



NbN-2 compliant keywords:
∙ pimavanserin: serotonin receptor inverse agonist/ antagonist (5-HT2A)
∙ clozapine: dopamine receptor antagonist (D2), serotonin receptor antagonist (5-HT2), norepinephrine receptor antagonist (α2)
∙ quetiapine: dopamine receptor antagonist (D2), serotonin receptor antagonist (5-HT2), norepinephrine reuptake inhibitor (metabolite)
Introduction
Parkinson’s disease psychosis (PDP) is an increasingly recognized nonmotor aspect of Parkinson’s disease (PD).Reference Weintraub and Stern 1 , Reference Ravina, Marder and Fernandez 2 Prior to 2016, recommendations for treating PDP focused primarily on off-label use of clozapine and quetiapine.Reference Friedman 3 , Reference Goetz, Diederich and Fénelon 4 Randomized, controlled trials demonstrated efficacy for clozapine at low doses (6.25 mg–50.0 mg/d) with limited adverse motor effects. Quetiapine failed to demonstrate clear efficacy in at least 4 of 5 clinical trials, but also had limited impact on motor function, and was very commonly prescribed in PDP.Reference Weintraub, Chen, Ignacio, Mamikonyan and Kales 5 , Reference Weintraub, Chiang and Kim 6 The limited impact of these drugs on PD motor symptoms may be attributed to the fact that both are weak D2 receptor antagonists. Both are also antagonists at serotonin 2A (5-HT2A) receptors, clozapine more potently than quetiapine (see Table 1), which may contribute to efficacy.Reference Stahl 7 Other antipsychotics that are potent D2 antagonists or partial agonists generally produce intolerable adverse motor effects and are not recommended for PDP.Reference Divac, Prostran, Jakovcevski and Cerovac 8 – Reference Black 10
Table 1 Receptor binding affinities for selected antipsychotic agentsReference Hacksell, Burstein, McFarland, Mills and Williams 46

+ weak binding affinity (100>Ki<1000);
++ moderate binding affinity (10>Ki<100);
+++ strong binding affinity (1>Ki<10);
++++ very strong binding affinity (Ki<1);
Abbreviations: 5-HT=serotonin; α=adrenergic; D=dopamine; H=histamine; M=muscarinic.
Ki (nM) values are derived from functional antagonist R-SAT™ assays (ACADIA, San Diego, CA, USA).
“-“ denotes no response.
Adapted from Hacksell et al. Neurochem Res 2014; 39: 2008–2017 and from data on file.
In 2016, the US FDA approved the first drug for treatment of psychotic symptoms in PDP.Reference Hawkins and Berman 11 Pimavanserin is a selective 5-HT2A receptor inverse agonist/antagonist with no appreciable affinity for dopaminergic receptors;Reference Vanover, Harvey and Son 12 – Reference Weiner, Burstein and Nash 14 it exerts clinically significant antipsychotic effects without impact on motor symptoms.Reference Meltzer, Mills and Revell 15 – Reference Friedman 18 Since the introduction of pimavanserin to the market, switching a PDP patient from a previous antipsychotic to pimavanserin is a common clinical consideration, motivated perhaps by lack of efficacy or side effect concerns with the previous agent. However, the details of how to implement such a switch have not been clear. An abrupt switch involves stopping a drug that may have provided partial benefit, potentially causing an interval of worsening psychosis or rebound insomnia before pimavanserin’s expected onset of action. Cross tapering, on the other hand, can raise concerns about drug interactions or QTc prolongation due to the lack of data on combined therapy with pimavanserin. In the absence of controlled trials directly comparing these approaches, we here review relevant principles to consider and provide expert suggestions for how to switch patients from atypical antipsychotics to pimavanserin.




