Introduction
The question of whether life arose on Mars has been widely discussed in the planetary community, partially because the conditions in its early history may have resembled those of the early Earth (McKay Reference McKay1997). However, the current harsh conditions on Mars, including energetic ultraviolet (UV) radiation, extreme dryness and the presumed oxidizing nature of the soil, are destructive to any organic material that is or was exposed to the Martian surface (Zent & McKay Reference Zent and McKay1994; Benner et al. Reference Benner, Devine, Matveeva and Powell2000). Although no organic material could be identified with the Viking Mars landers three decades ago (Biemann et al. Reference Biemann, Oro, Toulmin, Orgel, Nier, Anderson, Simmonds, Flory, Diaz and Rushneck1976; Biemann & Lavoie Reference Biemann and Lavoie1979), several space missions will visit the Martian surface in the coming decade to search for organic compounds as well as extinct and extant life. In this context, it is vital to obtain knowledge about the survival of organics in extreme conditions. Measuring the abundance and distribution of organic matter in Mars analogue samples collected in dry deserts is critical to our understanding of preservation.
Polycyclic aromatic hydrocarbons (PAHs) are among the most abundant organic molecules in space environments (Tielens Reference Tielens2008). Their high abundance and resistance to radiation and thermal degradation make them a key target in the search for organic material in the solar system. PAHs reach planetary surfaces through the impacts of small solar system bodies (Sephton & Botta Reference Sephton and Botta2005). Naphthalene, phenanthrene, pyrene and chrysene, for instance, have been found in the ALH84001 meteorite (McKay et al. Reference McKay, Gibson, Thomas-Keprta, Vali, Romanek, Clemett, Chillier, Maechling and Zare1996). PAHs are considered to be ubiquitous in terrestrial environments (Edwards Reference Edwards1983) and they originate from both natural and anthropogenic sources. PAHs are composed of polymerized six-member benzene rings and are probably the largest and the most structurally diverse class of organic compounds. The diversity of PAHs is caused in part by alkylation, mostly methylation (attachment of −CH3 groups) (Zolotov & Shock Reference Zolotov and Shock1999). The Gibbs energetic paths of PAH formation from pyrolysis, oxidation and dehydrogenation of other hydrocarbons are favourable, with dehydrogenation being the common path for PAH production from biogenic organic compounds. The complexity and diversity of environmental contaminants such as PAHs have resulted in the development of many different analytical techniques and protocols for their extraction and analysis. Analysis of organic compounds (including PAHs) in the environment often requires tedious sampling to achieve a proper picture of the level of contamination, and the complex matrices of soil samples have always been laborious to analyse. A liquid extraction may require large volumes of high-purity organic solvents, which must then be disposed of. Some of the more volatile substances may be lost during processing of the samples. With numerous sources of possible systematic errors inherent to complex analytical procedures, much of today's research is aimed at faster extractions, analysis methods and techniques for fast screening and field analysis.
Here we report the extraction and abundance measurements of PAHs from Mars soil analogues using solid-phase microextraction (SPME) and liquid extraction methods. SPME is a solvent-free extraction method invented by Pawliszyn (Berlardi & Pawliszyn Reference Berlardi and Pawliszyn1989; Pawliszyn Reference Pawliszyn1999) and applied in a variety of sampling-detection scenarios (Grote & Pawliszyn Reference Grote and Pawliszyn1997; Pawliszyn & Martos Reference Pawliszyn and Martos1997; Jia et al. Reference Jia, Koziel and Pawliszyn2000; Kataoka et al. Reference Kataoka, Lord and Pawliszyn2000; Lord & Pawliszyn Reference Lord and Pawliszyn2000; Pawliszyn Reference Pawliszyn2000; Wu et al. Reference Wu, Xie and Pawliszyn2000; Orzechowska & Paulson Reference Orzechowska and Paulson2005). When compared with traditional extraction methods, SPME may provide better analyte recoveries, less opportunity for rearrangement and decomposition of analytes, and faster analysis. Headspace analysis is a suitable method to determine volatile/semivolatile substances directly without chemical pretreatment of samples. We compare the analysis of PAHs in Mars soil analogues using SPME and liquid extraction methods and investigate PAH abundance and distribution in relation to soil properties.
Soil analogue samples and their geological context
Analogue samples had been collected in the vicinity of the Mars Desert Research Station (MDRS) by Crew 77 in February 2009 from different site locations (Table 1 and Fig. 1) as part of the EuroGeoMars campaign. Soils come from four geologic successions. In stratographic order, these are:
• Mancos Shale, Blue gate member
• Mancos Shale, Tununk member
• Dakota Sandstone
• Formation Bushy Basin member

Fig. 1. Map illustrating locations where soil samples were collected in the vicinity of Hanksville, Utah, February 2009. The exact positions of samples are in Table 1.
