



1. Introduction
Cell death is a predominant process in normal physiological development and the maintenance of homeostasis, and the dysregulation of cell death leads to various diseases, including cancer. Programmed cell death is characterised by necroptosis, apoptosis, autophagy and ferroptosis. The process of necroptosis is identified by swollen organelles and ruptured cell membranes. The main characteristics of apoptosis is cell shrinkage with chromatin condensation and DNA fragmentation. Intriguingly, the hallmarks of ferroptosis are condensed mitochondria, increased bilayer membrane density and a reduction in the number of mitochondrial cristae(Reference Dixon, Lemberg and Lamprecht1,Reference Yagoda, von Rechenberg and Zaganjor2) . Ferroptosis, a form of iron-dependent cell death, was first named in 2012. It is caused by the depletion of glutathione (GSH) and the inactivation of the phospholipid peroxidase glutathione peroxidase 4 (GPX4)(Reference Dixon, Lemberg and Lamprecht1,Reference Yang, SriRamaratnam and Welsch3) . GPX4 is the only major antioxidant enzyme known to convert toxic lipid hydroperoxides (L-OOH) into non-toxic lipid alcohols (L-OH)(Reference Yang, SriRamaratnam and Welsch3). Thus, the predominant characteristic of ferroptosis is the accumulation of oxidative phospholipids in the cell membrane, which could trigger cell death through unclear mechanism(Reference Conrad and Pratt4). Ferroptosis has attracted considerable interest from basic and clinical researchers since it has been implicated in the occurrence and development of multiple diseases.
Cellular biological functions are largely regulated by extracellular and intracellular nutrients and their metabolite concentrations. A rapid and efficient response to nutrient levels is crucial for maintaining cellular homeostasis. Recently, substantial progress has been made in the field of ferroptosis and nutrient sensing. In this review, we mainly focus on the potential cross talk between ferroptosis and nutrient signalling factors.
2. The general pathway of ferroptosis
As the hallmark of ferroptosis, lipid peroxidation is triggered by non-enzymatic and enzymatic processes(Reference Gaschler and Stockwell5). The non-enzymatic process is mainly catalysed by cellular free iron and oxygen and is known as the Fenton reaction (Figs. 1 and 2). Bis-allylic hydrogen atoms (the weakest C-H bond) in polyunsaturated fatty acyl moieties are vulnerable to oxidation by oxygen species and preliminarily form phospholipid radicals (PL•). After the initiation of lipid oxidation, PL• is converted into a phospholipid peroxyl radical (PLOO•), releasing one molecule of oxygen. Subsequently, PLOO• reacts with a PLH molecule to yield a molecule of hydroperoxide (PLOOH) and PL•(Reference Conrad and Pratt4). As a form of reactive oxygen species (ROS), PLOOH reacts with cellular ferrous iron to generate additional alkoxyl radicals (PLO•) and PLOO•. The reactions of lipid peroxidation lead to the accumulation of lipid-based oxidative secondary products such as 4-hydroxynonenal and malondialdehyde (MDA). This propagation process of lipid peroxidation is terminated when PLOOH is cleared in a timely manner by GPX4. The enzymatic process is also required to trigger ferroptosis (Fig. 2). Lipoxygenases (LOX) target PUFA located in lipid bilayers and regulate lipid peroxidation(Reference Kuhn, Banthiya and Van Leyen6). Inhibition of LOX makes cells more resistant to ferroptosis(Reference Li, Maher and Schubert7). Cytochrome P450 oxidoreductase (POR) is the other critical regulator of ferroptosis; it induces lipid peroxidation by directly transferring hydrogen from PUFA or by indirectly converting Fe3+ into Fe2+ (Reference Zou, Li and Graham8,Reference Ghosh, Mukhopadhyay and Chatterjee9) .
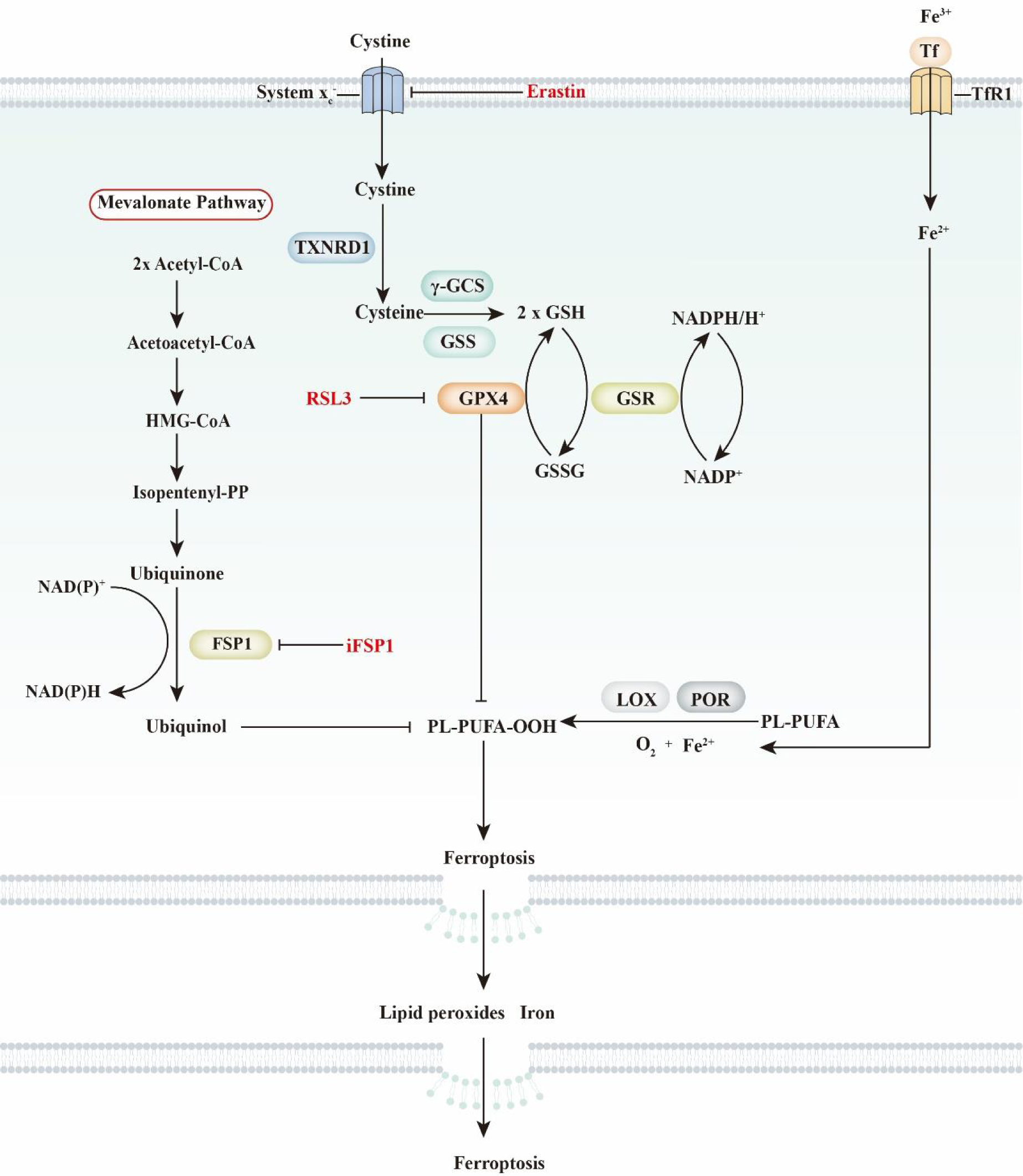
Fig. 1. General view of ferroptosis. Ferroptosis is characterised by iron-dependent phospholipid peroxidation. It is mainly regulated by selenium-dependent and CoQ10-dependent pathways. The selenium-dependent pathway is primarily dependent on the enzymatic activity of glutathione peroxidase 4 (GPX4), which is a unique selenoprotein that functions as a phospholipid peroxidase. Cellular cystine transportation has an intimate relationship with the selenium-dependent pathway. Briefly, cystine is transported by amino acid transporter system xc − and further oxidised into cysteine by thioredoxin reductase 1 (TXNRD1). Then, cysteine, glutamate and glycine are used to synthesise glutathione (GSH) by γ-glutamylcysteine synthetase (γ-GCS) and glutathione synthetase (GSS). As a cofactor of GPX4, GSH functions in conjunction with GPX4 to reduce L-OOH into L-OH. In the CoQ10-dependent pathway, the suppressor protein 1 (FSP1)–ubiquinone system is critical for lipid peroxidation. FSP1 reduces ubiquinone to ubiquinol, which further decreases lipid radicals to prevent lipid oxidation. Ubiquinone is derived from dietary CoQ10 intake or synthesis through the mevalonate pathway. FSP1, ferroptosis sensitising protein 1; GSH, glutathione; GSR, glutathione reductase; GSSG, glutathione disulfide; PL-PUFA, polyunsaturated fatty acid- containing phospholipid; Tf, transferrin; TfR1, transferrin receptor 1.
