


Introduction
The waters surrounding the Antarctic continent are characterized by a relatively low species diversity of fish (332 species approximately) given the size of the Southern Ocean and the global species diversity (c. 25000 fish species) (Eastman Reference Eastman2005). Notothenioids dominate the Antarctic fish fauna in terms of diversity, abundance and biomass. The genus Dissostichus is represented by two species, with similar life histories: the Antarctic toothfish, Dissostichus mawsoni (Norman), and the Patagonian toothfish, Dissostichus eleginoides (Smitt). The Patagonian toothfish is the largest member of the family Nototheniidae, growing up to 2 m in length and reaching 95 kg in weight. Toothfish are slow growing, mature at over 10 years and live for up to 50 years (Collins et al. Reference Collins, Brickle, Brown and Belchier2010, Welsford et al. Reference Welsford, Candy, Lamb, Nowara, Constable and Williams2011). Dissostichus eleginoides is a bentho-pelagic species, being found at depths between 200 and 2200 m. Adults mostly inhabit deep waters and juveniles are found closer to the surface. The Patagonian toothfish’s distribution is wide, with records from sub-Antarctic waters surrounding islands, seamounts and continental shelves of the Atlantic, Indian and Pacific ocean sectors. It is also found in slope waters off Chile and Argentina south of 30–35°S. The distribution of Patagonian toothfish is limited by water temperature as they lack antifreeze glycoproteins present in other Southern Ocean nototheniids (Collins et al. Reference Collins, Brickle, Brown and Belchier2010).
Trawl and longline fisheries for Patagonian toothfish commenced in the mid-1980s and toothfish are now a valuable food species. Despite careful management, illegal, unreported and unregulated fisheries are a threat to the sustainability of the Patagonian toothfish fishery. A rapid expansion in illegal longline fisheries occurred in 1997 with c. 10 000 tonnes of toothfish illegally fished, especially in the Indian Ocean sector. However, illegal fisheries have decreased since increased controls on harvesting and trade by the Commission for Conservation of Antarctic Marine Living Resources (CCAMLR; Duhamel & Williams Reference Duhamel and Williams2011). Hence knowledge of the population structure of this species has the potential to allow detection of mislabelled fish or illegally caught fish by analysing their geographical origin (assuming marker and alleles are geographically fixed).
The Patagonian toothfish does not appear to be a consistently migratory species. Tagging studies have demonstrated strong site fidelity in adults (Collins et al. Reference Collins, Brickle, Brown and Belchier2010, Welsford et al. Reference Welsford, Candy, Lamb, Nowara, Constable and Williams2011, Brown et al. Reference Brown, Brickle and Scott2013). These studies also highlighted that some individuals move greater distances, but this is not a major phenomenon (Williams et al. Reference Williams, Tuck, Constable and Lamb2002, Marlow et al. Reference Marlow, Agnew, Pruves and Everson2003, Welsford et al. Reference Welsford, McIvor, Candy and Nowara2012). Therefore, it can be expected that genetic differences between geographically separated toothfish populations exist. These differences are most likely where physical barriers between regions are present, such as deep waters or oceanic fronts (Knowlton Reference Knowlton2000, Shaw et al. Reference Shaw, Arkhipkin and Al-Khairulla2004). Several techniques are currently available to study stock structure but the best approach is generally to combine several methods that provide complementary information (Collins et al. Reference Collins, Brickle, Brown and Belchier2010).
Population genetics is a widely used stock identification method and has become more prevalent in the last 15 years. Population genetics techniques are still improving, especially since the advent of high throughput sequencing. This technology enables the application of greater numbers of markers from both the mitochondrial and nuclear genomes. The maternal inheritance of mitochondrial markers means that their frequencies are primarily determined by which females breed in a population or migrate into or away from it (Ballard & Rand Reference Ballard and Rand2005). Nuclear single nucleotide polymorphisms (SNPs) are biparentally inherited and are single base-pair differences (observed as substitutions or insertions/deletions) in DNA sequences (Seeb et al. Reference Seeb, Carvalho, Hauser, Naish, Roberts and Seeb2011). Therefore, in combination, mitochondrial and nuclear DNA markers provide different but complementary information about the population genetics of the species (Ballard & Rand Reference Ballard and Rand2005).