Table 1. Martian soil analogues analysed in this study; sampling locations, pH, texture and organic mass loss of the soil samples

* The preliminary soil analysis in this table employed a less sophisticated technique than that used in Kotler et al. 2011.
The Bushy Basin member – the first representative of the Morrison Formation in the area – was deposited in a massive flood plain (Williams & Hackman Reference Williams and Hackman1971; Fillmore Reference Fillmore2000). As the flooding waned, phyllosilicate and bentonite-rich shale were deposited on a massive mudflat that was occasionally cut by eastward-flowing streams. Sparse units of white, relatively fine-grained sandstone and brownish conglomerate record the passage of these streams through the mudflats. The Morrison Formation unit was deposited in the late Jurassic epoch and is fossiliferous, containing dinosaur remains in some locations. In the MDRS area, the Morrison Formation mudflats form rounded hills with a finely fractured bentonite surface resembling popcorn. The colourful hills exhibit outcrops of red, brown, purple and greyish-green layers on a dominantly buff-coloured base. The streambeds that transected the mudflats form prominent stacks, often capped with a conglomerate layer. Some examples of these form complete inverted streambed successions. Cross-bedded features are common with alternating lenses of fine-grained sandstone and conglomerates, reflecting the different flow velocities at different times. The fine-grained sandstones often contain round concretions 3–5 mm in diameter. The Dakota Sandstone caps sections of the Morrison Formation but is discontinuous in the MDRS area. This sandstone was deposited in the early Cretaceous period and constitutes the ancient marine shoreline of a seabed, with evidence of tidal currents and large agglomerations of fossil seashells (samples P-13 and P-14 were collected from the top of the hill above MDRS that contains shells down to 10 cm). Samples P-2 and P-3 are from the Morrison Formation/Dakota Sandstone boundary and soils may be derived from some mixture of these units. The Mancos Shale, deposited in a deep sea during the Cretaceous period, is up to 3000 ft thick and is dramatically exposed in the Factory Butte area where samples P-8 and P-10 were collected. The record shows four large-scale, sea-level fall and rise cycles. The Tununk member of the Mancos Shale was deposited in the deep reaches of a seaway, far from shore where a thick pile of black ooze accumulated. The Tununk was deposited as the sea deepened, whereas deposition of the Ferron Sandstone took place as the sea withdrew. In the next marine transgression, the Blue gate member of the Mancos Shale was deposited. Again, this was a deep-sea depositional environment where carbon-rich black ooze was deposited. The Mancos Shale is very rich in sulfides, which have weathered to sulfate near the surface. In many locations, gypsum crystals form on the surface of the soils and large (10–30 cm) concretions of gypsum are found embedded in the walls of the buttes.
The HOBO sample was collected from the Atacama Desert in the Arequipa region of southern Peru at an altitude of 1165 m (S16°44′34.7′W72°02′37.9′). Anorthite has been identified as the dominating mineral in this sample and a low CaSO4 content (5%) was measured. The samples are organic poor and contain only a very low amount of amino acids (Peeters et al. Reference Peeters, Quinn, Martins, Sephton, Becker, van Loosdrecht, Brucato, Grunthaner and Ehrenfreund2009).
A Salten Skov sample had been obtained from the Jutland region in Denmark. The Salten Skov sediment had been selected as a Mars analogue by Merrison et al. (Reference Merrison, Jensen, Kinch, Mugford and Nornberg2004) to study wind speed and orientation with respect to electrical charges and gravity on Mars. It is a natural sedimentary soil, extremely fine grained and containing a high concentration of various iron oxides (over 60% by weight). It is mainly composed of goethite with 5.5 wt% maghaemite and 12.8 wt% haematite (Nornberg et al. Reference Nornberg, Schwertmann, Stanjek, Andersen and Gunnlaugsson2004). Fresh Salten Skov samples have been reported to contain 1.5×106CFU g−1 under aerobic conditions (Hansen et al. Reference Hansen, Merrison, Nornberg, Lomstein and Finster2005). Properties of Salten Skov have been extensively discussed in Seiferlin et al. (Reference Seiferlin, Ehrenfreund, Garry, Gunderson, Hutter, Kargl, Maturilli and Merrison2008).
Experimental section
All soil samples were stored in sterile low-density polyethylene bags. PAHs of interest are shown in Table 2.