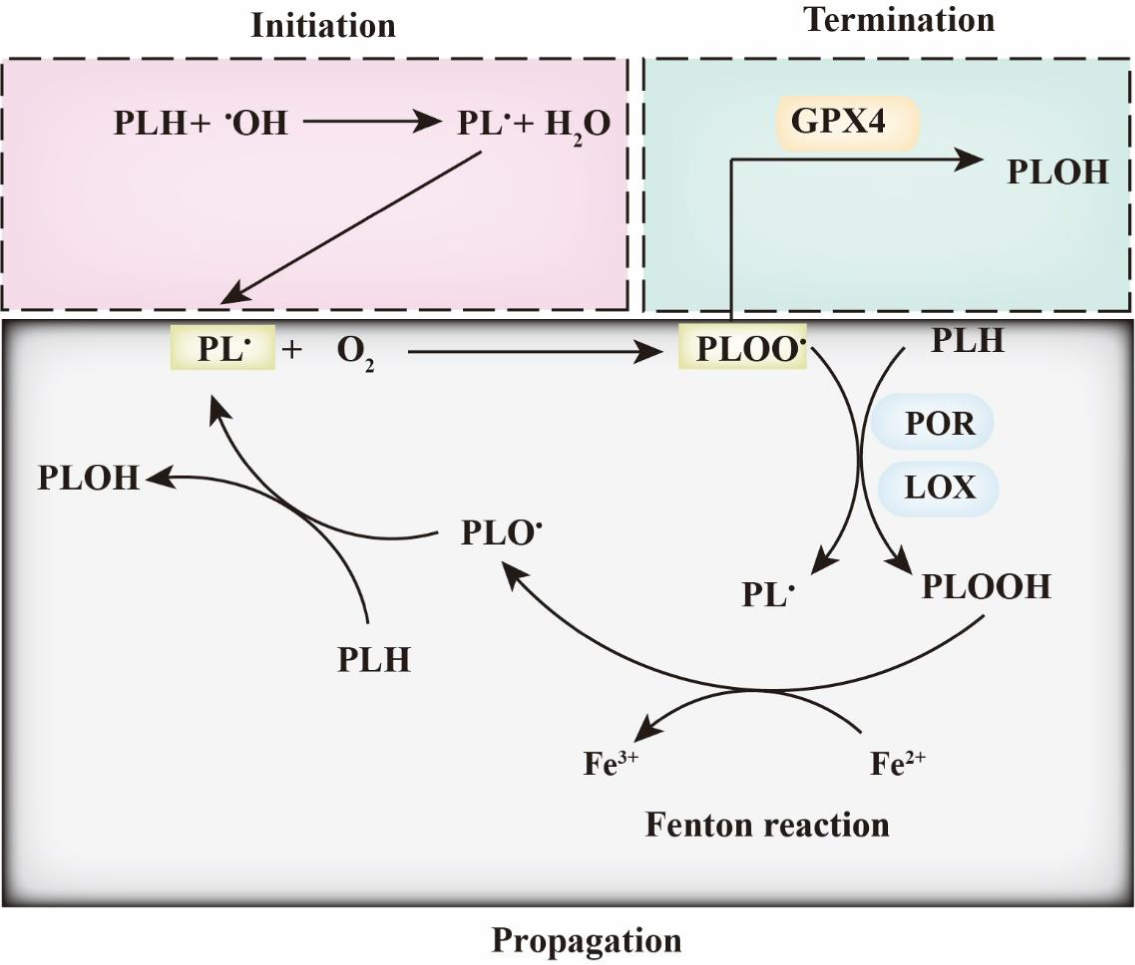
Fig. 2. Ferroptosis and phospholipid peroxidation. Polyunsaturated fatty acids contain bis-allylic hydrogen atoms, which are prone to removal, leading to the formation of phospholipid radicals (PL•). PL• reacts with O2 to form PLOO• and is subsequently converted into PLOOH by lipoxygenase (LOX) and/or cytochrome P450 oxidoreductase (POR). Then, the iron-dependent Fenton reaction yields PLO• and PL•, which leads to the induction of PLOOH production. This lipid propagation is terminated when PLOO• is reduced by glutathione peroxidase 4 (GPX4).
Cells have a protection program to safeguard them from ferroptosis. The canonical pathway to prevent ferroptosis in cells is mainly dependent on system xc – and GPX4 (Fig. 2). Mechanistically, system xc – regulates the cellular concentration of cystine, which could be converted into cysteine and further act as a critical substrate for GSH synthesis. GSH is a cofactor of GPX4, which reduces toxic PLOOH to corresponding non-toxic PLOHs. Inhibition of either system xc – or GPX4 induces ferroptosis. The system xc ––GSH–GPX4 axis is regarded as the most classical and is the best-studied pathway in the regulation of ferroptosis. Recently, studies identified a novel ubiquinone-dependent pathway in which FSP1 functions as an oxidoreductase to reduce ubiquinone to ubiquinol, which prevents lipid peroxidation(Reference Doll, Freitas and Shah10,Reference Bersuker, Hendricks and Li11) .
3. Ferroptosis and iron
Iron has been known as a critical component for haemoglobin synthesis since 1932(Reference Abbaspour, Hurrell and Kelishadi12). The role of iron in haemoglobin formation and oxygen transport has attracted the attention of nutritionists for many years. Insufficient iron intake has a direct connection to anaemia in many developing countries(Reference Dary and Hurrell13). Recently, the accumulation of iron has been found to be closely related to ferroptosis. Iron is proposed to enhance lipid peroxidation through the Fenton reaction or activation of enzymes such as LOX(Reference Yang, Kim and Gaschler14). Mechanistically, iron reacts with PLOOH to generate the free radicals PLO• and PLOO•, which propagate peroxidation chain reactions(Reference Conrad and Pratt4).
Feeding animals an iron-enriched diet increases cellular iron loading (Fig. 3)(Reference Yu, Jiang and Wang15). Iron is absorbed in the upper intestine in the form of ferrous iron (Fe2+), which is then quickly oxidised into a ferric ion (Fe3+). Once exported from enterocytes, Fe3+ binds with transferrin (TF) and circulates in the blood circulatory system. This complex is recognised and imported by the transferrin receptor (TFRC) located in the cell membrane. Once taken up, the iron bound to TF is reduced from Fe3+ to Fe2+ by the STEAP3 metalloreductase in the endosome and is then transferred to the cytosol via solute carrier family 11 member 2 (SLC11A2). The enrichment of iron further sensitises cells to ferroptosis.