Several population genetics studies have been performed on toothfish in the era before high throughput sequencing and some of these have identified differentiation between populations on regional or sub-regional scales (e.g. Appleyard et al. Reference Appleyard, Ward and Williams2002, Reference Appleyard, Williams and Ward2004, Kuhn Reference Kuhn2007). However, these studies were performed with a limited number of genetic markers, sometimes small sample sizes and were often focused on specific geographical areas. Moreover, they produced somewhat conflicting results depending on which marker was used (cf. allozyme, mtDNA or microsatellite loci) (e.g. Shaw et al. Reference Shaw, Arkhipkin and Al-Khairulla2004). Therefore, the overall genetic structure of this species is still not well understood.
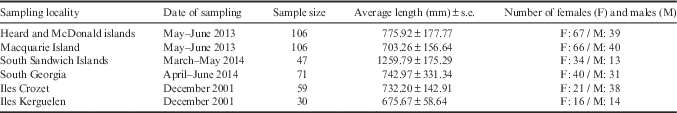
Sample sizes in previous studies have also been one of the main limitations because studies have previously depended upon acquisition of different tissues such as muscle or fin clips. The present study focused on using samples other than tissue for genetic investigations. Age determination in fish can be accomplished by examination of otoliths, which are aragonitic structures found in the inner ear that act as gravity and auditory receptors. The sectioned surface of otoliths can be examined under a microscope; the number of rings from the first annulus reflects the growth of the animal and an estimation of the age can be made (Collins et al. Reference Collins, Brickle, Brown and Belchier2010). Toothfish otolith samples have been collected from many areas for more than 20 years. For example, from the first fishing season in the Australian exclusive economic zone at Heard Island and McDonald Islands in 1997, biological data and otoliths were collected in order to determine the age structure and growth rate of the toothfish population (Welsford et al. Reference Welsford, Nowara, Candy, McKinlay, Verdouw and Hutchins2009).
While otoliths are acellular and do not contain DNA, several studies have highlighted the possibility of extracting DNA from tissue remaining on the surface of archived otoliths, for example in Gadus morhua (Poulsen et al. Reference Poulsen, Nielsen, Schierup, Loeschcke and Grønkjaer2006). Even if the DNA from archived material is often highly degraded, otoliths appear to be effective for genetic studies and if non-destructive protocols for DNA extraction are used, the same otoliths can be used for ageing and/or microchemistry experiments (Cuveliers et al. Reference Cuveliers, Bolle, Volckaert and Maes2009). Use of the large collections of toothfish otoliths stored at a number of Southern Ocean fisheries research institutes provides the potential for more definitive analysis of spatial and temporal population genetics studies.
We describe here methods for extracting DNA from archived toothfish otoliths as a resource for DNA-based population genetics studies. Our study focused on the investigation of Patagonian toothfish population genetic structure with large sample sizes and multiple markers. Population structure of Patagonian toothfish was studied from a combination of both mtDNA and nuclear DNA analyses. The use of multiplexed next-generation DNA sequencing methodologies as a high throughput approach allowed comparisons of samples from Heard and McDonald islands, iles Kerguelen and Crozet in the Indian Ocean, Macquarie Island in the Pacific Ocean, and South Georgia and the South Sandwich Islands in the Atlantic.
Methods
Samples
Adult toothfish otolith samples were collected in longline fisheries by observers from: Heard and McDonald islands (HIMI; 2013), Macquarie Island (MACCA; 2013), South Sandwich Islands (SSI; 2014) and South Georgia (SG; 2014) (Table I, Fig. 1). After collection, they were stored in paper envelopes (one otolith pair per envelope) at room temperature. Tissue samples (white muscle) from iles Crozet and Kerguelen were collected in 2001 from longline fisheries and stored in 70% ethanol at -20°C.

Fig. 1 Sampling localities of Dissostichus eleginoides in the Southern Ocean.
Table I Dissostichus eleginoides: characteristics of individuals sampled.