Methods
A panel of 14 experts agreed to establish consensus recommendations aimed at identifying optimal strategies for switching from an off-label antipsychotic, especially quetiapine or clozapine, to pimavanserin for patients with PDP. Members of the group had expertise with PDP (including movement disorders neurologists and neuropsychiatrists) and with the receptor pharmacology of antipsychotics, collectively treating thousands of patients with PD and PDP, including more than 150 patients with pimavanserin. Twelve responded to a brief survey about their PDP treatment experience and preferences. This expert panel established the following objectives:
∙ To present the pharmacology of atypical antipsychotics as it relates to switching strategies
∙ To provide insights into current practice patterns and potential challenges associated with switching patients with PDP from atypical antipsychotics, especially quetiapine or clozapine, to pimavanserin
∙ To seek consensus on recommendations for best practices for how to switch patients to pimavanserin
This manuscript will review relevant antipsychotic pharmacology, published recommendations on switching antipsychotics in primary psychiatric illnesses,Reference Takeuchi, Kantor, Uchida, Suzuki and Remington 19 , Reference Cerovecki, Musil and Klimke 20 and the pros and cons of various options from the perspective of substantial collective clinical experience. The manuscript was drafted by the authors, and all agreed to the final content.
PDP Diagnosis
The first step in treating PDP is to verify the diagnosis. While definitions of PDP have differed over the years, a National Institute of Neurological Disorders and Stroke–National Institute of Mental Health working group proposed diagnostic criteria for PDP in 2007 that require at least 1 of the following: illusions, false sense of presence, hallucinations, or delusions. These features should not begin prior to the onset of PD motor symptoms, and should be present for at least 1 month, either as recurrent or continuous symptoms. Importantly, acute delirium and other medical, neurological, or psychiatric causes should be excluded.Reference Ravina, Marder and Fernandez 2 Additionally, clinical judgment should be used in considering the need for treatment in patients with infrequent or non-bothersome symptoms.
Treatment of PDP with Atypical Antipsychotics
Prior to 1990, medications for psychosis were limited to antidopaminergic agents.Reference Li, Snyder and Vanover 21 However, administration of first-generation, or typical, antipsychotic drugs resulted in unacceptable exacerbation of motor symptoms due to the blockade of dopamine receptors. Therefore, these drugs and other strongly antidopaminergic treatments are generally not used to treat PDP, a recommendation supported by the Movement Disorder Society (MDS),Reference Seppi, Weintraub and Coelho 22 American Geriatrics Society, 23 and American Academy of Neurology.Reference Miyasaki, Shannon and Voon 24
Between 1993 and 2003, several second-generation, or atypical, antipsychotic drugs were evaluated in patients with PDP. Although these agents have been approved as effective in the treatment of schizophrenia, they vary greatly in their pharmacokinetic (PhK) and pharmacodynamic (PhD) properties. For example, receptor binding profiles include differing affinities for an array of receptors types, including dopamine, serotonin, acetylcholine, histamine, and adrenergic receptors (Table 1). The differing PhK and PhD properties in general, and the potency in binding dopamine receptors in particular, lead to variable side effect profiles. Clinical experience has shown that drugs such as risperidone,Reference Meco, Alessandria, Bonifati and Giustini 25 , Reference Workman, Orengo, Bakey, Molinari and Kunik 26 olanzapine,Reference Nichols, Hartlein, Eicken, Racette and Black 27 , Reference Ondo, Levy, Vuong, Hunter and Jankovic 28 aripiprazole,Reference Fernandez, Trieschmann and Friedman 29 and ziprasidoneReference Gray 30 , Reference Schindehütte and Trenkwalder 31 may result in intolerable motor side effects for PD patients even at low doses, and therefore are widely considered to be poor choices for treatment of PDP. However, despite lacking a US Food and Drug Administration (FDA) indication for use in treating PDP, 2 of the atypical antipsychotics, clozapine and quetiapine, have frequently been used to manage the disorder.
Clozapine is an atypical antipsychotic drug with a complex pharmacology (Table 1). It is a potent antagonist at the H1, 5-HT2B, 5-HT2C, and α1 receptors; a potent inverse agonist at the 5-HT2A receptors; and a relatively weak antagonist at the D2 receptors. Inverse agonists are defined as drugs that, when bound to a receptor, produce the opposite intracellular effect as the endogenous ligand, even in the absence of the endogenous ligand. Pivotal clinical studies showed that clozapine improved PDP without substantially worsening parkinsonism. 32 – Reference Morgante, Epifanio and Spina 35 However, clozapine carries the risk of neutropenia,Reference Alvir, Lieberman, Safferman, Schwimmer and Schaaf 36 which requires frequent blood testing. The use of clozapine in PDP is also potentially further constrained by other limiting side effects, such as sedation, orthostatic hypotension, or anticholinergic effects including constipation.
