, Coan and Sandovici22 In response to maternal undernutrition or restricted utero-placental blood flow, resulting in decreased placental transfer and limited fetal nutrient availability, the fetal demand model predicts that the fetus signals to the placenta to upregulate placental growth and nutrient transport (Fig. 2). This model represents a classical homeostatic mechanism by which the fetus compensates for changes in nutrient availability by regulating nutrient supply (i.e. placental transport) in the opposite direction.
Fig. 2 Placental nutrient transport in response to maternal undernutrition: two models. Schematic representation of the two models proposed for the regulation of placental function in response to maternal undernutrition (see text for details). The fetal demand model (bottom) predicts that the fetus signals to the placenta to upregulate placental growth and nutrient transport to meet fetal nutritional demands. In the placental nutrient sensing model (top), the placenta responds to maternal nutritional cues, resulting in downregulation of placental nutrient transporters, which leads to decreased fetal nutrient availability and intrauterine growth restriction.
In the subsequent sections, we will discuss the evidence for these two models and explore maternal and fetal nutritional cues that may be important regulating placental growth and nutrient transport. Subsequently, we will present a model in which fetal demand and placental nutrient sensing are integrated.
Decreased maternal nutrient availability
There is a wealth of information on the impact of impaired placental blood flow on placental transport functions in humans. However, no studies are available exploring the effects of maternal undernutrition on placental transport in pregnant women. In contrast, the placental response to maternal nutrient restriction has been investigated in some detail in animal models.
Studies in humans
In general, maternal undernutrition throughout pregnancy inhibits placental growth as shown by detailed studies of pregnancy outcomes during and after the Dutch famine 1944–1945.Reference Lumey23 However, maternal undernutrition restricted to first trimester resulted in increased placental weight at term.Reference Lumey23 The effects of maternal dietary restriction on placental transport in pregnant women are unknown. In contrast, there is an abundance of data, predominantly obtained in vitro, describing changes in placental transport capacity in pregnancies complicated by IUGR (Table 1).Reference Jansson and Powell19, Reference Sibley, Turner and Cetin24–Reference Cetin and Alvino26
Table 1 Placental transport in human IUGR
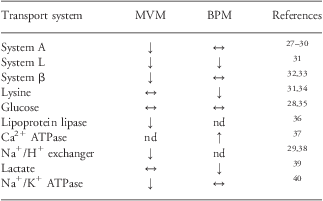
IUGR, intrauterine growth restriction; MVM, microvillous plasma membrane; BPM, basal plasma membranes; nd, not determined.
Increased (↑), unaltered (↔) or reduced (↓) transporter activity in MVM and BPM isolated from human pregnancies complicated by IUGR at term as compared with appropriate-for-gestational age controls.
In most of these studies, IUGR was caused by ‘placental insufficiency’, suggesting that the primary defect might have been a failure in the normal increase of utero-placental blood flow with advancing gestation. A subgroup of IUGR fetuses are hypoglycemic in utero,Reference Economides and Nicolaides41 however, this appears not to be due to a decreased transport capacity for glucose across the placental barrier.Reference Jansson, Ylvén, Wennergren and Powell28, Reference Jansson, Wennergren and Illsley35 In contrast, restricted fetal growth due to maternal hypoxemia at high altitude may be associated with decreased placental glucose transport capacity, as indicated by downregulation of glucose transporter (GLUT) expression in BPM.Reference Zamudio, Torricos and Fik42
System A is a Na+-dependent transporter mediating the cellular uptake of non-essential neutral amino acids.Reference Mackenzie and Erickson43 System A activity establishes the high intracellular concentration of amino acids like glycine, which is used to exchange for extracellular essential amino acids via System L. Thus, System A activity is critical for placental transport of both non-essential and essential amino acids. System A activity has consistently been reported to be decreased in the MVM, the rate-limiting step in transplacental amino acid transfer, isolated from IUGR placentas.Reference Dicke and Verges27–Reference Mahendran, Donnai and Glazier30 Furthermore, the most severe cases of IUGR, as defined by abnormal pulsatility index in the umbilical artery and abnormal fetal heart rate tracings, are associated with the most pronounced decreases in MVM System A activity.Reference Glazier, Cetin and Perugino29 In contrast to these findings in ‘idiopathic’ IUGR, Shibata et al.Reference Shibata, Hubel and Powers44 reported that placental System A activity, as measured in villous explants, was not altered in placentas of small-for-gestational age (SGA) babies in pregnancies complicated by preeclampsia.
The mechanisms underlying these interesting differences between IUGR/SGA pregnancies with and without preeclampsia remain to be established. However, the difference may be related to the observation that preeclampsia is characterized by increased maternal levels of hormones, including insulin and leptin, which are well established to stimulate placental System A activity in vitro.Reference Jansson, Greenwood, Johansson, Powell and Jansson45, Reference von Versen-Hoynck, Rajakumar, Parrott and Powers46 A recent report demonstrated that homocysteine is a competitive inhibitor of System A transport.Reference Tsitsiou, Sibley and D'Souza47 Thus, despite the unchanged in vitro System A activity in placentas of SGA babies from pregnancies complicated by preeclampsia,Reference Shibata, Hubel and Powers44 it is possible that increased circulating maternal levels of homocysteine observed in this syndrome may decrease placental System A activity in vivo.