Table 2. Chemical structure, quantitation and confirmation ions of PAHs analysed by GC/MS. Compound are listed in the order they eluted from the column
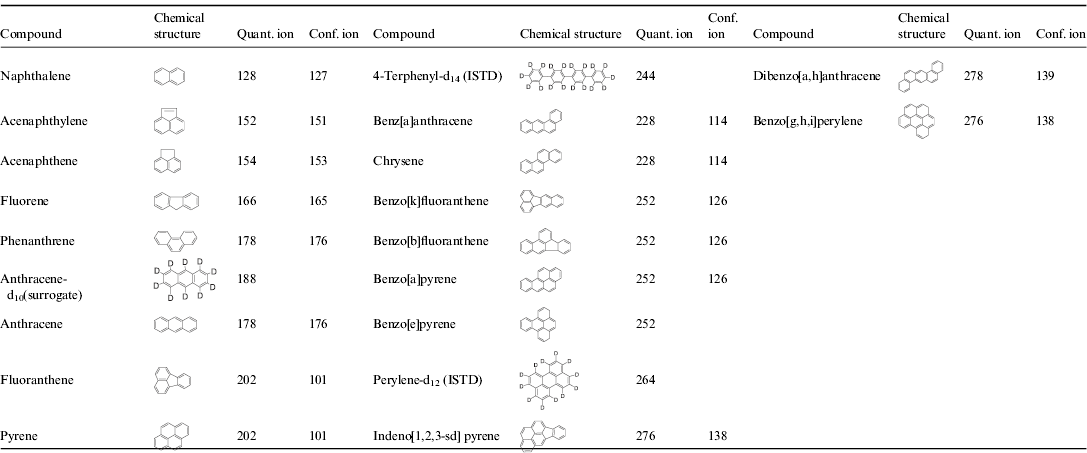
* As is standard practice, the compounds are ordered via ascending levels of the quantification ions. This order was kept for Tables 3–5.
Soil texture pH and organic matter content
A simple texture test was performed on each soil sample (using about 1 g for each). Samples were placed in glass vials and high-performance liquid chromatograph (HPLC)-grade water (Burdick & Jackson) was added to three-fourths of the vial volume and then capped. Samples were shaken vigorously for 2 min to bring soil particles into suspension in the water. The sample was allowed to settle for about 1 min and the top of the layer that settled was marked. This layer is comprised primarily of sand and larger particles. After 1 h of settling, the next layer (silt) was marked. Samples were then left to settle for 24 h and the last settled layer (clay) was marked. The percentage of each layer was then calculated (Table 1).
The pH of each soil sample was measured using an Accumet Excel XL20 pH meter (Fisher Scientific). Measurements were performed with samples that had been examined for soil texture after the soils had been soaked in water for 24 h and then again after 2 weeks (Table 1).
The amount of organic material was determined by placing weighed, dried (24 h at 105°C) soil samples in an oven heated to 600°C for 24 h. The organic matter in soil is evaporated as gases (CO2 and H2O) resulting in a change of the sample weight, which enabled calculations of the organic content of the soil sample (Table 1).
Chemicals and materials
Liquid standards were purchased from Supelco (Sigma-Aldrich): PAHs standard, 2000 μg ml−1 in dichloromethane:benzene (1:1); 4-terphenyl-d14, 2000 μg ml−1 in dichloromethane; perylene-d12, 2000 μg ml−1 in dichloromethane; anthracene-d10, 2000 μg ml−1 in dichloromethane; and benz[e]pyrene, 100 μg ml−1 in cyclohexane. Stock and diluted solutions of standards were prepared in a 1:1 mixture of dichloromethane:hexane.
Certified soil materials: Soil standard BCR-524 (Supelco 2008) used in this study was collected from a creosote-contaminated area. It contains relatively high levels of PAHs, including several PAHs not certified in this standard, and also contains a low level of PCP (pentachlorophenol). Loamy sand (CRM 171-050), clay (CRM 170-050) and sediment (CRM 104-100) soil standards were purchased from Resource Technology Corporation, Laramie, WA. The levels of PAHs in the soil standards are shown in Table 3.
Table 3. Certified amounts of PAH in CRM standard (Resource Technology Corporation, Laramie, WA, 2009) and BCR-524 (ATSDR 1995) soil samples

Chemicals used with the liquid extraction procedure: Gas chromatograph/mass spectrometer (GC/MS)-grade dichloromethane (Across Organics), HPLC-grade hexane and LC/MS-grade methanol (Burdick & Jackson) were all purchased from VWR, and activated silica gel for column chromatography, 60–200 μm, 60 Å (Across Organics), was purchased from Fisher-Scientific.