Quetiapine, another atypical antipsychotic medication that has been evaluated in PDP, exerts potent antagonism at the H1 receptor and relatively weaker binding at D2 and 5-HT2A receptors (Table 1). Quetiapine showed lesser antipsychotic benefit than clozapine in some trials,Reference Merims, Balas, Peretz, Shabtai and Giladi 37 , Reference Klein, Prokhorov, Miniovich, Dobronevsky and Rabey 38 but did not differ significantly in another.Reference Morgante, Epifanio and Spina 35 However, in 4 of 5 randomized controlled trials, quetiapine did not significantly reduce psychotic symptoms compared with placebo when objectively assessed using the Brief Psychotic Rating Scale.Reference Fernandez, Okun and Rodriguez 39 – Reference Shotbolt, Samuel, Fox and David 43 The fifth study specifically excluded patients with delusions, and enrolled 16 patients, 5 of whom discontinued prematurely (including 4 of the 8 patients assigned to quetiapine). The treatment groups differed significantly only on secondary outcome measures.Reference Fernandez, Okun and Rodriguez 39 In terms of safety, reported side effects in these studies included sedation, confusion, and orthostatic hypotension, which sometimes led to treatment discontinuation.Reference Fernandez, Okun and Rodriguez 39 – Reference Shotbolt, Samuel, Fox and David 43 Among our panel, sedation was the quetiapine side effect that most commonly motivated a switch to pimavanserin (cited by all respondents), followed by hypotension (cited by two-thirds). Nevertheless, many experts continue to prescribe quetiapine in PDP, based in part on their clinical experience.Reference Friedman 44 For instance, most of our expert panel reported that pimavanserin was their first-line medication for PDP, but one third chose quetiapine.
The Evidence-Based Medicine Review from the MDS, last published in 2011,Reference Seppi, Weintraub and Coelho 22 , Reference Miyasaki, Shannon and Voon 24 concluded that clozapine is efficacious for treating PDP with a safety risk that requires specialty monitoring, while quetiapine has an acceptable safety risk but may not be tolerated in this population at doses needed for benefit. Due to the monitoring requirements and safety risks of clozapine, and the lack of convincing efficacy associated with quetiapine, pimavanserin may be considered a first option for the treatment of PDP. A recent evidence-based review recommended pimavanserin and clozapine as first-line agents,Reference Wilby, Johnson, Johnson and Ensom 45 and the most recent American Academy of Neurology classification recommended pimavanserin and clozapine at level B, and quetiapine at level C.Reference Hawkins and Berman 11
Treatment of PDP with Pimavanserin
In 2016, pimavanserin (NUPLAZID®, ACADIA Pharmaceuticals Inc.) became the first drug approved by the FDA for the treatment of hallucinations and delusions associated with PDP. 13 Pimavanserin binds with high affinity to 5-HT2A receptors as an inverse agonist, with a 5-fold lower affinity to 5-HT2C receptors, and has no appreciable affinity for 5-HT2B, dopaminergic, muscarinic, histaminergic, or adrenergic receptors or for calcium channels.Reference Vanover, Harvey and Son 12 , Reference Hacksell, Burstein, McFarland, Mills and Williams 46 An early phase 3 clinical trial with subjects randomized to placebo, 8.5, or 34 mg/d showed nonsignificant benefit for the highest dose (N=298), as did a trial with a maximum daily dose of 17 mg (N=123) and a small, ascending dose, phase 2 study (https://clinicaltrials.gov/ct2/results?term=pimavanserin).Reference Meltzer, Mills and Revell 15 , Reference Hacksell, Burstein, McFarland, Mills and Williams 46 The pivotal clinical trial was a randomized, double-blind, 2-arm, phase 3 study in 199 patients. It showed that pimavanserin 34 mg daily was significantly better than placebo at reducing frequency and/or severity of hallucinations and delusions in patients with PDP as measured using the Scale for the Assessment of Positive Symptoms–Parkinson’s Disease (SAPS-PD, p=0.0014) and the SAPS Hallucinations and Delusions subscales (p=0.0011).Reference Cummings, Isaacson and Mills 16 Sleep quality, caregiver burden, and other exploratory outcome measures also significantly improved with pimavanserin. Of course, treatment response varies; in the pivotal clinical trial, 68% of pimavanserin patients had a clinically meaningful responseReference Voss, Bahr, Cummings, Mills, Ravina and Williams 47 (vs. 43% on placebo), 49% responded on the CGI-I (Clinical Global Impressions–Improvement) scale (vs. 26% on placebo), and partial response was typical (14% experienced a complete remission vs. 1% on placebo).Reference Cummings, Isaacson and Mills 16 , Reference Citrome, Norton, Chi-Burris and Demos 48 One quarter of our panel reported concern for breakthrough hallucinations or delusions as a reason for not switching some PDP patients to pimavanserin.