The activities of transporters of essential amino acids, such as System β (transporting taurine) and System L (mediating the uptake of a range of essential amino acids including leucine) are reduced in MVM and/or BPM isolated from IUGR placentas (Table 1). These in vitro findings are consistent with stable isotope studies in pregnant women demonstrating that placental transfer of the essential amino acids leucine and phenylalanine is reduced in IUGR at term.Reference Marconi, Paolini and Stramare48, Reference Paolini, Marconi and Ronzoni49 Furthermore, a reduced placental capacity to transport amino acids is in agreement with studies showing reduced circulating amino acids, in particular essential amino acids, in IUGR fetuses.Reference Cetin, Corbetta and Sereni50–Reference Economides, Nicolaides, Gahl, Bernadini and Evans52 The activity of MVM lipoprotein lipase (LPL), which mediates the first critical step in transplacental transfer of free fatty acids (FFA), is reduced in IUGR.Reference Magnusson, Waterman, Wennergren, Jansson and Powell36 These data are in line with clinical studies showing lower fetal/maternal plasma ratios for long-chain polyunsaturated fatty acids (LCPUFAs) in IUGR.Reference Cetin, Giovannini and Alvino53
Key placental ion transporters are also affected when fetal growth is restricted. The activities of Na+/K+-ATPase, the Na+/H+ exchanger and lactate transporters are downregulated in IUGR.Reference Glazier, Cetin and Perugino29, Reference Johansson, Jansson, Glazier and Powell38–Reference Johansson, Karlsson, Wennergren, Jansson and Powell40 These membrane transport systems are involved in pH regulation, vectorial Na+ transport and maintenance of the Na+ gradient that drives the transport of other vital nutrients such as amino acids. Some ions, however, appear to be regulated quite differently. In particular, Ca2+-ATPase is upregulated in BPM isolated from IUGR placentas.Reference Strid, Bucht, Jansson, Wennergren and Powell37
In summary, these studies show a downregulation of key placental transporters for amino acids, lipids and ions in human IUGR. However, most of these studies were performed at term, or in a few cases using tissue obtained from pre-term deliveries in third trimester,Reference Jansson, Ylvén, Wennergren and Powell28, Reference Johansson, Jansson, Glazier and Powell38 and it is possible that compensatory changes consistent with fetal demand signals may be present earlier in pregnancy. Furthermore, the distinct upregulation of BPM Ca2+-ATPase activity in IUGR placentasReference Strid, Bucht, Jansson, Wennergren and Powell37 may represent a compensatory activation of the placental calcium transport system stimulated by an increased fetal demand. Despite these caveats, the available information from IUGR in humans is in general agreement with the placental nutrient sensing model for regulation of placental transporters.
Studies in animal models
The effect of maternal undernutrition on placental growth in animal models appears to depend on the species under study and the timing, duration, type and degree of nutrient restriction. For example, in sheep, a 50% calorie restriction during the first half of pregnancy increased placental weights at term.Reference Heasman, Clarke, Firth, Stephenson and Symonds54 Similarly, a 50% reduction in protein intake in rats starting 2 weeks before pregnancy and maintained throughout gestation resulted in higher placental weights close to term.Reference Langley-Evans, Gardner and Jackson55 In contrast, 30% calorie restriction throughout pregnancy in the baboon reduced placental weights by 18% near term.Reference Schlabritz-Loutsevitch, Ballesteros and Dudley56 Similarly, 40% calorie restriction from gestational days (GDs) 25 to 65 in the guinea pig,Reference Dwyer, Madgwick, Crook and Stickland57 50% reduction in calorie intake in the second half of pregnancy in the ratReference Belkacemi, Chen, Ross and Desai58 and 75% protein restriction in the rat caused placental growth restriction.Reference Jansson, Pettersson and Haafiz3, Reference Malandro, Beveridge, Kihlberg and Novak4
Studies in the non-human primate and in the rat indicate that maternal undernutrition downregulates placental nutrient transporter expression and activity. Preliminary observations show that 30% global maternal nutrient restriction from GD 30 in the baboon results in downregulation of MVM amino acid and GLUT isoforms close to term (GD 165, term = 184) and decreased circulating fetal levels of essential amino acids.Reference Kavitha, Nathanielsz and McDonald59 A number of studies in the rat, employing in vivo measurements of transplacental transfer of isotope-labeled substrate analogs, have shown that placental capacity to transport neutral amino acids and glucose in response to calorie or protein restriction is decreased in late pregnancy.Reference Rosso60–Reference Varma and Ramakrishnan63 In contrast, Ahokas et al.Reference Ahokas, Lahaye, Anderson and Lipshitz64 found no significant change in in vivo placental amino acid transport near term in rats subjected to 50% calorie restriction. However, other investigators using a similar protocol have reported downregulation of placental GLUT3Reference Belkacemi, Jelks, Chen, Ross and Desai65, Reference Lesage, Hahn and Leonhardt66 and sodium-dependent neutral amino acid transporter (SNAT)1 and 2 protein expressionReference Belkacemi, Jelks, Chen, Ross and Desai65 and upregulation of placental SNAT4 protein expression.Reference Belkacemi, Jelks, Chen, Ross and Desai65
Protein restriction in pregnant rats have been shown to decrease the in vitro activity of specific placental amino acid transporters close to term.Reference Malandro, Beveridge, Kihlberg and Novak4 Using the same model, we studied placental transport in the unstressed chronically catheterized animal at GDs 15, 18, 19 and 21 (term at GD 23), and reported that downregulation of the placental System A transporter activity precedes the occurrence of IUGR.Reference Jansson, Pettersson and Haafiz3 These findings suggest that, in this model, decreased placental amino acid transport is a cause of IUGR, rather than a consequence. Furthermore, MVM protein expression of specific System A (SNAT1 and 2) and System L (LAT1 and 2) amino acid transporter isoforms was decreased in response to a low-protein diet.Reference Rosario, Jansson and Kanai8 In contrast, maternal protein restriction did not affect placental glucose transport.Reference Jansson, Pettersson and Haafiz3 Notably, downregulation of placental amino acid transport was observed at GD 19, and there was no evidence of compensatory upregulation before this gestational age.Reference Jansson, Pettersson and Haafiz3, Reference Rosario, Jansson and Kanai8 These data indicate that fetal demand signals may not be present in this model, at least not from GD 15 and onwards. Overall, these observations in the baboon and rat are consistent with the placental nutrient sensing model for regulation of placental transporters.