SPME device and fibres: A Supelco SPME device containing a syringe-like holder with a stainless-steel needle was purchased from Sigma-Aldrich. Analytes are absorbed into a thin polymer phase, which had been deposited onto a silica fibre housed in a syringe needle. Sampling is accomplished by penetrating the septum of a reaction vessel with the syringe needle, depressing the plunger and exposing the fibre to the target mixture. The fibre is allowed to absorb the analytes for a specified time, after which the needle containing the fibre is withdrawn and then inserted into a hot GC injector, desorbing the analytes in splitless mode for GC/MS analysis. The selection of a fibre is based on overall fibre performance and compatibility with target analytes. A manual SPME device with a 100 μm polydimethylsiloxane (PDMS)-coated fibre was chosen for all experiments. A 30 μm fibre was also tested. Although the 30 μm fibre had the better desorption efficiency, the 100 μm fibre provided a better response because of the higher extraction levels. We noticed approximately 3-fold higher sampling efficiencies for the 100 μm compared to the 30 μm PDMS fibres for naphthalene through fluorene and 15×higher efficiencies for phenanthrene and larger PAHs under similar sampling conditions. The biggest drawbacks of using the 100 μm vs. 30 μm fibres for large semi-volatile compounds are the longer equilibration times and an increased risk for carryover (more difficult to completely desorb analytes from the fibres between runs) due to the high affinity for the stationary phase. To eliminate carryover the fibre was left in the hot injection port for the entire GC run.
Procedure of liquid extraction with organic solvents
Extractions were performed on soil samples (on average 3.1 g), certified reference material (CRM) standard soils (1 g on average), BCR-524 standard soil (0.3 g on average) and liquid standard (48905-U, diluted in 1:1 dichloromethane:hexane) to obtain 2 ng μl−1 concentration in the final volume of the extract (250 μl). A mixture of 5 ml dichloromethane:methanol (93:7, v/v) was added to samples placed in Kimble test tubes with screw caps. Then a surrogate PAH, anthracene-d10 (diluted in 1:1 dichloromethane:hexane, v/v), was added to the mixtures to obtain a final concentration of 2 ng μl−1 in the extraction volume. The mixtures were shaken for 2 min and then extracted thrice using a sonication bath for 25 min at 25°C.
After each extraction the sample was centrifuged for 15 min at 90 rpm and then the liquid phase was transferred to a Pyrex test tube using a Pasteur pipette. The collected extracts were evaporated almost to dryness (<0.5 ml sample left) using a model RC 1022 (Jouan Inc.) desktop vacuum concentrator or in a stream of dry nitrogen gas. We did not notice significant differences in extraction efficiencies for samples dried under either condition. The vacuum concentrator, however, allows faster sample processing. Around 40 mg of activated silica was added to each test tube and allowed to dry under nitrogen. Then the dried silica+sample was transferred onto a column filled with about 0.8 g of activated silica. The columns had been previously prepared using long-stem Pasteur pipettes with a small piece of quartz wool placed on the bottom of the pipette column to stabilize the silica bed in the column. The columns were rinsed with 5 ml of hexane before the sample was transferred onto the column. Organics were eluted using a sequence of the following solvents: hexane (1 ml), 1:1 dichloromethane:hexane (1 ml), dichloromethane (2 ml) and 93:7 dichloromethane:methanol (1 ml). Eluants of respective samples were collected (combined) in Pyrex test tubes and then evaporated to dryness under a stream of dry nitrogen. The internal standard solution mixture of 4-terphenyl-d14 and perylene-d12 was then added to each sample to obtain 1 ng μl−1 as the final concentration. Then the sample was reconstituted to a final volume of 250 μl in dichloromethane:hexane (1:1). The GC/MS analyses (described below) were performed with 60 μl of the reconstituted sample placed in a GC vial that had an attached conical insert (volume of 150 μl) and then capped with a screw cap with septa. The amount of PAHs in each sample was calculated from the respective calibration curves obtained with the MS quantization method described below. All glassware used in the experiments was washed and then baked at 500°C for 24 h, and stored in oven-baked aluminium foil. The quartz wool was also baked and stored in aluminium foil. The extraction procedure adapts steps described in the Lab Manual 2007/2008 of the Imperial College Organic Geochemistry, Imperial College London (UK) (Imperial College 2008).
Headspace SPME sampling procedure
Operating principles of SPME can be found in the monograph by Pawliszyn (Reference Pawliszyn1999). In this study, we have chosen a headspace non-equilibrium condition with a relatively long time-frame (40 min) and slightly elevated temperature (40°C) for extraction. In practice, the temperature chosen is a compromise between sensitivity and sufficiently short extraction time. Our data from liquid extraction indicated very low levels of target analytes in our samples; therefore, we allowed longer times for the SPME extractions. A few samplings of the CRM standard soils with 30 μm PDMS fibres at 60°C showed that the 20°C temperature change resulted in smaller amounts of PAHs adsorbed on the PDMS fibre. Sampling at 60°C for 10 min with 100 μm fibre, however, gave a faster analysis turnaround and amounts of PAHs adsorbed were close to that from sampling at 40°C for 40 min. The smaller partitioning of PAHs into the 30 μm PDMS fibre, with increasing temperature, and the uptake processes were reported by Muijs & Jonker (Reference Muijs and Jonker2009) and they were attributed to an enthalpy change.