Importantly, pimavanserin did not worsen motor function compared with placebo, which is a major concern when treating PDP. Serious adverse events were uncommon in the clinical trials cited above, but the pimavanserin label shares with other drugs for psychosis a boxed warning of an increased risk of death in elderly patients with dementia.
Initiating Pimavanserin in PDP Patients Not on an Antipsychotic
When the decision has been made to start a patient on pimavanserin, the recommended dose is 34 mg by mouth, once daily (Figure 1). 13 Given the long half-life of pimavanserin (approximately 57 hours), starting 34 mg immediately will likely produce a gradual increase in blood concentration over approximately 2 weeks. The only exceptions to the use of the 34 mg dose included in the label are for patients who are taking strong inhibitors of cytochrome P450 (CYP) 3A4, in which case a 10 mg daily dose is recommended, and in patients taking CYP3A4 inducers, in whom “an increase in dosage may be needed.” 13 , Reference Mathis 49 In the pivotal, randomized, placebo-controlled, phase 3 trial, a decrease in psychotic symptoms from baseline was evident at 2 weeks for pimavanserin and placebo. Improvement in the pimavanserin arm separated from placebo beginning at 4 weeks, with further improvement at 6 weeks.Reference Cummings, Isaacson and Mills 16 The improvement at 6 weeks was significant and clinically meaningful.Reference Cummings, Isaacson and Mills 16 This time to onset of therapeutic action is key to bear in mind when considering switching to pimavanserin, especially when switching from drugs like quetiapine or clozapine with much shorter half-lives. Efficacy of pimavanserin was maintained at 10 weeks during an open-label extension phase of the study.Reference Mills, Isaacson and Azulay 50 Therefore, a minimum of a 6-week trial period with the 34 mg dose of pimavanserin is needed to fully assess efficacy.

Figure 1 Patients not currently taking antipsychotic medication. Start full dose of pimavanserin immediately.
Pharmacologic Principles to Consider When Switching PDP Patients from an Antipsychotic to Pimavanserin
There are a number of factors that should be considered to increase success in antipsychotic switches, including PhK and PhD characteristics, receptor selectivity, reasons for the switch (efficacy or adverse events), and urgency of the switch. Although much of the data come from studies in other populations, abrupt discontinuation of antipsychotic drugs is known to cause both withdrawal and rebound effects (Table 2). The appearance of specific withdrawal symptoms depends on the target receptor. For example, abrupt withdrawal from antipsychotic drugs targeting dopamine D2 receptors, 5-HT1A, and/or 5-HT2A receptors has been linked to withdrawal dyskinesias and rebound psychosis (Table 2), whereas abrupt withdrawal from drugs targeting histamine H1 and muscarinic M1 receptors has been linked to anxiety, agitation, and insomnia, and sometimes autonomic symptoms such as sweating, tremor, diarrhea, and other gastrointestinal symptoms (Table 2).Reference Stahl 7 , Reference Cerovecki, Musil and Klimke 20 , Reference Correll 51 The potential for rebound and withdrawal phenomena is greatest when the pre- and post-switch antipsychotics differ considerably with regard to binding affinity for specific receptors, as these effects typically involve exposure of previously blocked receptors that are no longer targeted by the post-switch antipsychotic (PhD rebound). Differences in half-lives, time to steady state, and metabolism between pre- and post-switch antipsychotics (PhK rebound) may also contribute to rebound and withdrawal symptoms (Table 2), especially if loss of 5-HT2A receptor occupancy occurs in the transition between 2 drugs.Reference Correll 51 Withdrawal effects in general may be less severe when discontinuing the lower doses of quetiapine or clozapine typically prescribed to patients with PDP than when discontinuing the higher doses of antipsychotic drugs used in patients with schizophrenia.