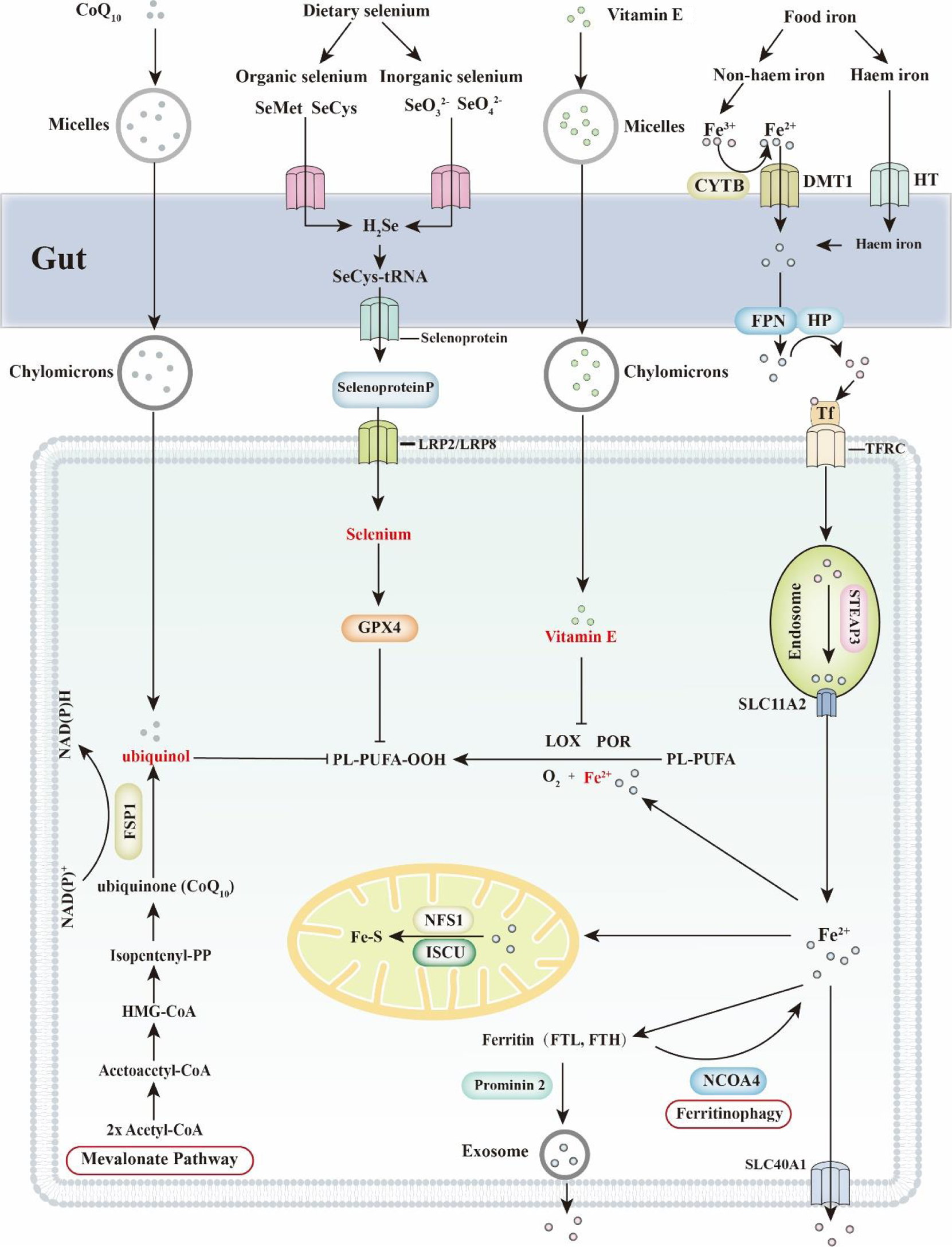
Fig. 3. Ferroptosis and dietary nutrients (CoQ10, selenium, vitamin E and iron). Ferroptosis is an iron-dependent form of cell death. Thus, regulation of cellular iron concentration is critical to ferroptosis. Dietary iron includes haem iron and non-haem iron. Haem iron is directly transported into enterocytes through haem transporters (HT). Dietary ferric iron (Fe3+) is reduced into ferritin iron (Fe2+) by the ferric reductase enzyme cytochrome b (CYTB) and then transported into cells via divalent metal transporter 1 (DMT1). Subsequently, Fe2+ is exported by FPN1 and oxidised into Fe3+ by hephaestin (HP). Finally, Fe3+ is combined with transferrin (TF) and transported to targeted organs, which are recognised by the transferrin receptor (TFRC) in the cell membrane. Selenium is classified into organic selenium and inorganic selenium, both of which are absorbed in the small intestine. SeO3 2− is passively absorbed by the gut, while SeO4 2− seems to be transported by a sodium-mediated carrier. Organic forms of selenium are not recognised and are transported as minerals. For instance, selenomethionine (SeMet) and selenocysteine (SeCys) are transported in the form of amino acids. More than 50 % of selenium is converted into selenoprotein P (SELENOP) and transported to the targeted organs through blood. Low-density lipoprotein receptor-related protein 2 and 8 (LRP2/LRP8) are critical for receptor-dependent selenium uptake in different tissues. Cellular selenium is subsequently synthesised into GPX4 and protects cells from ferroptosis. Vitamin E and CoQ10 are both regarded as fat-soluble vitamins. Dietary vitamin E and CoQ10 are incorporated into micelles and absorbed by the small intestine. In enterocytes, vitamin E and CoQ10 are attached to chylomicrons and then transported to target organs through the lymph and blood circulation systems. CoQ10 (ubiquinone) is largely transformed into ubiquinol in lymph. In blood vessels, CoQ10 is attached to low-density lipoprotein (LDL) and very-low-density lipoprotein (VLDL), while vitamin E is attached to high-density lipoprotein (HDL). Mechanistically, ubiquinol directly prevents lipid peroxidation, whereas vitamin E regulates ferroptosis mainly by competing with lipoxygenases (LOX) for PUFA substrate-binding sites. FSP1, ferroptosis sensitising protein 1; FTH, ferritin heavy chain; FTL, ferritin light chain; FPN, fragipain; GPX4, glutathione peroxidase 4; HMG-CoA, β-Hydroxy β-methylglutaryl-CoA; ISCU, iron–sulphur cluster assembly enzyme; NCOA4, nuclear receptor coactivator 4; NFS1, nitrogen fixation 1 homologue; PL-PUFA, polyunsaturated fatty acid- containing phospholipid; POR, cytochrome P450 oxidoreductase; SLC40A1, solute carrier family 40 member 1; SLC11A2, solute carrier family 11, member 2.