A series of studies in mice have provided evidence for compensatory upregulation of placental nutrient transporters in response to maternal undernutrition.Reference Sferruzzi-Perri, Vaughan and Coan67–Reference Coan, Vaughan and McCarthy69 A 20% reduction in calorie intake from embryonic day (E)3 resulted in decreased placental but not fetal weight at E16 and reductions in both placental and fetal weights at E19. Placental gene expression of GLUT1 was decreased at E16, but increased at E19. At E19, placental gene expression of SNAT2 was found to be increased but SNAT4 gene expression was decreased.Reference Sferruzzi-Perri, Vaughan and Coan67, Reference Coan, Vaughan and Sekita68 Whereas placental transport capacity for glucose was maintained at E16 and E19, placental capacity to transport neutral amino acids was increased at E19.Reference Sferruzzi-Perri, Vaughan and Coan67, Reference Coan, Vaughan and Sekita68 In addition, Coan et al.Reference Coan, Vaughan and McCarthy69 explored the effect of a moderate (−22%) and severe (−61%) reduction in protein intake on placental transport function in mice in vivo. Whereas placental capacity to transport glucose was increased at E16 in both protein restriction groups, at E19 it was elevated only in the group subjected to severe protein restriction. In contrast, placental amino acid transport capacity was unchanged at E16 but decreased in the moderate protein restriction group at E19. Placental gene expression of GLUT1 was increased at E16 in the moderate, but not in the severe, protein restriction group, but was unaltered at E19. At E16 placental gene expression of SNAT2 was found to be increased in the severe protein restriction group, whereas at E19, SNAT1 gene expression was decreased in the severe restriction group and SNAT4 gene expression was reduced in both protein restriction groups.Reference Coan, Vaughan and McCarthy69 These studies suggest that placental nutrient transport appears to be regulated differently by maternal undernutrition in the mouse as compared with the non-human primate and the rat.
The distinct placental responses to maternal undernutrition in the mouse and the rat could reflect true species differences, but may also be related to subtle differences in the feeding paradigms. In addition, the tracer methodology used in all these studies is sensitive to differences in circulating concentrations of the endogenous substrate for the transporter under study. Thus, the marked hypoglycemia (27–58% lower glucose levels than controls) reported for mice subjected to 20% calorie restrictionReference Sferruzzi-Perri, Vaughan and Coan67, Reference Coan, Vaughan and Sekita68 or moderate/severe protein restriction,Reference Coan, Vaughan and McCarthy69 as well as a 32% reduction in maternal α-amino nitrogen in response to calorie restriction,Reference Sferruzzi-Perri, Vaughan and Coan67 could result in significant overestimation of transplacental transport of glucose and amino acids. Collectively, these studies in the mouse are in general agreement with the model that fetal demand signals play an important role in modulating placental nutrient transport in response to changes in maternal nutrition.
Because compromized utero-placental blood flow is believed to be involved in many clinical cases of IUGR secondary to placental insufficiency,Reference Miller, Turan and Baschat70 fetal outcomes and developmental programming have been extensively studied in animal models of restricted utero-placental blood flow. In some of these studies, placental transport functions have been assessed. Uterine artery ligation in the rat resulted in IUGR and decreased transplacental transport of glucose and amino acids in vivo.Reference Nitzan, Orloff and Schulman71 In contrast, neither the activity of the System A transporter measured in vitro in the maternal-facing plasma membrane of rat syncytiotrophoblastReference Glazier, Sibley and Carter72 nor the placental expression of GLUT1 and GLUT3Reference Reid, Lane, Flozak and Simmons73 were altered in this model. In guinea pigs, we performed unilateral uterine artery ligation in mid-pregnancy (GD 35) and determined placental blood flows and transport of neutral amino acids and glucose at GDs 44, 50 and 63 (term at GD 68) in chronically catheterized non-stressed animals.Reference Jansson and Persson74 At GD 44, modest IUGR was observed and placental capacity to transfer glucose and amino acids was maintained, whereas IUGR was more severe and placental capacity to transport amino acids was decreased at GD 50 and 63.Reference Jansson and Persson74 Saintonge and RossoReference Saintonge and Rosso75 studied placental blood flow and placental transport in relation to normal variations in fetal and placental growth in the guinea pig. They reported that placental capacity to transport glucose and amino acids was maintained over the range of fetal weights with the important exception of the smallest fetuses in which placental capacity to transport amino acids was decreased.Reference Saintonge and Rosso75 Naturally occurring ‘runts’ in the guinea pig therefore have the same decrease in placental amino acid transport capacity as experimentally induced IUGR.Reference Jansson and Persson74 These observations are in contrast to intra-litter variations in placental nutrient transport and fetal growth in mice, where placental amino acid transport capacity and SNAT2 expression have been reported to be increased in the smallest placentas.Reference Coan, Angiolini and Sandovici76
There are numerous approaches to induce IUGR in the sheep. A model involving exposure of the ewe to high ambient temperature, which decreases utero-placental blood flow and placental growth resulting in asymmetric IUGR, resembles placental insufficiency in humans.Reference Barry, Rozance and Anthony77 Because maternal and fetal vessels in the sheep are accessible to chronic catheterization, allowing for precise measurements of nutrient fluxes across the placenta, a body of information on placental nutrient transport in this model is available. For example, the placental capacity to transport glucose,Reference Thureen, Trembler, Meschia, Makowski and Wilkening78 leucine,Reference Ross, Fennessey, Wilkening, Battaglia and Meschia79 threonineReference Anderson, Fennessey, Meschia, Wilkening and Battaglia80 and aminocyclopentane-1-carboxylic acid (ACP)Reference de Vrijer, Regnault, Wilkening, Meschia and Battaglia81 (a branched-chain amino acid analog) is reduced in this IUGR model. Taken together, studies of utero-placental insufficiency and IUGR in a range of animal models show that placental nutrient transport is downregulated. These findings are reminiscent of the human data and support the placental nutrient sensing model.