Table 2 Effects of blockade of receptors and potential side effects from withdrawal/rebound during switching

Abbreviations: 5-HT=serotonin; α, adrenergic; D=dopamine; EPS=extrapyramidal symptoms; H=histamine; M=muscarinic.
Adapted from Correll 2010.Reference Correll 51
Potential Risk of Concomitant Therapy with Antipsychotics and Pimavanserin During Cross Taper
Pimavanserin is generally well tolerated with no worsening of motor function.Reference Meltzer, Mills and Revell 15 – Reference Mills, Friedman and Ondo 17 The safety profile of pimavanserin is consistent with its receptor pharmacology (Table 1) and differentiates this medication from current antipsychotic drugs. Based on the tolerability profile of pimavanserin, it is anticipated that there is a low risk of exacerbating the side effects of other antipsychotic medications when pimavanserin is administered concurrently. However, as with all drug combinations, clinicians should consider history of cardiac risk factors and/or ECG data when pimavanserin is taken concurrently with other antipsychotics, as each carries a risk of modest (mean 5–8 msec) prolongation of the QTc interval.
Recommendations: Switching to Pimavanserin in Patients Currently Taking an Antipsychotic with Suboptimal Efficacy and/or Tolerability
The goals of switching a patient with PDP from a current antipsychotic medication, especially quetiapine or clozapine, to pimavanserin are (1) to avoid dopamine D2 antagonism, (2) to maintain a stable level of 5-HT2A antagonism during the transition so that efficacy is maintained throughout the switch, and (3) to avoid rebound effects whenever possible by monitoring the pharmacology of the medications involved, usually by slow down-titration of agents with anticholinergic and antihistaminic actions, such as quetiapine and clozapine.
Several principles should be considered when initiating pimavanserin in patients with PDP who are already taking an antipsychotic medication. Typical or atypical antipsychotic drugs with a high risk of motor side effects, such as haloperidol, risperidone, or olanzapine, are not recommended for patients with PDP, and these medications should be stopped as quickly as possible (Figure 2). Motor worsening may persist for 30 days, and often much longer, after these medications have been discontinued. Clozapine and quetiapine (at doses ≤100 mg) have a low propensity for worsening motor symptoms of PD.
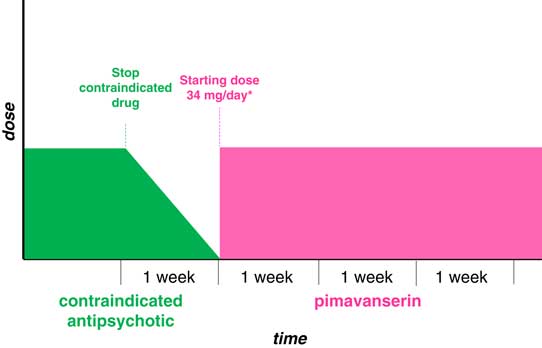
Figure 2 Patients currently taking contraindicated antipsychotic. Stop as quickly as possible, then start pimavanserin after washout.