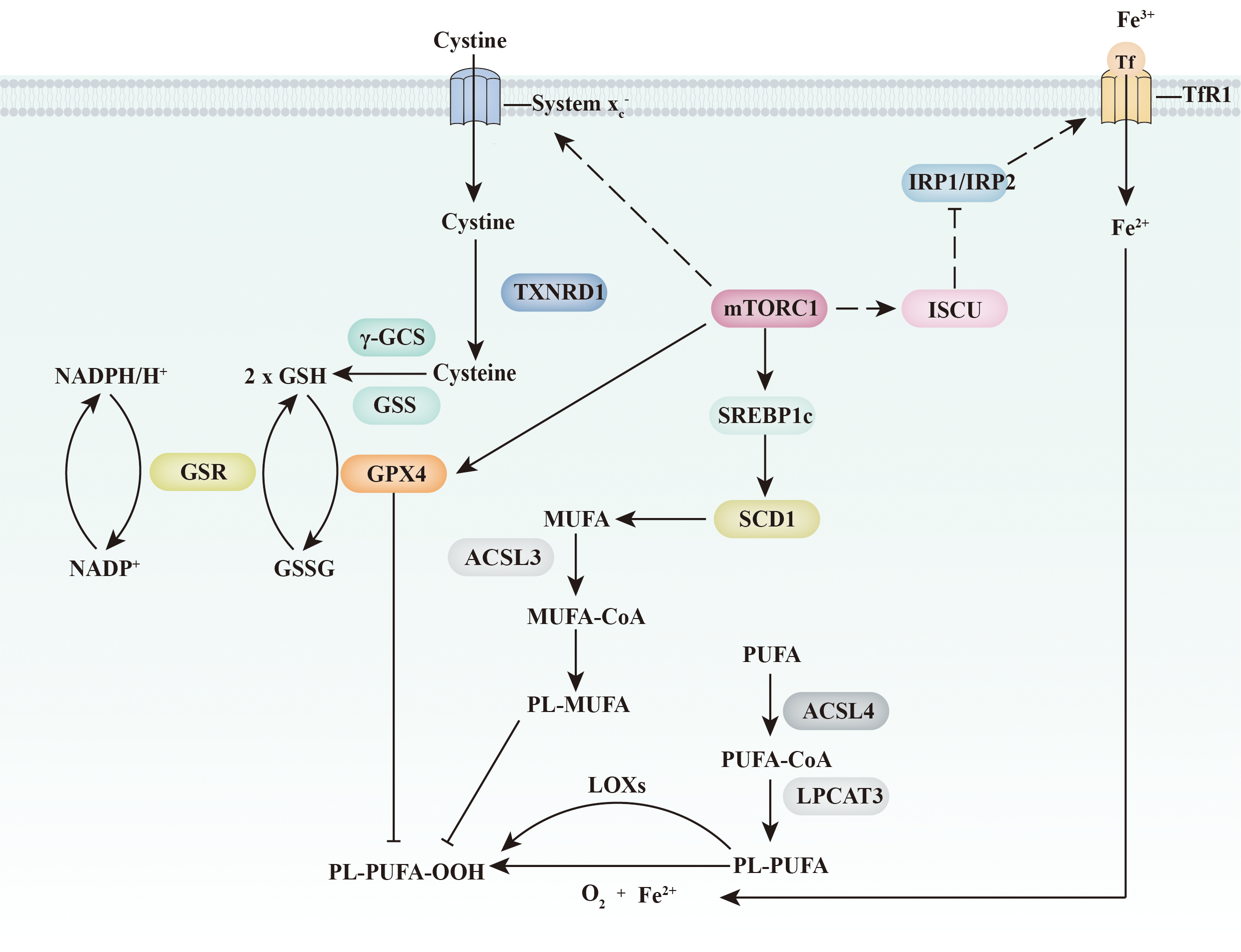
Fig. 4. Ferroptosis and the mTORC1 signalling pathway. mTORC1 inhibits ferroptosis through three independent pathways. On the one hand, mTORC1 increases the expression of GPX4 and the abundance of GSH (through the up-regulation of amino acid transport system xc −). On the other hand, mTORC1 increases MUFA abundance in the plasma membrane through the SREBP1c/SCD1 pathway. Furthermore, mTORC1 might directly inhibit an increase the intracellular iron concentration through the ISCU/IRP/TfR1 signalling pathway. ACSL 3/4, acyl-CoA synthetase long-chain 3 and 4; γ-GCS, γ-glutamylcysteine synthetase; GPX4, glutathione peroxidase 4; GSH, glutathione; GSR, glutathione reductase; GSS, glutathione synthetase; GSSG, glutathione disulfide; IRP1/IRP2, iron regulatory protein 1 and 2; ISCU, iron–sulphur cluster assembly enzyme; LOX, lipoxygenases; LPCAT3, lysophospholipid O-acyltransferase 3; mTORC1, mechanistic target of rapamycin complex 1; PL-MUFA, monounsaturated fatty acids-containing phospholipid; PL-PUFA, polyunsaturated fatty acid-containing phospholipid; SCD1, stearoyl-CoA desaturase-1; SREBP1c, sterol regulatory element binding protein-1c; Tf, transferrin; TfR1, transferrin receptor 1; TXNRD1, thioredoxin reductase 1.
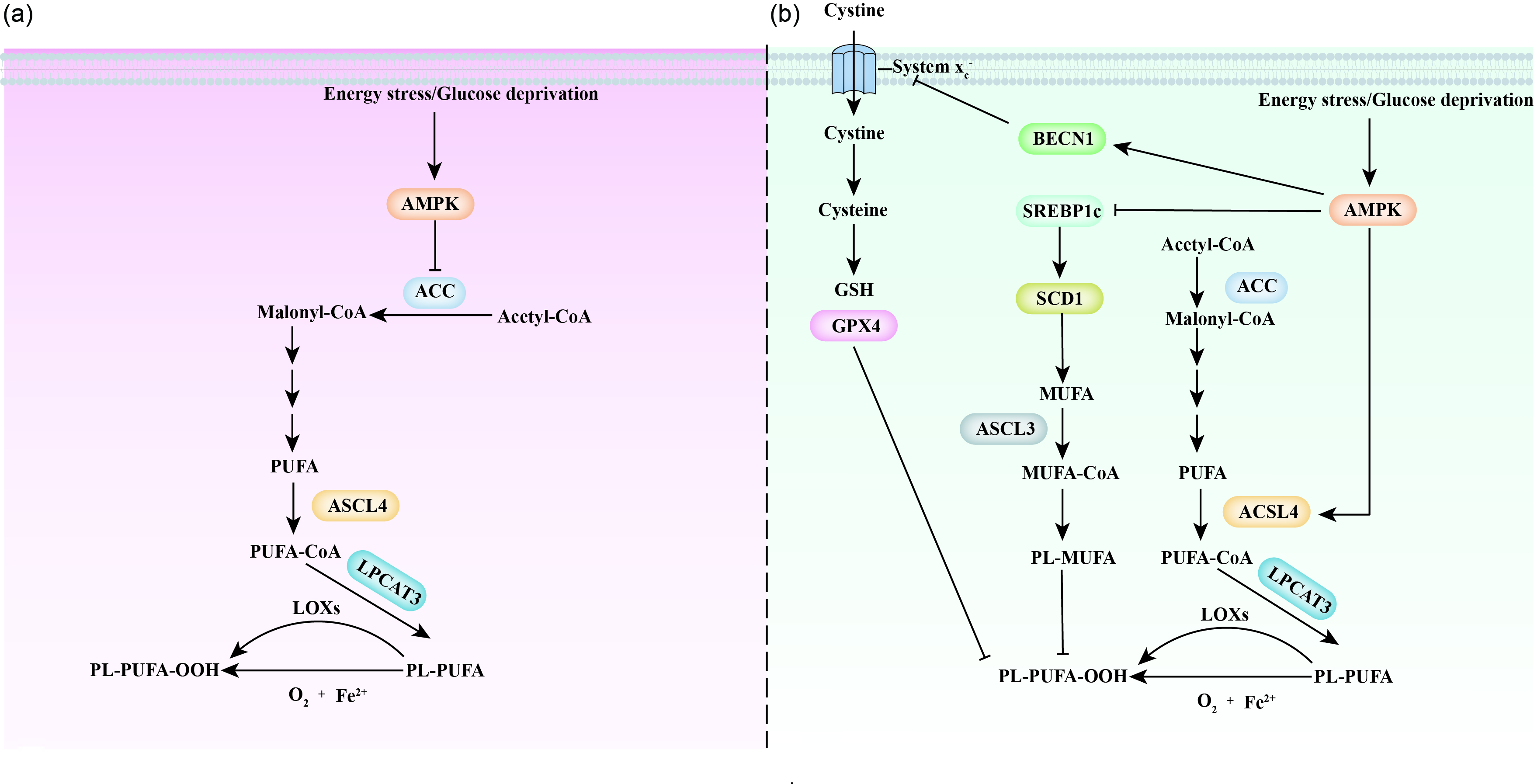
Fig. 5. Ferroptosis and the AMPK signalling pathway. Inconsistent and contradictory results have been obtained in studies of AMPK and ferroptosis. (a) A group of scientists found that AMPK prevents ferroptosis by decreasing PUFA levels and increasing MUFA levels through the inhibition of ACC function. (b) Another group of scientists promotes an alternative view, suggesting that AMPK promotes ferroptosis with an increase in PUFA levels and a decrease in MUFA levels through activation of ACSL4 and inactivation of the SREBP1c/SCD1 pathway. In addition, AMPK also inhibits the activity of amino acid transport system xc − via BECN1, which further triggers the initiation of ferroptosis. ACC, Acetyl-CoA carboxylase; ACSL 3/4, acyl-CoA synthetase long-chain 3 and 4; AMPK, 5'AMP- activated protein kinase; BECN1, beclin-1; GPX4, glutathione peroxidase 4; GSH, glutathione; LOXs, lipoxygenases; LPCAT3, lysophospholipid O-acyltransferase 3; PL-MUFA, monounsaturated fatty acids-containing phospholipid; PL-PUFA, polyunsaturated fatty acid-containing phospholipid; SCD1, stearoyl-CoA desaturase-1; SREBP1c, sterol regulatory element binding protein-1c.