DNA extraction
DNA was extracted from the otolith’s surrounding tissue for a total of 419 individuals (Table I). A bone incubation buffer was used containing NaCl (1 M), Tris with 0.1 ethylene diamine tetraacetic acid (EDTA; 1 M), EDTA (0.5 M), sodium dodecylsulphate (SDS; 20%) and milliQ® water. The otolith was incubated overnight at 56°C in the bone incubation buffer and proteinase K (18 mg µl-1). After centrifugation (2 min, 13 rpm), the supernatant was removed for extraction in the Maxwell® 16 automated DNA extraction instrument (Promega, USA). The otolith was also removed from the tube, rinsed and dried. Additionally, muscle DNA (from 90 individuals) was directly extracted in the same machine. Otolith and muscle tissue DNA was extracted using the Maxwell® 16 Tissue DNA Purification Kit (Promega). DNA was resuspended in Elution Buffer and stored at -20°C. DNA quality and quantity was measured with a Nanodrop 1000® (Thermo Scientific, USA).
Selection of mitochondrial and nuclear DNA markers
Four mitochondrial markers were selected to assess genetic variation between individuals. Amplification of two parts of the control region (Ctr1 and CR4; AB723627.1), one region of the cytochrome B gene (CytB3; AB723627.1) and the cytochrome oxidase subunit I gene (COI; AB723627.1) were carried out by using the primers designed and presented in Table II. Small amplicons were used because of the degraded otolith DNA. Four nuclear markers were designed for the following genes: the endoplasmic reticulum membrane protein translocator Sec61 alpha (Sec61a; EF088426), the myoglobin (Mb; EF589658), the dystrophin 6a (Dyst6a; EF589655) and the L-lactate dehydrogenase A intron 5 (LDHA; EF589657). To facilitate Illumina sequencing, each forward primer had at its 5' extremity a MiSeq-F-tail (5'-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG-3') and each reverse primer a MiSeq-R-tail (5'-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G-3'). The nuclear markers were selected after amplifying and sequencing several possible markers in order to identify polymorphisms.
Table II Dissostichus eleginoides: PCR mitochondrial and nuclear primer sets.

SNP=single nucleotide polymorphisms.
The reaction (10 µl) components were: 1x Phusion® High Fidelity PCR Master Mix, 10x BSA Purified, 0.1 µM of each primer and template DNA. Reaction conditions were: 98°C for 2 min, 40 cycles of 98°C for 5 s, 59–67°C (depending on primer) for 20 s and 72°C for 20 s, then 72°C for 1 min. All amplifications were carried out in a Mastercycler® (Eppendorf, Germany). Negative and positive (muscle) controls were performed in each run. Polymorphisms were initially checked by Sanger sequencing. PCR products from eight individuals were randomly selected for SNP discovery. They were purified using AMPure® beads according to the manufacturer’s protocol (Agencourt Bioscience, USA). Cycle sequencing components were: 5x Sequencing Buffer® (Life Technologies, USA), BigDye® Terminator v3.1 Ready Reaction Mix (Life Technologies), 3.2 µM primer forward or reverse, nuclease free water and template. Reactions were run in both directions with the following amplification conditions: 96°C for 5 min, 25 cycles of 96°C for 10 s, 50°C for 5 s and 60°C for 4 min. Products from sequencing reactions were purified using Agencourt CleanSEQ® according to the manufacturer’s protocol (Agencourt Bioscience) and run on a 3130 Genetic Analyzer® (Applied Biosystems, USA). Sequences were aligned in the BioEdit program (Hall Reference Hall1999) and polymorphisms were checked.