Effects of altered levels of micronutrients on placental transport have received little attention, with the possible exception of maternal iron deficiency, which results in maternal and fetal anemia and IUGR.Reference Crowe, Dandekar and Fox82, Reference Godfrey and Barker83 However, fetal anemia typically is less severe than maternal anemia suggesting compensatory mechanisms, possibly at the placental level. Indeed, maternal iron deficiency in the rat results in upregulation of the placental transferrin receptor, which is expressed in the trophoblast maternal-facing plasma membrane and mediates iron uptake into the placenta. Furthermore, maternal iron deficiency increases the expression of placental divalent metal transporter 1 (DMT1), which transports iron out of the lysosome into the cytoplasm of the trophoblast.Reference Gambling, Danzeisen and Gair84 It is likely that iron itself represents the signal mediating these changes in placental expression because iron-responsive elements are present in both the transferrin receptor and DMT1 genes. However, whether other signals, such as local hypoxia or signals originating in the fetus, are also involved remain to be established.
Increased maternal nutrient availability
Most human and animal studies of the effect of increased maternal nutrient availability on placental transport have been focused on diabetes, whereas maternal obesity has attracted much less attention.
Studies in humans
Diabetes in pregnancy, especially if poorly controlled, is associated with intermittently elevated maternal levels of glucose, amino acids and FFA and can therefore be regarded as a condition of increased nutrient availability. Although many studies in pregnant women with diabetes indicate an increased placental capacity to transfer nutrients, data is less consistent than for decreased maternal nutrient availability. Pregnancy can be complicated by type 1, type 2 or gestational diabetes (GDM), and of these conditions GDM is the most common affecting 2–10% of all pregnancies in the United States. However, the prevalence of GDM is expected to increase by two- to three-fold if the new diagnostic criteria of the Hyperglycemia and Adverse Pregnancy Outcome study is fully adopted.Reference Metzger, Lowe and Dyer85 With the exception of subgroups of women with type-1 diabetes who develop vascular complications, diabetes in pregnancy, in particular GDM, is associated with fetal overgrowth.Reference Metzger, Lowe and Dyer85 Placental nutrient transport capacity in diabetes associated with fetal overgrowth has been studied in isolated syncytiotrophoblast plasma membranes (Table 2).
Table 2 Placental transport in fetal overgrowth in association to maternal diabetes
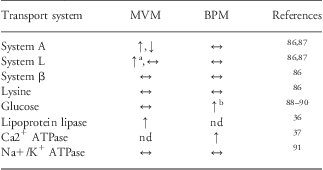
MVM, microvillous plasma membrane; BPM, basal plasma membrane; nd, not determined.
Transporter activity per mg of membrane protein was measured in isolated MVM and BPM vesicles. The table shows the transport activity in cases of fetal overgrowth in relation to gestational age matched appropriately grown controls: increased (↑), unaltered (↔) or reduced (↓) transporter activity.
aOnly gestational diabetes.
bOnly type-1 diabetes.
Available data on trophoblast amino acid transporter activities in pregnancies complicated by maternal diabetes are inconsistent. Dicke and HendersonReference Dicke and Henderson92 found no differences in the uptake of neutral amino acids into MVM isolated from GDM pregnancies as compared with controls, however, these subjects did not give birth to larger babies. System A amino acid transport activity was reduced and System L transport activity unaltered in MVM isolated from pregnancies with type-1 diabetes and fetal overgrowth.Reference Kuruvilla, D'Souza and Glazier87 In contrast, we found that the activity of MVM System A transporter was increased in type-1 diabetes, independent of fetal overgrowth, and placental transport of leucine was increased in GDM.Reference Jansson, Ekstrand, Björn, Wennergren and Powell86 These discrepant findings may be related to differences in methodology or in study populations. Notably, although birth weights were similar in the two latter reports, placental weights were 100–300 g higher in the diabetic groups in the Swedish study.Reference Jansson, Ekstrand, Björn, Wennergren and Powell86 This may indicate that the two study populations differ in some fundamental way with regard to, for example, ethnicity, nutrition or clinical management.
BPM glucose transport activity and GLUT1 expression are increased in type-1 diabetes,Reference Jansson, Wennergren and Powell89, Reference Gaither, Quraishi and Illsley90 which could enhance placental glucose transport even during normoglycemia. Indeed, these changes have been proposed to contribute to fetal overgrowth in type-1 diabetes with apparent optimal glucose control.Reference Jansson, Wennergren and Powell89 Recently, it was reported that the protein expression of GLUT9 is upregulated in MVM and BPM isolated from placentas of women with diabetes,Reference Bibee, Illsley and Moley93 adding to the evidence of increased placental glucose transport capacity in this pregnancy complication. On the other hand, using placental lobuli perfused in vitro, Osmond et al.Reference Osmond, Nolan, King, Brennecke and Gude94 showed that placental glucose transport was decreased in GDM pregnancies with normal fetal growth; however, these changes were normalized in GDM women treated with insulin.Reference Osmond, King, Brennecke and Gude95 It has been suggested that GLUT abundance in the placental barrier does not affect transplacental glucose transport because glucose uptake varies with placental and umbilical blood flow.Reference Desoye, Gauster and Wadsack96 Notwithstanding that changes in blood flow can alter placental glucose transport, this view may be too simplistic. BPM has much lower surface area and GLUT1 expression as compared with MVM, and it has therefore been proposed that the transfer across BPM, at least to some extent, limits the diffusion of glucose across the barrier.Reference Jansson, Wennergren and Illsley35 Therefore, with all other factors kept constant, any alterations in GLUT expression/activity in the BPM is likely to alter glucose flux across the barrier.