Iron storage and export are both critical processes that regulate intracellular iron levels (Fig. 3). Cellular free iron is stored by ferritins, which are composed of ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1). Knocking down Ftl and Fth1 increases the concentration of free iron, sensitising cells to ferroptosis triggered by GPX4 inhibition(Reference Brown, Amante and Chhoy16). Similarly, overexpression of mitochondrial ferritin inhibits erastin-induced ferroptosis(Reference Wang, Chang and Wu17). In addition to ferritin, multiple mitochondrial proteins (such as NFS1 cysteine desulphurase, CDGSH iron sulphur domain 1 and CDGSH iron sulphur domain 2) also regulate cellular iron concentrations by manipulating iron–sulphur cluster biogenesis(Reference Alvarez, Sviderskiy and Terzi18–Reference Kim, Shin and Lee21). Iron chelators (such as deferoxamine, deferasirox, deferiprone, ciclopirox and dexrazoxane) mimic the effects of ferritin and are widely used to treat iron overload diseases(Reference Chen, Yu and Kang22). The other way to relieve iron overloading is through the regulation of iron export. Solute carrier family 40 member 1 (SLC40A1), which is also known as ferroportin, is the only known transporter of iron exportation. Siramesine is a sigma-2 receptor ligand, which is a lysosomotropic agent that can be used to treat depression(Reference Heading23), while lapatinib is originally identified as a dual tyrosine kinase inhibitor of ErbB1 and ErbB2 tyrosine kinase receptors(Reference Xia, Gerard and Liu24). The combination of siramesine and lapatinib has been reported to synergically regulate ferroptosis through iron regulatory proteins(Reference Ma, Henson and Chen25). Overexpression of SLC40A1 inhibits siramesine- and lapatinib-induced ferroptosis, whereas knockdown of SLC40A1 promotes ferroptosis(Reference Ma, Henson and Chen25). Consistently, prominin 2 (PROM2), a member of the prominin family of pentaspan membrane glycoproteins, regulates exosome-dependent iron exportation and desensitises cells to ferroptosis(Reference Brown, Amante and Chhoy16). Some regulators participate in the modulation of cellular iron homeostasis. Iron-regulatory proteins 1 and 2 (IRP1 and IRP2) regulate the genes involved in iron uptake, export, storage and utilisation(Reference Zhang, Ghosh and Rouault26,Reference Volz27) .
4. Ferroptosis and selenium
Selenium was discovered approximately 200 years ago by Swedish scientists. This element was originally regarded as a toxin in animals because livestock fed selenium-enriched plants suffered hair loss and a necrotic hoof malady(Reference Franke28). With the gradual understanding of selenium, scientists have found that selenium is toxic at high levels but essential at low levels(Reference Hatfield, Tsuji and Carlson29). Insufficient selenium induces various diseases, such as cancer, inflammation, immune disorders and nervous system diseases.
Selenium is a critical component of selenoproteins. Briefly, selenide (HSe−) is converted into selenophosphate (H2SePO3 −), which is incorporated into P-Ser-tRNA[Ser]Sec and used for selenoprotein synthesis(Reference Conrad and Proneth30). Knockout of the Sec-specific tRNA gene Trsp (nuclear encoded tRNA selenocysteine 2) leads to lethality in mice during embryonic development(Reference Bosl, Takaku and Oshima31). To date, approximately twenty-five selenoproteins have been identified in humans(Reference Gladyshev, Arner and Berry32,Reference Kryukov, Castellano and Novoselov33) . Among these selenoproteins, GPX4 is the only antioxidant enzyme protecting cells from lipid peroxidation and ferroptosis. Similar to Trsp deficiency, knocking out GPX4 in mice is lethal during the early embryonic period. Importantly, selenium utilisation by GPX4 is critical to inhibit ferroptosis(Reference Ingold, Berndt and Schmitt34). The replacement of the catalytically active selenium to a cysteine in GPX4 makes cells highly sensitive to peroxide-induced ferroptosis(Reference Ingold, Berndt and Schmitt34). The evidence obtained from this mutation indicates that selenolate-based catalysis of GPX4 is critical to prevent ferroptosis in cells.
Increasing selenium intake is an efficient way to increase the cell selenium concentration and protect cells from ferroptosis (Fig. 3). Dietary selenium is usually taken up as selenoproteins. Selenomethionine (SeMet, organic form) is absorbed most rapidly by all the intestinal segment, while selenate and selenite (inorganic form) can only be efficiently absorbed by the ileum(Reference Vendeland, Butler and Whanger35). Selenium salts are required to be converted into selenoamino acids before being absorbed by the intestine(Reference Wolffram, Ardüser and Scharrer36). Thus, for dietary sources of selenoproteins, organic forms of selenium are absorbed more efficiently than inorganic forms. Selenoproteins can then be transported into the liver for selenoprotein P (SELENOP) synthesis(Reference Gladyshev, Arner and Berry32). Subsequently, SELENOP is delivered to target organs, which are recognised by low-density lipoprotein receptor-related protein 2 and 8 (LRP2 and LRP8)(Reference Burk and Hill37,Reference Chiu-Ugalde, Theilig and Behrends38) .
Ingestion of selenocysteine-containing peptides activates homeostatic transcription to inhibit ferroptosis and alleviate haemorrhagic or ischaemic stroke(Reference Alim, Caulfield and Chen39). Notably, ingested selenium not only acts as a component in GPX4 synthesis but also drives the transcription of GPX4 through coordinated activation of the transcription factors TFAP2c and Sp1 in neurons(Reference Alim, Caulfield and Chen39). Interestingly, selenium deficiency drives adaptive transcription to attenuate ferroptotic insults, but this adaptation appears to be insufficient to protect cells from undergoing ferroptosis(Reference Alim, Caulfield and Chen39). When dietary selenium is limited, selenium is preferentially enriched in the brain and testes, which indicates the predominant function of selenium-dependent enzymes in these organs(Reference Schomburg and Schweizer40). This selective selenium distribution might partially explain why Parkinson’s disease, Alzheimer’s disease, dementia, seizures, coordination and cognitive decline are commonly identified in selenium-deficient individuals(Reference Rayman41). Together, nutritional supplementation with selenium is an efficient way to protect the cell from ferroptosis and its related metabolic diseases through the regulation of GPX4 in selenium-deficient individuals.
5. Ferroptosis and CoQ10
Coenzyme Q10 (CoQ10, also known as ubiquinone), a vitamin-like substance, is a lipid-soluble antioxidant used for the treatment of cardiovascular disease. CoQ10 maintains the mitochondrial membrane potential, promotes ATP synthesis and inhibits ROS generation. CoQ10 protects neuronal cells from oxidative stress and prevents related neurodegenerative diseases, including Parkinson’s and Alzheimer’s diseases(Reference McCarthy, Somayajulu and Sikorska42,Reference Flint Beal and Shults43) .
Recently, CoQ10 has been reported to prevent the production of lipid peroxides. In addition to the canonical GSH-based GPX4 pathway, the non-mitochondrial CoQ10 antioxidant system acts in parallel to protect the cell from ferroptosis(Reference Bersuker, Hendricks and Li11). Ferroptosis suppressor protein 1 (FSP1) has been identified by two individual groups as a crucial ferroptosis-resistance factor(Reference Doll, Freitas and Shah10,Reference Bersuker, Hendricks and Li11) . In the plasma membrane, FSP1 functions as an oxidoreductase to reduce ubiquinone to ubiquinol and prevents lipid peroxidation. In addition, CoQ10 increases the phosphorylation of AMPK(Reference Xie, Wang and Jin44–Reference Lee, Lee and Kim46), which might be the other pathway that prevents ferroptosis. However, since the biological effects of AMPK signalling on ferroptosis are still unclear (additional details are discussed in the AMPK and ferroptosis sections), more research is warranted to determine whether CoQ10 is connected to ferroptosis through AMPK.