Soil samples were ground in an agate mortar before weighing and sampling. On average, 0.8 g of soil sample was weighed into a standard 2 ml GC vial, which was then sealed with a PTFE septum cap. The sample amounts of Salten Skov and standard BCR-524 soils were smaller, 0.4 and 0.3 g, respectively. Vials were set into a heating block at 40°C for at least 2 h for thermal equilibrium to be reached throughout the sample. Afterwards, the fibre was exposed for 40 min to the headspace without making contact with the soil. Immediately after sampling, the fibre was inserted into the GC injector for thermal desorption and GC/MS analyses. The fibre was then left in the injector (for continuous cleaning) for 18 min (the entire chromatographic run) before it was removed for a new extraction. Blank injections between extractions (empty fibre) were performed to monitor possible carryover from the exposed fibre. The CRM soil standards (on average 0.95 g of the soil) were headspace sampled at 40°C for 40 min.
Instrumentation
Analyses of SPME and liquid extraction samples were performed using a Varian GC/MS system consisting of a Saturn model 3800 gas chromatograph (injection port model 1079) that was coupled to a Saturn model 2000 ion trap mass spectrometer. A 0.25 mm ID×30 m long Zebron ZB-5MS fused silica capillary column (0.25 μm film thickness) (Phenomenex) was used for all GC/MS experiments. The helium carrier gas was maintained at a constant flow of 1 ml min−1. The MS was operated in electron impact (EI) mode, with trap temperature set at 220°C, transfer line at 300 °C and manifold at 80°C. The ion storage level for all segments was set at 55 m/z. To increase the sensitivity of our GC/MS system, the target compounds were detected in the selected segments, which were timed according to the retention times and molecular ions (Table 2) of the individual analytes. The MS data handling software included a compound table with respective retention times and mass spectra for all target analytes.
Instrument parameters for the SPME runs were as follows: the programmable injection port temperature was set to 285°C with the initial split set at 20:1. After the SPME fibre was introduced into the injection port, the split mode was turned off for 5 min (thermal desorption of analytes). After this time had elapsed, the injection port temperature was set to 250°C and the split was activated again and remained 20:1 for the entire run. To eliminate carryover of compounds between extractions, the fibre was left in the injection port for the entire run. The fibre depth in the injector was set at 3 on the Supelco SPME device scale; this had been optimized to allow desorption of analytes from the fibre in the hottest part of our GC injection port. The GC oven temperature was programmed as follows: 75°C – put on hold for 1 min, ramped to 280°C at a rate of 25°C min−1 and then to 300°C at 10°C min−1 and, finally, put on hold for 3 min. The total chromatographic run was 17.9 min long. External calibration curves were used to determine the levels of target analytes. Although the total run lasted only ∼18 min, all peaks were sufficiently resolved to be positively identified by their mass fractional pattern and even by their retention times.
Instrument parameters for liquid injections were as follows: the programmable injection port temperature was set to 250°C with the initial split turned on (20:1). After the 0.0 min mark, the split mode was turned off for 1 min. The split was activated after 1 min and remained at a 20:1 ratio for the entire run. Liquid samples (1 μl) were injected using a Varian model 8200 auto-sampler equipped with a 48-vial carousel. The user-defined programme of the auto-sampler included solvent flush sampling, syringe wash with dichloromethane (20 s), solvent plug (0.2 μl), upper and lower air gap, injection rate (0.2 μl s−1) and needle residence time (0.02 min). The GC oven temperature was programmed as follows: 60°C – put on hold for 1 min, ramped to 250°C at rate 10°C min−1, ramped to 280°C at rate 5°C min−1 and then put on hold for 4 min. The total chromatographic run was 50 min long. The MS data handling included a quantification method of an internal standard calibration using 4-terphenyl-d14 and perylene-d12 as internal standards and anthracene-d10 as a surrogate compound. The calibration curves (five replicates) for each PAH were obtained for the concentration range of 0.25–2 ng μl−1.