Maternal lipoproteins are the predominant source for fetal supply of FFA. Triglyceride hydrolases in the MVM of the syncytiotrophoblast release FFA from maternal lipoproteins, allowing them to be transported across the placental barrier mediated by plasma membrane fatty acid transporters (FATP) and cytosolic fatty acid-binding proteins (FABP).Reference Gil-Sanchez, Koletzko and Larque97 Although there is some controversy with respect to which type of triglyceride hydrolase constitutes the major MVM lipase activity, LPL and endothelial lipase (EL) are probably the two key hydrolases.Reference Desoye, Gauster and Wadsack96, Reference Gil-Sanchez, Koletzko and Larque97 The activity of placental LPL has been reported to be increased in type-1 diabetes associated with fetal overgrowth.Reference Magnusson, Waterman, Wennergren, Jansson and Powell36 Furthermore, FABP1 protein expression was upregulated in the placenta of both GDM and type-1 diabetic women giving birth to large babies.Reference Magnusson, Waterman, Wennergren, Jansson and Powell36 Lindegaard et al. reported increased placental mRNA expression for EL and hormone-sensitive lipase, but not for LPL, in type-1 diabetes associated with poor metabolic control and fetal overgrowth.Reference Lindegaard, Damm, Mathiesen and Nielsen98 Moreover, placental expression of FABP4Reference Scifres, Chen, Nelson and Sadovsky99 and ELReference Gauster, Hiden and van Poppel100 is elevated in pregnancies of obese women with GDM. These observations are consistent with an increased placental capacity to supply lipids to the fetus in maternal diabetes, however, considering the complexity of placental lipid transport much more work is needed to draw firm conclusions. In addition to the total amount, the FFA composition of lipids made available to the fetus is of critical importance for fetal development. Indeed, the content of LCPUFAs in plasma phospholipids has been reported to be decreased in fetuses of mothers with GDM,Reference Wijendran, Bendel and Couch101 implicating a decreased supply of these fatty acids. Altogether, the data on placental nutrient transport in pregnancies complicated by diabetes is variable. However, the capacity to transport FFA and, possibly, glucose may be increased in diabetic women, in broad agreement with the placental nutrient sensing model.
The effect of maternal overweight and obesity on placental function in women without diabetes remains largely unknown.Reference Higgins, Greenwood, Wareing, Sibley and Mills102 More than half of all US women enter pregnancy overweight or obese,Reference Chu, Kim and Bish103 representing one of the most daunting problem in obstetrical practice of today. It is well established that high pre-pregnancy body mass index (BMI) is strongly associated to fetal overgrowth.Reference Sebire, Jolly and Harris104–Reference Ehrenberg, Mercer and Catalano106 Farley et al.Reference Farley, Choi and Dudley107 reported decreased System A amino acid transport activity in placental villous fragments isolated from placentas of obese Hispanic women giving birth to normal sized babies. In contrast, preliminary studies in our laboratory show that System A activity is unaltered in MVM isolated from placentas of women with high BMI in the same population.Reference Gaccioli, Jansson and Powell108 Furthermore, our preliminary data on Swedish women with varying pre-pregnancy BMI indicate that System A, but not System L, amino acid transport activity is increased in MVM isolated from placentas of obese women giving birth to large babies.Reference Jansson, Jones and Schumacher109 Dube et al.Reference Dube, Gravel and Martin110 recently reported increased placental LPL activity and gene and protein expression of CD36 in obese mothers giving birth to normal sized babies. On the other hand, placental expression of FATP4, FABP1 and 3 was decreased in placentas of obese women.Reference Dube, Gravel and Martin110 However, protein expression studies and LPL activity measurements in this study were done using placental homogenates, which may not represent changes in syncytiotrophoblast plasma membranes. Taken together, additional data is needed to allow firm conclusions with respect to the impact of maternal obesity on placental nutrient transport.
Studies in animal models
Reports on placental nutrient transport in animal models of diabetes lack consistency. Diabetes in pregnancy has been extensively studied in rodent models using surgical, chemical and genetic approaches to induce the disease.Reference Jawerbaum and White111 Of these methods, administration of streptozotocin (STZ), which selectively destroys pancreatic β-cells and reduces circulating insulin resulting in hyperglycemia, has been widely employed as a model of type-1 diabetes. However, at least in earlier studies, this model was associated with severe maternal hyperglycemia raising questions with respect to its relevance to pregnant women with diabetes. Furthermore, utero-placental blood flow has been reported to be reduced in rats with STZ-induced diabetesReference Palacin, Lasuncion, Martin and Herrera112, Reference Eriksson and Jansson113 sometimes resulting in IUGR, complicating the interpretation of placental nutrient transport measurements in the context of increased maternal nutrient availability. Nevertheless, placental transport capacity for neutral amino acids has been shown to be decreased in STZ-treated rats.Reference Copeland and Porterfield114 Placental expression of GLUT1 is downregulatedReference Ogura, Sakata, Yamaguchi, Kurachi and Murata115 or unchangedReference Devaskar, Devaskar and Schroeder116 in mice with STZ-induced diabetes, whereas placental GLUT3 expression is increased in this model in rats.Reference Boileau, Mrejen, Girard and Hauguel-de-Mouzon117 Transplacental glucose transport capacity in STZ rats in vivo has been reported to be decreased, unchanged or increased.Reference Palacin, Lasuncion, Martin and Herrera112, Reference Herrera, Palacin, Martin and Lasuncion118, Reference Thomas and Lowy119 In addition, fatty acid transfer in STZ rats has been shown to be increased or decreased.Reference Honda, Lowy and Thomas120–Reference Goldstein, Levy and Shafrir122 It is likely that the variable results on placental transport in STZ-treated rodents are related to differences in the severity of metabolic disturbance, variable effects on utero-placental blood flow and differences in methodological approaches between studies.