To date, no direct in vivo or in vitro evidence has indicated that administration of CoQ10 can rescue cells from ferroptosis. However, over many decades, the pharmaceutical use of CoQ10 has been demonstrated to restore respiratory chain activities and prevent oxidative stress. Dietary supplementation with CoQ10 prevents the initiation of lipid peroxidation(Reference Mohr, Bowry and Stocker47,Reference Choy, Deng and Hou48) , which is consistent with the results observed in vitro (Reference Ahmadvand, Mabuchi and Nohara49). Similar to CoQ10, the CoQ10 analogue idebenone also halts cellular lipid peroxidation(Reference Shimada, Skouta and Kaplan50). In addition to dietary supplementation, CoQ10 is also synthesised through the mevalonate pathway. It has been reported that an inhibitor of HMG-CoA reductase (cerivastatin) or a scavenger of CoQ10 (compound FIN56) sensitises cells to ferroptosis(Reference Shimada, Skouta and Kaplan50). Similarly, 4-chlorobenzoate inhibits CoQ10 production and triggers ferroptosis(Reference Bersuker, Hendricks and Li11). Thus, dietary supplementation with CoQ10 or an increase in CoQ10 de novo synthesis might efficiently protect cells from ferroptosis.
6. Vitamin E and ferroptosis
Discovered approximately 100 years ago, vitamin E is one of most predominant inhibitors of ferroptosis(Reference Evans and Bishop51). Dietary vitamin E comprises eight homologues: α-, β-, γ- and δ-tocopherols and α-, β-, γ- and δ-tocotrienols. All of these isoforms are found to break the autoxidation chain reaction, especially α-tocopherol, which has the highest antioxidant activity(Reference Burton and Ingold52,Reference Khanna, Roy and Ryu53) . Thus, clinial symptoms of vitamin E deficiency are similar to those of selenium deficiency. LOX-mediated lipid peroxidation has been implicated as a central event in ferroptosis. Tocotrienols and tocopherols regulate ferroptosis mainly through their competition with LOX (through a corking mechanism) for PUFA-substrate binding sites(Reference Kagan, Mao and Qu54). Tocotrienols are more effective than tocopherols in protecting cells from ferroptosis, as they are more effective LOX inhibitors(Reference Kagan, Mao and Qu54). Mechanistically, vitamin E inhibits LOX by reducing the non-haem iron in the enzyme (converts Fe3+ to Fe2+).
Dietary supplementation with the lipophilic antioxidant vitamin E is an effective way to prevent ferroptosis and its related diseases. In an in vivo study, Gpx4-knockout mice fed a vitamin-E-deficient diet exhibited increased lipid peroxidation and hippocampal neurodegeneration(Reference Hambright, Fonseca and Chen55). Similarly, mice with endothelial cell knockout of Gpx4 appear normal when fed a vitamin-E-sufficient diet but abnormal when fed a vitamin-E-insufficient diet(Reference Wortmann, Schneider and Pircher56). Dietary supplementation with vitamin E restores the dysfunction of T cells induced by Gpx4 deficiency(Reference Matsushita, Freigang and Schneider57). In addition, severe embryo lipid peroxidation is observed when vitamin E is absent. Dietary supplementation with vitamin E during gestation prevents disruption in mouse embryonic fibroblasts, cortical neurons and even fetal lethality caused by GPX4 knockout(Reference Carlson, Tobe and Yefremova58,Reference Seiler, Schneider and Forster59) . Recently, α-tocopherol hydroquinone, an endogenous metabolite of vitamin E, was implicated as a more powerful inhibitor of ferroptosis than vitamin E(Reference Hinman, Holst and Latham60).
To improve the biological function of vitamin E as ferroptosis inhibitors, scientists have attempted to develop better analogues of vitamin E. Tetrahydronaphthyridinols (THN) constitute a specific group of aza-phenols, which are considered the most promising synthetic compounds to inhibit ferroptosis(Reference Valgimigli and Pratt61). The speed at which THN react with peroxyl radicals is almost 30-fold faster than that of α-tocopherol(Reference Nam, Rector and Kim62). THN with alkyl chains ranging from twelve to fifteen carbons are more effective in protecting cells from ferroptosis than the classical ferroptosis inhibitors Fer-1 and Lip-1(Reference Zilka, Shah and Li63). This evidence indicates that THN can efficiently inhibit ferroptosis.
7. Fatty acids and ferroptosis
Fatty acids (FA) are lipids that not only act as an energy sources and constitute portions of cellular membranes but also play multiple physiological roles. FA can be classified into saturated fatty acids (SFA), MUFA and PUFA based on the degree of hydrocarbon chain saturation. PUFA are predominant components in cellular, mitochondrial and retinal membranes. Cells (such as neurons) with higher levels of PUFA are more sensitive to ferroptosis(Reference Doll, Proneth and Tyurina64).
By using quantitative redox lipidomics, a study made clear that ferroptosis occurs in cells with high levels of plasma membrane phosphatidylethanolamines (PE), which are a class of specific phospholipids with two fatty acid chains – arachidonic acid (AA) and adrenic acid (AdA)(Reference Kagan, Mao and Qu54). Fatty acid elongation by very-long-chain fatty acid protein 5 (ELOVL5) and fatty acid desaturase 1 (FADS1) is required to maintain the intracellular levels of AA and AdA(Reference Lee, Nam and Son65). Blockage of AA or AdA esterification into PE efficiently prevents ferroptosis in cells. FA must be activated by acyl-CoA synthetase (ACS) before they can be incorporated into triacylglycerol (TAG)(Reference Tang, Zhou and Hooi66). Among the five isoforms of acyl-CoA synthetase long-chain family members (ACSL), ACSL4 is specific for the activation of AA and AdA(Reference Doll, Proneth and Tyurina67). Intriguingly, ACSL4 seems to serve as a double-edged sword in terms of cell survival. On the one hand, ACSL4 decreases fatty acid-induced lipotoxicity(Reference Das68). On the other hand, ACSL4 drives esterification of AA and AdA into PE, which triggers ferroptosis(Reference Kagan, Mao and Qu54). Excess AA increases Fenton reaction-induced oxidative stress with the up-regulation of anti-inflammatory lipoxinA4 (LXA4) and down-regulation of prostaglandin E2 (PGE2) (LXA4 and PGE2 are proposed to promote and inhibit cell growth, respectively), which further induce cell death(Reference Das68). Thus, the effects of PUFA (AA and AdA) to promote cell survival or induce ferroptosis may be dose dependent(Reference Tang, Zhou and Hooi66).
Administration of cells with PUFA is an efficient way to promote ferroptosis. PUFA, especially AA, can induce inflammatory bowel disease (IBD) by triggering ferroptosis in Gpx4-deficient mice(Reference Mayr, Grabherr and Schwärzler69). Dietary ingestion of the PUFA dihomo-γ-linolenic acid (DGLA; 20:3 n-6) induces ferroptosis in the nematode Caenorhabditis elegans and human cells(Reference Perez, Magtanong and Dixon70). These experiments propose that the enrichment of PUFA in the plasma membrane makes cells more susceptible to ferroptosis.
In contrast to PUFA, the administration of MUFA causes cells to enter a ferroptosis-insensitive state. Exogenous MUFA decrease lipid ROS accumulation and inhibit ferroptosis in an acyl-CoA synthetase long-chain family member 3 (ACSL3)-dependent manner(Reference Magtanong, Ko and To71). Mechanistically, MUFA are converted into MUFA-PL by ACSL3, and they displace PUFA in the plasma membrane and hinder lipid ROS accumulation(Reference Magtanong, Ko and To71). Clinically, increased serum MUFA prevent ferroptosis induced by neurodegeneration or kidney failure(Reference Stockwell, Friedmann Angeli and Bayir72). Overloading intracellular SFA triggers a specified apoptosis process called lipotoxicity(Reference Garbarino and Sturley73). Intriguingly, MUFA also protect cells from apoptotic lipotoxicity caused by SFA. Thus, modification of dietary MUFA levels can be an efficient way to regulate ferroptosis and lipotoxicity.