The nature, severity, and frequency of side effects and the current dose should always be considered before discontinuing medications. If one switches to pimavanserin because of the lack of efficacy with low-to-moderate doses of quetiapine (≤100 mg/day), for most cases quetiapine may be tapered over 2–4 weeks starting 4–6 weeks after adding pimavanserin, with the goal of minimizing rebound effects (eg, insomnia) and any loss of antipsychotic benefit that might occur from abrupt discontinuation of quetiapine (Box 1, Figure 3).Reference Stahl 7 , Reference Correll 51 The duration of taper should depend on the current dose of quetiapine; in cases of treatment with quetiapine at >100 mg/day, a longer period of down-titration of the quetiapine is suggested (Box 2, Figure 4). However, if a patient is experiencing sedation during the day that affects daily activities or increases the risk of falling, reducing or eliminating the daytime dose of quetiapine more abruptly may be considered. If, on the other hand, a patient is deriving a nocturnal sleep benefit from quetiapine due to sedation, removing the bedtime dose may worsen sleep and lead to an increase in psychotic symptoms. Given the low level of urgency for this type of switching, the authors recommend avoiding 2 changes in dosing at the same time (ie, increasing the new treatment while decreasing the older treatment) in order to reduce the risk of rebound agitation. Instead, it is recommended to allow the pimavanserin being initiated to attain the efficacy demonstrated at 4–6 weeks in controlled clinical trials prior to a slow down-titration of quetiapine over a few weeks. Persistent insomnia after withdrawal of quetiapine may require adjustments to this plan.

Figure 3 Patients currently taking quetiapine (≤100 mg/day).
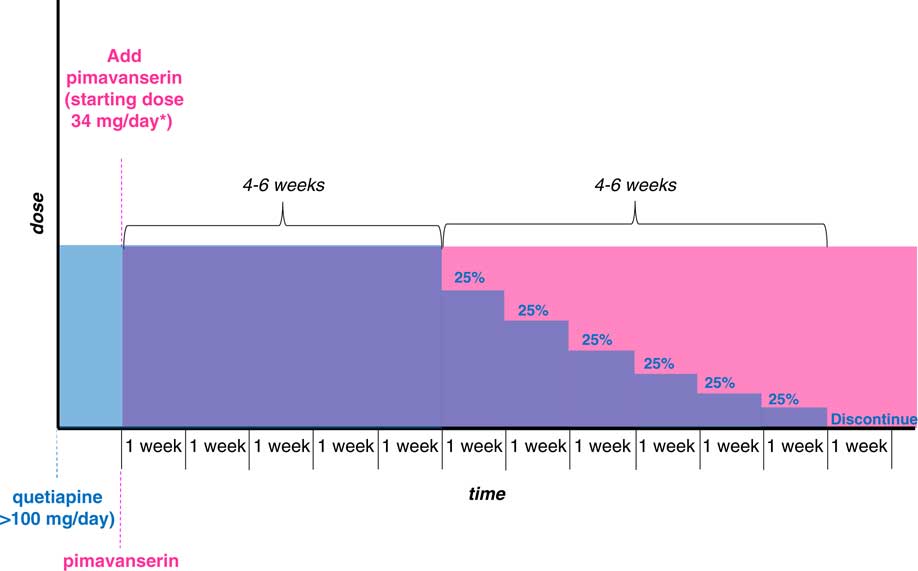
Figure 4 Patients currently taking quetiapine (>100 mg/day).
When switching from concomitant clozapine, for most cases, clozapine may be continued at the current dose for 6 weeks with pimavanserin 34 mg daily. At 6 weeks, total daily clozapine dose may be reduced by 6.25 mg to 25 mg weekly (Box 3, Figure 5), depending on the starting dose (eg, reduce by 25 mg if starting dose is >100 mg/day) and physician preference (Box 4, Figure 6). Absolute neutrophil counts should be monitored for 2 weeks after clozapine discontinuation (see clozapine manufacturer prescribing information). The patient may be continued in the clozapine registry for at least 2 months so that the patient will not have to be re-enrolled if clozapine is restarted.

Figure 5 Patients currently taking clozapine (≤100 mg/day).

Figure 6 Patients currently taking clozapine (>100 mg/day).
As a general rule, if efficacy is reduced at any time during the down-titration of quetiapine or clozapine, return to the previous dose level and wait. Reduction can be attempted again after 1 week, or if added benefit of the combined therapy is clear, the patient may need to continue both medications.