The impact of maternal obesity on placental transport has yet to be systematically described in well-characterized animal models. The effect of a maternal high-fat diet and/or obesity on fetal development has been explored extensively in a variety of animal models.Reference Li, Sloboda and Vickers123, Reference Armitage, Khan, Taylor, Nathanielsz and Poston124 However, the maternal phenotype of these studies has received very little attention and it is therefore not entirely clear to which extent these models resemble obesity in pregnant women. Indeed, in many of these paradigms fetal growth is restricted, which is not the typical clinical outcome in humans.Reference Sebire, Jolly and Harris104, Reference Baeten, Bukusi and Lambe105 One explanation for the development of IUGR in animal models of obesity is reduced utero-placental blood flow, which has been reported for overnourished adolescent sheepReference Wallace, Bourke, Aitken, Milne and Hay125 and in chronically high-fat-fed non-human primates.Reference Frias, Morgan and Evans126 Overnutrition of the adolescent sheep is associated with an unaltered placental glucose transport capacity.Reference Wallace, Bourke, Aitken, Milne and Hay125 In adult obese pregnant sheep provided 150% of the normal calorie intake, fetal growth was enhanced at mid-gestation but fetal weight was not different as compared with the controls close to term.Reference Zhu, Ma, Long, Du and Ford7 Interestingly, there was a marked upregulation of placental expression of FATP and increased fetal blood triglycerides in this model, in particular at mid-gestation.Reference Zhu, Ma, Long, Du and Ford7
We explored a mouse model in which female mice were given a high-fat diet (32%) for 8 weeks and subsequently mated.Reference Jones, Woollett and Barbour127 Dams continued their diet during pregnancy and they were studied at GD 18.5. It was demonstrated that this approach resulted in a modest increase in maternal adiposity but not obesity, a metabolic profile resembling the obese pregnant woman, without evidence of diabetes. Importantly, this paradigm resulted in a fetal overgrowth and in vivo transport studies demonstrated marked increases in placental clearances of both 3H-methyl-glucose and 14C-MeAIB in response to the high-fat diet. The increase in placental clearance rates was associated with a significant increase in GLUT1 and SNAT2 expression.Reference Jones, Woollett and Barbour127 In a slightly different approach, Rebholz et al.Reference Rebholz, Burke, Yang, Tso and Woollett128 fed female mice a diet containing 16% fat diet for 4 weeks and animals were subsequently mated, which did not affect the adiposity or leptin levels of the dam but resulted in increased fetal weights close to term without affecting MVM GLUT1 expression. Collectively, placental transport data from animal models of obesity is still too scant to be applied to the fetal demand and placental nutrient sensing models.
Mechanisms regulating placental transport in response to changes in maternal nutrition
A detailed and full account of the mechanisms known to regulate placental transport is beyond the scope of this overview and the reader is referred to recent reviews.Reference Atkinson, Boyd and Sibley18, Reference Jones, Powell and Jansson129, Reference Lager and Powell130 Instead, we will briefly discuss factors reported to be altered in response to changes in maternal nutrition and also shown to regulate placental transport.
Under and overnutrition elicit changes in maternal metabolism and levels of circulating hormones, which may regulate placental function. Maternal protein restriction in the ratReference Jansson, Pettersson and Haafiz3 and calorie restriction in the mouseReference Sferruzzi-Perri, Vaughan and Coan67 are associated with decreased maternal plasma insulin, insulin-growth factor (IGF)-I and leptin. Furthermore, Sferruzzi-Perri et al.Reference Sferruzzi-Perri, Vaughan and Coan67 demonstrated that a 20% restriction in total calorie intake in mice elevated maternal corticosterone levels. Calorie restriction in non-pregnant humans and animals typically increases serum concentrations of adiponectin.Reference Longo and Fontana131 Maternal serum concentrations of IGF-I are decreased in human IUGRReference Holmes, Montemagno and Jones132 and some studies indicate that maternal serum leptin concentrations are reduced in this pregnancy complication.Reference Yildiz, Avci and Ingec133 In addition, placental insulin receptor number,Reference Potau, Riudor and Ballabriga134 placental insulin/IGF-I signaling activityReference Laviola, Perrini and Belsanti135 and placental leptin productionReference Lea, Howe and Hannah136 are reduced in IUGR. On the other hand, maternal overnutrition appears to result in the opposite hormonal changes. For example, obese pregnant women typically have higher serum levels of leptin, insulin, IGF-I and interleukin-6 and decreased serum concentrations of adiponectin as compared with pregnant women with normal pre-pregnancy BMIReference Ramsay, Ferrell and Crawford137, Reference Jansson, Nilsfelt and Gellerstedt138 and similar changes are observed in GDM.Reference Lauszus, Klebe and Flyvbjerg139 Furthermore, circulating maternal leptin was found to be increased and adiponectin decreased in our pregnant mice fed a high-fat diet,Reference Jones, Woollett and Barbour127 consistent with obese pregnant women.Reference Jansson, Nilsfelt and Gellerstedt138 Thus, maternal undernutrition results in a catabolic hormonal profile, whereas overnutrition causes changes in maternal hormones that promote anabolism.