In addition to replacing PUFA with MUFA in the plasma membrane, increasing ether lipid abundance is an alternative way to protect cells from ferroptosis. Endogenous ether lipids are regarded as oxidative ‘sinks’ that protect PUFA chains from oxidation(Reference Shi, Tarazona and Brock74,Reference Wallner and Schmitz75) . Infants with inherited ether lipid synthesis enzyme deficiency present with severe growth and mental disorders(Reference Wanders and Waterham76). The synthesis of ether lipids protects C. elegans and human cancer cells from undergoing ferroptosis(Reference Perez, Magtanong and Dixon70). Future studies need to clarify the potential methods to manipulate the synthesis of ether lipids.
8. mTOR signalling and ferroptosis
mTORC1 is a master regulator of protein synthesis, which is triggered by multiple stimuli, such as growth factors, energy levels and amino acids(Reference Jewell, Russell and Guan77–Reference Gomes and Blenis79). Amino acids regulate mTORC1 lysosomal localisation and subsequently activate mTORC1 through Rag GTPase-dependent and Rag GTPase-independent pathways(Reference Liu and Sabatini80–Reference Jewell, Kim and Russell82). The small G-protein Ras homologue enriched in the brain (Rheb) contains a CAAX (C = Cys, A = aliphatic, X = terminal amino acid) box, which facilitates its localisation to lysosomes(Reference Menon, Dibble and Talbott83,Reference Sancak, Bar-Peled and Zoncu84) . Rheb directly interacts with mTORC1 and triggers conformational changes in mTOR, triggering its activation(Reference Yang, Jiang and Li85). In contrast to amino acids, growth factors activate mTORC1 by stimulating the dissociation of the tuberous sclerosis complex (TSC) from the lysosome and Rheb(Reference Menon, Dibble and Talbott83). TSC is a negative regulator of mTORC1, and its mutation constitutively activates mTORC1(Reference Carsillo, Astrinidis and Henske86–Reference Kwiatkowski and Manning88). mTORC1 is composed of three components: the catalytic component mTOR, the regulatory-associated protein of mTOR (Raptor) and mammalian lethal with Sec13 protein 8 (mLST8, also known as GβL). Raptor recognises mTORC1 substrates, and mLST8 acts as a positive regulator of mTOR activity. Activation of mTORC1 regulates mRNA translation and protein transcription through the phosphorylation of eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) and S6 kinase (S6K1), which are the two key downstream substrates of the mTORC1 complex.
Recently, activation of mTORC1 was reported to prevent lipid peroxide accumulation and suppress ferroptosis(Reference Yi, Zhu and Wu89). This effect was partially due to mTORC1 signalling participating in the regulation of lipid metabolism. As a transcriptional target of sterol regulatory element-binding protein 1 (SREBP1), stearoyl-coenzyme A desaturase-1 (SCD1) is critical for the conversion of SFA into MUFA. Synthesised MUFA displace PUFA in the plasma membrane and inhibit the accumulation of lipid ROS(Reference Magtanong, Ko and To71). Knocking out either SREBP1 or SCD1 impaired the effects of mTORC1 on ferroptosis(Reference Yi, Zhu and Wu89). Exogenous MUFA palmitoleic acid (16:1, PO) or oleate acid (18:1, OA) supplementation also restores ferroptosis resistance when mTORC1 signalling is depressed. This evidence indicates that mTORC1 signalling protects cells from ferroptosis through the SREBP1/SCD1-mediated synthesis of MUFA (Fig. 4).
Another connection between mTORC1 signalling and ferroptosis is GPX4 (Fig. 4). Han et al. (Reference Han, Jiang and Gu90) also observed that inhibition of mTORC1 was accompanied by a decrease in GPX4 level. In addition, some preliminary evidence suggests that mTOR signalling might be required to maintain the expression of SLC7A11(Reference Zhang, Liu and Liu91). SLC7A11 is critical for cystine transportation, which is required for GSH synthesis and GPX4 activity.
In addition, mTORC1 is proposed to regulate ferroptosis directly through the modulation of the iron transport system (Fig. 4). Some evidence indicates that the activation of mTORC1 decreases the cellular iron concentration. mTORC1 phosphorylates iron–sulphur cluster assembly enzyme (ISCU) at serine 14 and increases iron–sulphur cluster (ISC) assembly(Reference Guan and Wang92,Reference La, Yang and Dennery93) . Stabilisation of ISCU further inhibits IRP1/IRP2 function, promoting ferroportin 1 (Fpn1, a protein that mainly mediates iron export) and decreasing cellular iron concentration(Reference Bayeva, Khechaduri and Puig94). Notably, however, the effect of mTORC1 on cellular iron concentration is inconsistent. Bayeva et al. (Reference Bayeva, Khechaduri and Puig94) reported that activation of mTORC1 negatively regulates the expression of tristetraprolin, which binds to Transferrin receptor 1 (TfR1) mRNA to enhance its degradation. TfR1 is a protein that mainly mediates iron import. Thus, activation of mTORC1 is proposed to increase cellular iron concentration. Future experiments are needed to clarify the effect of mTOR on iron transport.
In clinical research, mTORC1 plays a vital role in iron metabolism in cardiomyocytes. Approximately 80 % of mTOR-treated orthotopic heart transplant patients have functional iron deficiency(Reference Przybylowski, Malyszko and Macdougall95). In cardiomyocytes, activation of mTOR protects against ferroptosis partially by decreasing ROS production(Reference Baba, Higa and Shimada96). An in-depth understanding of the cross talk between mTOR signalling and ferroptosis will lead to a novel therapy for patients with myocardial infarction.
9. Energy sensing and ferroptosis
Energy homeostasis is important to sustain the biological function of cells. AMPK acts as the predominant energy sensor in eukaryotic cells(Reference Hardie97). The adenylation system (ATP + ADP + AMP) has been measured to estimate cellular energy levels since 1976(Reference Atkinson and Fall98,Reference Atkinson and Walton99) . Increased ADP and AMP indicate a decrease in cellular energy status. AMPK is a heterotrimer that contains an α catalytic subunit, β regulatory subunit and γ regulatory subunit(Reference Xiao, Sanders and Underwood100). AMPK subunits include multiple isoforms, such as α1, α2, β1, β2, γ1, γ2 and γ3(Reference Hardie, Hawley and Scott101). The classical pathway to AMPK activation (phosphorylation at Thr172) is dependent on increased levels of ADP and AMP(Reference Hawley, Boudeau and Reid102).
Recently, AMPK has been shown to be a critical regulator of ferroptosis (Fig. 5). Cells with AMPK activation are more resistant to ferroptosis, while those with AMPK inactivation are more sensitive to ferroptosis(Reference Lee, Zandkarimi and Zhang103–Reference Currais, Huang and Goldberg105). This AMPK effect is due to the energy stress-impaired biosynthesis of PUFA, especially arachidonic acid (C20:4) and adrenic acid (C22:4), which are required for ferroptosis(Reference Kagan, Mao and Qu54). Mechanistically, AMPK regulates ferroptosis partially through phosphorylation of ACC, which inhibits PUFA-containing lipid biosynthesis and makes cells more resistant to ferroptosis(Reference Lee, Zandkarimi and Zhang103,Reference Currais, Huang and Goldberg105) . Inhibition (treatment with 5-(tetradecyloxy)-2-furoic acid) or site mutation (mutation of ACC1 Ser79 to ACC1 Ala79; mutation of ACC2 Ser212 to ACC Ala212) of ACC mimics the protective role of AMPK activation in ferroptosis(Reference Lee, Zandkarimi and Zhang103). This evidence proposes a protective mechanism that protects cells from organ injury induced by energy failure. For instance, energy stress was found to protect against renal ischaemia–reperfusion injury(Reference Lee, Zandkarimi and Zhang103).