Conclusions
Antipsychotic drugs are commonly used by clinicians to treat patients with PDP. The goal of this review was to summarize the challenges of switching antipsychotic drugs in patients with PDP, and to provide consensus guidance on approaches to switching from atypical antipsychotics to pimavanserin. Understanding the receptor binding and safety profile of antipsychotics should guide providers in treating symptoms associated with PDP. In the absence of controlled clinical trials aimed at evaluating the optimal conditions for switching, a challenge for clinicians is to determine the type of discontinuation (abrupt vs tapered) for the current antipsychotic drug and the timing and starting dose of the new medication in order to maintain optimal receptor occupancy and limit the risks of rebound effects. Most commonly, according to clinical experience, when switching from quetiapine to pimavanserin, reducing the quetiapine before the pimavanserin has started to work may cause the patient to deteriorate during the switch. This can be avoided by waiting to taper quetiapine for 4 to 6 weeks after starting pimavanserin. The overall algorithm suggested as guidance for switching to pimavanserin for PDP patients taking an antipsychotic is summarized in Figure 7. Ultimately, the optimal methods for switching will depend not only on the pharmacology and safety of antipsychotic drugs, but also on individual patient profiles (ie, a precision medicine approach).
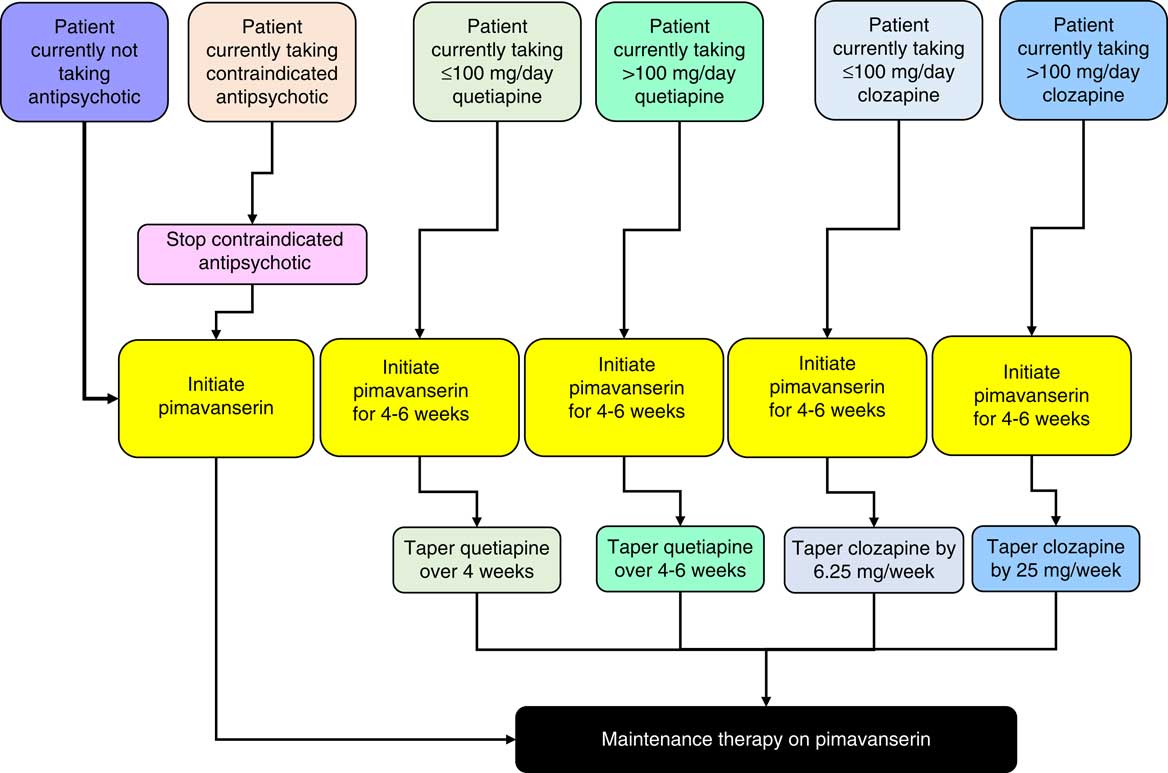
Figure 7 Algorithm for initiating pimavanserin.