The significance of these changes in the levels of maternal hormones and cytokines in response to nutrition is that these factors have been shown to regulate placental nutrient transport. For example, IGF-I,Reference Karl140 insulin,Reference Jansson, Greenwood, Johansson, Powell and Jansson45, Reference Karl, Alpy and Fischer141 leptinReference Jansson, Greenwood, Johansson, Powell and Jansson45 and cytokinesReference Jones, Jansson and Powell142 stimulate, whereas adiponectin inhibits trophoblast amino acid transporter activity.Reference Jones, Jansson and Powell143 For IGF-I and adiponectin, these findings have also been confirmed in vivo in the rodent.Reference Rosario, Schumacher and Jiang144, Reference Sferruzzi-Perri, Owens and Standen145 Furthermore, administration of corticosteroids to pregnant mice inhibits placental System A activity.Reference Audette, Challis, Jones, Sibley and Matthews146 It is important to note that receptors for many polypeptide hormones on the syncytiotrophoblast cell, including receptors for insulin, IGF-I and leptin,Reference Desoye, Hartmann and Blaschitz147–Reference Fang, Furesz, Lurent, Smith and Fant149 are predominantly expressed in the MVM, and therefore directly exposed to maternal blood. Thus, it is likely that syncytiotrophoblast nutrient transporters are mainly regulated by maternal rather than fetal hormones.
It is reasonable to assume that maternal under and overnutrition are associated with changes in placental nutrient, oxygen and energy levels, which can regulate nutrient sensors in the placenta. Signaling pathways involved in placental nutrient sensing may include the amino acid response signal transduction pathway, AMP-activated kinase, glycogen synthase-3, the hexosamine signaling pathway and mammalian target of rapamycin complex 1.Reference Jansson, Aye and Goberdhan150 Of these nutrient sensors, mTORC1 signaling may be of particular importance in linking maternal nutrition to placental nutrient transport. First, placental insulin/IGF-I signaling and fetal levels of oxygen, glucose and amino acids are altered in pregnancy complications such as IUGR,Reference Economides and Nicolaides41, Reference Cetin, Corbetta and Sereni50, Reference Laviola, Perrini and Belsanti135, Reference Yung, Calabrese and Hynx151 and all these factors are well-established upstream regulators of mTORC1.Reference Zoncu, Efeyan and Sabatini152 Furthermore, mTORC1 is a positive regulator of placental amino acid transporters,Reference Roos, Kanai, Prasad, Powell and Jansson153, Reference Roos, Jansson and Palmberg154 suggesting that trophoblast mTORC1 modulates amino acid transfer across the placenta. In addition, placental mTORC1 signaling activity is changed in pregnancy complications associated with altered fetal growth and in animal models in which maternal nutrient availability has been altered experimentally. For example, placental mTORC1 activity is inhibited in human IUGRReference Yung, Calabrese and Hynx151, Reference Roos, Jansson and Palmberg154 and preliminary studies indicate an activation of placental mTORC1 signaling in association with maternal obesity.Reference Jansson, Jones and Schumacher109, Reference Gaccioli, Jansson and Powell155 Furthermore, placental mTORC1 activity has been reported to be decreased in hyperthermia-induced IUGR in the sheep,Reference Arroyo, Brown and Galan156 in response to a maternal low-protein diet in the ratReference Rosario, Jansson and Kanai8 and maternal calorie restriction in the baboon.Reference Kavitha, Nathanielsz and McDonald59 Taken together, this evidence implicates mTORC1 signaling as an important placental nutrient sensor, which may constitute a critical link between maternal nutrient availability and fetal growth.
Placental signals originating from imprinted genes regulate nutrient transport in the mouse placenta.Reference Coan, Burton and Ferguson-Smith157 Imprinted genes are predominantly expressed from one of two parental alleles and in mice more than 70 imprinted genes have been discovered. A subgroup of these genes are imprinted only in the placenta and are involved in regulation of fetal and placental growth.Reference Coan, Burton and Ferguson-Smith157 An example of a paternally expressed/maternally repressed placental gene is igf-2. Reference Constancia, Hemberger and Hughes5 IGF-II regulates placental growth and therefore indirectly its transport capacity. Interestingly, Sferruzzi-Perri et al.Reference Sferruzzi-Perri, Vaughan and Coan67 have provided evidence to suggest that placental igf-2 plays a role in the placental response to maternal undernutrition in mice.Reference Sferruzzi-Perri, Vaughan and Coan67
Significant support for fetal demand signals regulating placental amino acid transport comes from studies of mice with placenta-specific knockout of igf-2. In this model, placental growth restriction occurs in mid-gestation and there is a temporary upregulation of placental System A amino acid transporter activity. This increased nutrient transport maintains fetal growth in the normal range until late pregnancy when compensatory mechanisms fail and IUGR develops.Reference Constancia, Hemberger and Hughes5, Reference Constancia, Angiolini and Sandovici21 Based on a comparison of the placental phenotype in complete igf-2 knockout mice and in mice with knockout of the placental-specific igf-2 only, it has been suggested that fetal IGF-II may be an important fetal demand signal.Reference Coan, Fowden and Constancia158 However, at least some studies in humans have shown that IGF-II levels are reduced in IUGR fetusesReference Smerieri, Petraroli and Ziveri159 and higher in large-for-gestational age fetuses,Reference Christou, Connors and Ziotopoulou160 which is not entirely consistent with IGF-II as a fetal demand signal. In human pregnancy, it is possible that fetal parathyroid hormone-related peptide regulates the activity of the calcium pump in the syncytiotrophoblast BPM.Reference Strid, Bucht, Jansson, Wennergren and Powell37, Reference Strid, Care, Jansson and Powell161 Additional indirect evidence for fetal regulation of placental transport functions comes from a study by Godfrey et al.Reference Godfrey, Matthews and Glazier162 showing that MVM System A amino acid transporter activity is inversely correlated to fetal size within the normal range of birth weights. Collectively, these observations are consistent with the model proposing that placental nutrient transporters are regulated by fetal demand; however, the nature and identity of the fetal signals remain to be fully established.