Notably, there is a fierce debate regarding the role of AMPK in the regulation of ferroptosis (Fig. 5). Cells under energy stress commonly show increased ROS production, which indicates the promotion of ferroptosis(Reference Hay106). Song et al. (Reference Song, Zhu and Chen107) observed that activation of AMPK is required for BECN1-induced ferroptosis, a finding in opposition to the results shown by Lee et al. (Reference Lee, Zandkarimi and Zhang103). BECN1 is a crucial regulator of macroautophagy/autophagy(Reference Liang, Jackson and Seaman108). Mechanistically, activation of AMPK phosphorylates BECN1 at Ser90/93/96 and promotes BECN1–SLC7A11 complex formation; this complex blocks system xc −-regulated homeostasis in GSH and induces ferroptosis(Reference Song, Zhu and Chen107,Reference Zhong, Tian and Ma109) . In addition, Zhao et al. (Reference Zhao, Li and Yao110) found that inactivation of AMPK promotes SREBP1/SCD1 expression, which inhibits ferroptosis with the production of MUFAs. However, it is worth noting that activation of SREBP1, a critical transcriptional regulator of lipogenesis, is proposed to promote the production of both MUFA and PUFA, but not specific MUFA. Thus, more studies are required to confirm this finding. Notably, AMPK is the upstream and negative regulator of mTORC1(Reference Xu, Ji and Yan111). As activation of mTORC1 protects the cell from ferroptosis, it is reasonable to hypothesise that the activation of AMPK promotes ferroptosis.
Mitochondria are predominant organelles that participate in energy production and the regulation of ferroptosis. It is worth noting that the potential mechanism of mitochondria-triggered ferroptosis is also controversial. Some evidence indicates that the mitochondrial TCA cycle and electron transfer chain increase lipid peroxide accumulation and promote ferroptosis induced by cysteine deprivation(Reference Gao, Yi and Zhu112,Reference Basit, Van Oppen and Schöckel113) . However, one study reported that functional mitochondrial electron transfer chain is not required in erastin-induced cell death(Reference Dixon, Lemberg and Lamprecht1). In addition, mitochondria have many other functions independent of the electron transfer chain that could contribute to ferroptosis, such as tumor suppressor protein p53(Reference Zhang, Gai and Ding114) and enzymes involved in mitochondrial respiration(Reference Zhang, Gai and Ding114,Reference Venkatesh, Li and Saito115) . Interestingly, mitochondria were involved only in ferroptosis caused by cysteine deprivation but not by GPX4 inhibition (induced by RSL3)(Reference Gao, Yi and Zhu112). This result might be due to inhibited GPX4 quickly enhancing the accumulation of lipid ROS in other pathways that remain active, a supposition that requires further study. Clinically, MitoTEMPO (a superoxide scavenger targeted to the mitochondria) decreases lipid peroxidation and protects the heart from ischemia/reperfusion (IR)-induced damage(Reference Fang and Wang116). Understanding the physiological functions of mitochondria and AMPK in the regulation of ferroptosis will help in identifying novel pharmacological targets for ferroptosis-related diseases.
10. Conclusions and perspectives
Tremendous progress has been made in the field of ferroptosis in recent years. Multiple nutrients have been reported to regulate redox homeostasis and lipid peroxidation. To date, nutrients participating in ferroptosis regulation are classified into three categories: (1) selenium, iron, vitamin E and CoQ10, which mediate the production of PLOOH generated in cells; (2) exogenous fatty acids that manipulate the fatty acid profiles of cellular membranes (mainly in monogastric animals); and (3) amino acids and energy stress, which trigger the activation of mTORC1 and AMPK signalling pathways related to ferroptosis. However, our understanding of ferroptosis and the cross talk of its factors with nutrient signalling factors is still limited but is expected to explode in the coming years. Critical issues in this field that must be addressed are listed below.
First, ferroptosis is not only involved in diverse diseases but might also be critical to regulate normal physiology. Under physiological conditions, iron and ROS are involved in the regulation of cellular biological functions. Once a threshold is crossed, iron overload and oxidative damage can cause ferroptotic cell death. MDA, a critical marker of lipid peroxidation, is generally increased in animals under heat and oxidative stress(Reference Gaweł, Wardas and Niedworok117,Reference Gonzalez-Rivas, Chauhan and Ha118) . In contrast to classical ferroptosis, the accumulation of MDA under heat and oxidative stress is usually insufficient to induce a notable increase in the cell death rate. Thus, the possibility that sublethal ferroptosis can reprogram cell metabolism and biological functions in different cells and/or organs needs to be studied in the future. In addition, the thresholds for inducing sublethal ferroptosis-related damage are unknown. Nutritional strategies may be an efficient way to manipulate sublethal ferroptosis-related disorders.
Second, current findings on the cross talk between ferroptosis and nutrient sensing factors are not consistent, especially results related to ferroptosis and AMPK. To date, an intense debate rages regarding the biological function of AMPK in cancer cells(Reference Ross, MacKintosh and Hardie119,Reference Hardie120) . The signalling model of AMPK and ferroptosis might be more complicated than the current model. For instance, AMPK regulates not only lipid metabolism through the phosphorylation of ACC1 and ACC2 but also sterol regulatory element-binding protein 1c (SREBP1c) and PPAR γ coactivator 1α (PGC1α)(Reference Kohjima, Higuchi and Kato121,Reference Li, Wong and Zhang122) . The expression and effects of SREBP1c and PGC1α vary by cell type. Therefore, the effects of AMPK-regulated ferroptosis could be cell type- or tissue-dependent.
Third, iron is not the only ion to trigger the Fenton reaction, which raises the question, why is ferroptosis more sensitive to liable iron concentrations? Recently, some studies have indicated that other ions also participate in the regulation of ferroptosis. For instance, under specific conditions, copper regulates the initiation of ferroptosis(Reference Maher123). Endoplasmic reticulum zinc homeostasis is closely related to ferroptosis(Reference Chen, Wu and Xu124). Thus, future studies are required to determine whether current findings are missing critical information regarding other ions with respect to ferroptosis.
In addition to the unclear connections between nutrient sensing and ferroptosis, precise biological markers for ferroptosis need to be clarified in the future. Although the expression of some genes (for instance, prostaglandin-endoperoxide synthase 2 (PTGS2/COX2), ChaC glutathione-specific γ-glutamylcyclotransferase 1 (CHAC1/BOTCH), nuclear factor erythroid 2-like 2 (NFE2L2/NRF2)) is related to the process of ferroptosis, these genes are not uniquely involved in ferroptosis(Reference Chen, Comish and Tang125). Furthermore, potential connections among ferroptosis, apoptosis and autophagy need to be explored. It is difficult to separate ferroptosis from other types of cell death. Nutrient signalling regulates not only ferroptosis but also apoptosis(Reference Pintus, Floris and Rufini126,Reference Molina, Wikstrom and Stiles127) and autophagy(Reference Lee, Wagner and Xiao128,Reference Russell, Yuan and Guan129) . Future studies need to answer the following questions: What is the precise relationship among different types of cell death? Does nutrient signalling individually regulate different types of cell death, or does it regulate cell death processes in combination?
In summary, an in-depth understanding of ferroptosis and nutrient signalling might lead to a new approach for the therapeutic intervention of ferroptosis-related diseases, such as cancer, neurodegeneration and inflammation. Further study should focus on ferroptosis in both non-pathological and the normal physiology contexts.
Financial Support
This work was supported by National Natural Science Foundation of the P.R. of China (no. 31802067 and 31872364) and the Natural Science Foundation of Guangdong Province (No. 2018A030310201).
Authors’ contributions
Yingao Qi and Shihai Zhang initiated the idea, the scope and the outline of this review paper. Yingao Qi, Xiaoli zhang, Min Tian, Shihai Zhang, Zhihui Wu, Jinghui Heng and Fang Chen studied and analysed all of the publications cited in this paper and were involved in the manuscript preparation. Shihai Zhang and Wutai Guan conducted the final editing and proofreading. All authors read and approved the final manuscript.
Conflicts of interest
The authors declare that they have no competing interests.