Results and discussion
Texture, pH and organic matter content of soil samples
We performed tests for texture (type), pH and organic mass loss on all collected soil samples (Table 1). The three major categories of soil are sandy (typically comprised of approximately 80–100% sand, 0–10% silt and 0–10% clay), loam (typically comprised of approximately 25–50% sand, 30–50%, silt and 10–30% clay) and clay (typically comprised of approximately 0–45% sand, 0–45% silt and 50–100% clay). Different proportions of sand, silt and clay give rise to different types of loam soils: sandy loam, silty loam, clay loam, sandy clay loam, silty clay loam and loam. A soil that meets the textural definition of loam, however, can lose its characteristics when it is compacted, depleted of organic matter or has clay dispersed throughout its earth fraction. Our soil samples exhibit different ratios of sand, silt and clay (Table 1). We classified samples P-2, P-8 and Salten Skov to be the 100% silt. The highest 75% of clay was determined in sample P-6 and 50% in sample P-5. The HOBO sample shows 50% sand and 50% silt, and this is in good agreement with reports on the loamy sand and loamy silt types of soils found in the vicinity of the HOBO site (Squeo et al. Reference Squeo, Holmgren, Jimenez, Alban, Reyes and Gutierrez2007). Analyses of Utah soil samples, including clay and mineral composition, are reported by Kotler et al. (Reference Kotler, Martins, Foing and Ehrenfreund2011).
After soils were soaked in water for 24 h, all Utah (P), HOBO and Salten Skov samples showed moderately basic pH, with the most alkaline being P-7 and P-5 at pH 10.01 and pH 9.28, respectively. The pH dropped on average by 0.7 pH units to constant levels after 2 weeks (Table 1). The HOBO soil was found to be slightly basic to neutral with a measured pH range of 7.18–7.04. The sampling site for HOBO was located around 60 km from the coast close to the Meija region (S-17°00′W-71°59′) in the southern part of Peru. Alkaline soil pH 8.4 is reported for Piura in the northern part of the Atacama Desert in southern Peru. The slightly acidic soils (pH 6.79) are in the Fray–Jorge area in north-central Chile (Squeo et al. Reference Squeo, Holmgren, Jimenez, Alban, Reyes and Gutierrez2007). It has been suggested that on Mars the chemical weathering processes are dominated by low pH, acidic environment and dry deposition (Hurowitz & McLennan Reference Hurowitz and McLennan2007; Skelley et al. Reference Skelley, Aubrey, Willis, Amashukeli, Ehrenfreund, Bada, Grunthaner and Mathies2007). In contrast, Quinn & Orenberg (1993) argue that in order to explain the results of the Viking gas exchange (GEX) experiment, the soil should be slightly to moderately basic (pH 7.4–8.7). Also, the recent in situ data from the Phoenix Mars lander indicate moderately alkaline (pH 7.7) soil (Hecht et al. Reference Hecht, Kounaves, Quinn, West, Young, Ming, Catling, Clark, Boynton and Hoffman2009). The Salten Skov soil, on the other hand, was slightly acidic with a pH range of 6.01–5.8; this pH value is higher than that reported by Peeters et al. (Reference Peeters, Quinn, Martins, Sephton, Becker, van Loosdrecht, Brucato, Grunthaner and Ehrenfreund2009).
The highest concentration of organic matter (12%) was found in the Salten Skov sample. All other samples show relatively low 1–3% organic matter content except for P-8 and P-10 (4–5%). Organic matter levels of 0.5 and 3% are reported in soil samples from the Atacama Desert in southern Peru in the Piura and Mejia regions, respectively (Squeo et al. Reference Squeo, Holmgren, Jimenez, Alban, Reyes and Gutierrez2007). Navarro-Gonzalez et al. (Reference Navarro-Gonzalez, Navarro, de la Rosa, Iniguez, Molina, Miranda, Morales, Cienfuegos, Coll and Raulin2006) report 10–1500 μg C g−1 of total organic matter (TOM) in different desert soils and 20–400 μg C g−1 in Atacama soils in Chile.
PAH abundance in soil samples
There are no data available on the overall comparison of extraction procedures using different soils with variations in abiotic properties (e.g. texture, pH) and PAH concentration (Song et al. Reference Song, Jing, Fleischmann and Wilke2002). In order to understand the possible relationship between the texture, pH and PAH content in our samples, we have chosen soil standards that echo those properties (Table 3). We used certified amounts of PAHs found in the European Commission BCR CRM standards for quantification of SPME sampling, efficacy of the liquid extraction procedure and calculation of the recovery of the surrogate PAH compound (anthracene-d10).