Placental nutrient sensing and fetal demand: an integrated model
In this review, we have focused on maternal, placental and fetal signals that may regulate placental transport in response to changes in maternal nutrition, which (when defined broadly) also can include compromized utero-placental blood flow. Because placental nutrient uptake/transport is intimately related to the growth of the placenta, it is likely that the signals that regulate nutrient uptake and transport in the placenta also affect placental growth. Furthermore, by releasing an array of hormones into the maternal circulation, the placenta governs the maternal physiological adaptation to pregnancy. It is therefore plausible that changes in placental endocrine function in response to altered maternal nutrition may regulate placental growth or transport functions indirectly by affecting maternal physiology, adding an additional level of complexity. In support of this concept, emerging evidence shows that placental-specific deletion of igf-2 increases maternal corticosterone and insulin levels and decreases plasma α-amino nitrogen.Reference Sferruzzi-Perri, Vaughan and Coan67
We propose a model in which the placenta integrates a multitude of maternal and fetal nutritional cues with information from intrinsic nutrient-sensing signaling pathways to balance fetal demand with the ability of the mother to support the pregnancy by regulating maternal physiology, placental growth and nutrient transport (Fig. 3). We argue that these mechanisms have evolved due to the evolutionary pressures of maternal undernutrition. Although these regulatory loops may function in the ‘reverse’ direction in response to overnutrition, it is possible that these responses may not be as readily apparent in maternal obesity or diabetes as in response to maternal undernutrition. Fetal demand signals are predicted to compensate for reduced nutrient availability by upregulation of placental nutrient capacity, which represents a homeostatic regulatory mechanism that is a sound strategy from an evolutionary perspective. However, the existence of maternal signals that in response to undernutrition will inhibit placental growth and nutrient transport (placental nutrient sensing) is equally important from an evolutionary point of view. Matching fetal growth to maternal resources in response to maternal undernutrition will produce an offspring that is smaller in size but who, in most instances, will survive and be able to reproduce. This reduced fetal growth is sometimes a better alternative than the fetus extracting all the nutrients needed for normal growth from an already deprived mother, thereby potentially jeopardizing both maternal and fetal survival. We speculate that the relative importance of placental nutrient sensing and fetal demand signals for the regulation of placental function may differ between species and depend on the type, duration and severity of the nutritional perturbation. For example, it is plausible that regulation by fetal demand signals dominates when the nutritional challenge is moderate and brief, whereas regulation by placental nutrient sensing may override fetal demand if the nutritional challenge is severe and prolonged.
Fig. 3 Placental nutrient sensing and fetal demand: an integrated model. We propose that the placenta integrates maternal and fetal nutritional cues with information from intrinsic nutrient sensors, such as mammalian target of rapamycin (mTOR) signaling. These signals then regulate placental growth and nutrient transport to balance fetal demand with the ability of the mother to support pregnancy. Thus, the placenta plays a critical role in modulating maternal–fetal resource allocation, thereby affecting fetal growth and the long-term health of the offspring. See text for detailed explanation. IGF-II, insulin-like growth factor II; PTHrp, parathyroid hormone-related peptide.
Conclusion and future perspectives
Our long-term health is critically dependent on the availability of nutrients during fetal life, which is determined by placental transport. The understanding of the role of the placenta in fetal nutrition has evolved from the view that the placenta constitutes a selective but passive filter to the recognition that the placenta adapts to changes in maternal nutrition by responding to maternal nutritional cues, fetal demand signals and intrinsic nutrient-sensing signaling pathways. The complexity of these regulatory pathways is only beginning to be appreciated. A better understanding of the molecular mechanisms regulating placental transport functions may help to identify critical links between maternal nutrition, fetal growth and developmental programming. In addition, this knowledge is essential when designing novel intervention strategies. However, currently our understanding of these processes is limited, at best, presenting great challenges and opportunities for the future. For example, there is a lack of information on the (1) molecular identity of fetal demand signals, (2) the mechanisms by which lipids are transported across the placenta and the role of placental lipid transport in programming of obesity and diabetes, (3) how multiple placental nutrient-sensing signaling pathways are integrated and (4) how signals between the placenta and the mother influence maternal–fetal resource allocation. Furthermore, additional animal models that are relevant for the human condition are needed, in particular for GDM and maternal obesity. Finally, attention on the influence of fetal sex, ethnicity, maternal age and parity on placental function is required in future studies.
Acknowledgments
Figure 1 is reproduced by permission from Elsevier Ltd; this figure was published in the chapter ‘Placental function and materno-fetal exchange’ in Fetal Medicine: Basic Science and Clinical Practice, 2 Ed, 2008, ISSN/ISBN 978-0-443-10408-4. Supported by DK089989 (TLP), HD065007 (TJ and TLP), HD068370 (TJ) and HD071306 (TJ).