We used the headspace SPME/PDMS-GC/MS technique as an expedited screening method to determine the presence of PAH in our Mars analogue soil samples. We did not perform detailed studies on a fibre-headspace distribution (Havenga & Rohwer Reference Havenga and Rohwer1999) or extensive calibration procedures (Eriksson et al. Reference Eriksson, Faldt, Dalhammar and Borg-Karlson2001). The BCR-524 soil standard was used as a cross-check sample for the SPME and liquid extraction procedures (Table 4). The BCR-524 standard soil represents a sample of highly contaminated industrial soil with a high concentration of heavy PAHs. The heaviest compounds in molecular weight found with SPME extraction were pyrene, benz[a]anthracene, and chrysene while compounds with more than four rings were extracted via liquid extraction. These data demonstrate the limitation of the headspace SPME sampling for high-molecular-weight PAHs. In contrast to this limitation, SPME data show its high sensitivity for volatile PAHs such as two- and three-ring compounds, e.g. naphthalene, acenaphthylene, acenaphthene and fluorene. Losses of these volatile PAHs during the liquid extraction process are identified (Martens et al. Reference Martens, Maguhn, Spitzauer and Kettrup1997; Song et al. Reference Song, Jing, Fleischmann and Wilke2002), and they are reflected here in the % recovery of a three-ring surrogate, anthracene-d10 (Tables 5a and 5b). It is apparent that in order to achieve a more coherent data set, both liquid extraction and SPME methods are needed to provide complementary information. The SPME results presented in Tables 5a and 5b are from a single calibration replicate. Data are obtained from a headspace sampling using a 100 μm PDMS fibre at 40°C for 40 min. Quantification of analyte concentrations is based on an external calibration method using CRM soil standards (Table 3). The amounts of PAHs measured in the soil analogue samples are very low, up to ∼60 ng g−1 at the most, and they were calculated from the extrapolation ranges of the calibration curves. Our data show that analytes that could be extracted with the headspace SPME technique, at the concentration levels present in the studied soils, are naphthalene through pyrene, except for the Salten Skov sample where even benz[a]anthracene and chrysene were detected.
Table 4. Detection of PAHs in the standard soil BCR-524 using SPME and liquid extraction procedures

Table 5. Amounts of target PAHs detected in Mars analogue samples. Samples ‘P’-analytes extracted with the SPME/PDMS; samples ‘PE’-analytes extracted through liquid extraction

a STDEV (n=3) given in parenthesis ; all samples ‘PE’.
Our results show that the number of aromatic rings and analyte concentration significantly influence sampling efficiency. The larger the ring formation of a polyaromatic hydrocarbon, the lower the vapour pressure and, therefore, the lower the efficacy of headspace sampling. Headspace SPME sampling of PAHs up to a four-ring structure had been previously reported by Havenga & Rohwer (Reference Havenga and Rohwer1999) and Eriksson et al. (Reference Eriksson, Faldt, Dalhammar and Borg-Karlson2001), among others. While naphthalene was detected in all samples by both the SPME and liquid extraction methods, benzo[k]fluoranthene was not determined in any of the samples by either method. Anthracene was only determined in the P-1 and P-7 samples, where SPME analysis showed 0.3 and 0.4 ng g−1, respectively, and in the Salten Skov sample (1.1 ng g−1). Anthracene, however, was not found in those samples when processed with the liquid-extraction method. A high concentration (9000±6000 ppm) of anthracene was reported in a soil sample from the Atacama Desert (Young Hill) using the Mars Organic Analyzer-Capillary Electrophoresis (MOA-CE) system. The authors, nevertheless, conclude that their result is probably an overestimate due to the experimental design (Stockton et al. Reference Stockton, Chiesl, Scherer and Mathies2009). Our analysis shows that when phenanthrene is present in higher quantities, careful inspection of chromatograms and manual integration need to be performed in spite of the programmed calibration of the GC/MS method. Phenanthrene and anthracene have the same quantity of ions and they elute within 10 s.
The evidence for heavier PAHs in our samples was obtained from the liquid extraction analyses. The amounts of PAHs presented in Tables 5a and 5b are given with the standard deviation of the results (n=3), which are shown in parentheses. The recoveries of the surrogate PAH, anthracene-d10, are also shown for each analysis. Data point towards an analysis error of 25–30% for naphthalene through fluorene and 10–15% for heavier PAHs. The recovery of the surrogate (50–125%) in our analyses varies for different samples, but we cannot establish a relationship between sample texture, pH and surrogate yield. Recovery rates for the target PAHs were obtained from liquid extractions of standard soil samples (Table 3) and a liquid standard in which the concentration of each compound was 1 μg ml−1. Recoveries of naphthalene, acenaphthylene, acenaphthene and fluorene were consistently the lowest in all samples (40–60%), while the heavier PAHs were extracted with a yield of 80–100%. In contrast to headspace SPME sampling, heavier PAHs are more efficiently extracted with organic solvent. Instead, more volatile PAHs are lost due to evaporation processes involved in the liquid-extraction procedure. Our results agree with the reported data where the recovery rates range from 41 to 56% for 2- and 3-ring PAHs and from 81 to 96% for larger-ring compounds (Martens et al. Reference Martens, Maguhn, Spitzauer and Kettrup1997; Song et al. Reference Song, Jing, Fleischmann and Wilke2002). The recovery of the surrogate, anthracene-d10, was 55±5% for all standards except, for BCR-524 where it was 74%. The GC/MS chromatograms of the P-13, P-10, HOBO and Salten Skov samples show additional peaks to those of target PAHs. These compounds seem to be oxygenated polyaromatic molecules with polyfunctional groups but their proper identification was not possible due to the focal point of our GC/MS method, which was targeted for PAHs.
The concentrations of PAHs in our samples are at least four orders of magnitude lower than those determined in some industrial soils polluted with coal tar and petroleum products, collected at an old gasworks, around the airport in Delhi (India) as well as from those found in sediment samples from the Niger Delta region (Havenga & Rohwer Reference Havenga and Rohwer1999; Eriksson et al. Reference Eriksson, Faldt, Dalhammar and Borg-Karlson2001; Anyakora et al. Reference Anyakora, Ogbeche, Palmer, Coker, Ukpo and Ogah2005; Ray et al. Reference Ray, Khillare, Agarwal and Shridhar2008). The total amounts of PAHs in those samples are reported to be from 1 to 100 μg g−1 sample, which correlates to the level and duration of influx of PAH contamination in a given environment.
To the best of our knowledge there are no reports in the literature regarding the contents of PAHs in the desert soils of Utah. Atmospheric deposition is the most common source of soil contamination, and so the deposition of particulates associated with different PAH sources could result in spatial variability and enhanced levels at certain areas. PAHs may undergo adsorption, volatilization, photolysis and chemical degradation; microbial activity can be an effective catabolic process. Degradation of PAHs requires a consortium of micro-organisms and the rate depends on the environmental conditions and the number and type of micro-organisms. Changes in the microbial community are still unpredictable and so, consequently, are the rates of bioremediation. The metabolic breakdown of pyrene and phenanthrene to cyclic carboxylic acids along with other functional groups within the molecules has been observed (Haritash & Kaushik Reference Haritash and Kaushik2009). The culture-independent molecular characterization of microbial community structure and diversity has been performed on several of the samples analysed in this paper. All three domains of life (Archaea, Bacteria and Eukarya) were present, with most diversity in Bacteria. A wide variety of putative extremophiles were also observed in these analogue soils (Direito et al. Reference Direito, Ehrenfreund, Mareas, Staats, Foing and Roling2011). A relation between the microbial distribution and the geological context (minerals and elemental composition), organic content and environmental parameters is discussed in Ehrenfreund et al. (Reference Ehrenfreund, Röling, Thiel, Quinn, Septhon, Stoker, Direito, Kotler, Martins, Orzechowska, Kidd and Foing2011), this volume.
Conclusion
In the context of Mars exploration, it is vital to obtain knowledge about the survival of organic molecules in extreme conditions. Measuring the abundance and distribution of organic matter in Mars analogue samples collected in terrestrial dry deserts is critical to our understanding of preservation. The aim of this study was to optimize the SPME method for fast screening and determination of PAHs in soil samples. Current organic detection instruments for Mars missions are specialized for small molecules and small PAHs, and therefore our analysis method is important since we optimize for small compounds. The measured concentrations of PAHs are, in general, very low, ranging from 1 to 60 ng g−1. We found that 100 μm SPME fibres provided a better response because of the higher extraction levels compared to 30 μm fibres. We noticed approximately 3-fold higher sampling efficiencies for the 100 μm compared to the 30 μm PDMS fibres for naphthalene through fluorene and 15× higher efficiencies for phenanthrene and larger PAHs (under similar sampling conditions). The heaviest compounds found with the SPME extraction method were pyrene, benz[a]anthracene and chrysene, while compounds with more than four rings were extracted using the liquid-extraction method. This demonstrates the limitation of headspace SPME sampling for high-molecular-weight PAHs; however, the loss of 2- and 3-ring volatile PAHs during liquid extraction is not insignificant. PAHs and organic matter constitute potential sources of carbon for growth and survival of microorganisms. However, PAHs and organic matter contents did not correlate with DNA yield or detection (Ehrenfreund et al. Reference Ray, Khillare, Agarwal and Shridhar2011) indicating that the levels and variety of PAHs detected were not significantly affected by microbes. SPME appears to be a rapid, viable field sampling technique with implications for use on planetary missions.
Acknowledgements
Grazyna E. Orzechowska and Pascale Ehrenfreund were supported by the NASA Astrobiology Institute (NAI). The research described in this paper was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with the National Aeronautics and Space Administration. The EuroGeoMars 2009 campaign was organized and supported by the International Lunar Exploration working group (ILEWG), NASA Ames Research Center and ESA/ESTEC. We acknowledge the contribution of the EuroGeoMars 2009 campaign crew and the mission support team.