Multiplexing and sequencing
To allow a large number of samples to be processed, primers were multiplexed. Reaction conditions were: 1x QIAGEN® Multiplex PCR Master Mix, 0.5x Q-solution, 0.1–0.3 µM of a pooled primer mix and template. Reaction conditions were: 95°C for 15 min, 35–40 cycles of 94°C for 30 s, the annealing temperature (Table II) for 90 s and 72°C for 90 s, then 72°C for 15 min. All amplifications were carried out in a Mastercycler® (Eppendorf), and negative and positive (muscle) controls were conducted with each run. Quality and quantity of several random PCR products were checked by electrophoresis (2%, 80 V, 1 h) and on an Agilent® 2100 Bioanalyzer (Agilent Technologies, USA). Multiplex products were pooled per otolith, mitochondrial and nuclear SNPs separately, and diluted 1:10 in MilliQ water. In some cases mitochondrial analyses were multiplexed with smaller numbers of amplicons in order to improve results: Ctr1 was coupled with CytB3, CR4 run on its own (using the Phusion protocol outlined in the previous paragraph). A second round of PCR to add the specific tags to each sample was run: 1X Phusion® High Fidelity PCR Master Mix, 0.1 µM of each tag and template DNA. Reaction conditions were: 98°C for 2 min, 10 cycles of 98°C for 5 s, 67°C for 20 s and 72°C for 20 s, then 72°C for 1 min. All samples were then combined into a single pool for Illumina sequencing. The pool of amplicons was purified using Agencourt AMPure® according to the manufacturer’s protocol (Agencourt Bioscience). The final concentration was measured by using the Qubit® dsDNA BR Assay Kit (Life Technologies). Concentration was then adjusted and sequencing was run on a MiSeq Desktop Sequencer® (Illumina) with each fragment sequenced from both ends in 150 bp reactions. All individuals from each population were analysed, except for MACCA and HIMI where subsamples of 110 individuals were sequenced. In order to estimate genotyping errors, 10% of all samples were genotyped twice for all gene regions.
Data analysis
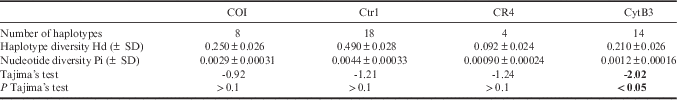
For mitochondrial markers, primary sequence processing was performed with USEARCH v7 (Edgar Reference Edgar2010). Each pair of primary .fastq sequences was filtered for quality and read length, and merged with the equivalent sequence from the opposite strand. Once all sequences had been converted to .fasta format, they were sorted into groups based on the sequences of the PCR primers for each amplicon. The amplicon-specific pools of sequences were then sorted into individual pools for each fish by unique combinations of two indexes attached in the second-round PCR. The primary sequence present in each mitochondrial amplicon pool for each fish was identified by clustering the sequences by similarity and selecting the larger cluster to discard minor sequence variants. Minor variant sequences caused by PCR error, sequencing error or potentially low-level homoplasy were thus ignored. A minimum coverage of three sequences was chosen. Polymorphic sites and their occurrence were identified in MEGA (Tamura et al. Reference Tamura, Stecher, Peterson, Filipski and Kumar2013) and mitochondrial sequences collected in previous population genetics studies were downloaded from GenBank to allow comparisons between datasets. Further analyses were made on haplotypes rather than on SNPs to allow better assessment of genotype diversity. Haplotype number and diversity (Hd), nucleotide diversity (π) and Tajima’s D for selectively neutral evolution were calculated using DnaSP v5 (Librado & Rozas Reference Librado and Rozas2009). Haplotype frequencies, number of effective haplotypes and unbiased diversity were calculated for each marker using the software GenAIEx (version 6.501; Peakall & Smouse Reference Peakall and Smouse2012). A non-hierarchical statistical parsimony network was also constructed for the combined sequences from mitochondrial markers (excluding CR4 due to lower recovery from samples) using the R script TempNet (Prost & Anderson Reference Prost and Anderson2011).
The genotyping procedure for the nuclear markers involved processing steps similar to those for the mitochondrial sequences. However, firstly SNPs were identified in MEGA (Tamura et al. Reference Tamura, Stecher, Peterson, Filipski and Kumar2013) from sequences obtained. Then, instead of clustering, all sequences were scanned for exact matches of the whole region and were compared with nuclear sequences collected in Kuhn (Reference Kuhn2007). A minimum coverage of three occurrences of a SNP was chosen to be required for each allele to be scored. Allele frequencies, number of effective alleles, observed and expected heterozygosities and Wright’s F statistics were calculated to estimate Hardy-Weinberg equilibrium for each marker using the software GenAIEx. Further analyses were made with all markers combined. Genotyping error per haplotype or per allele was estimated for mitochondrial and nuclear markers by comparing haplotypes/genotypes of samples that were replicated.
In order to investigate differentiation analyses, the variation within and among populations was investigated for both nuclear and mitochondrial markers through an analysis of molecular variance (AMOVA). For mitochondrial markers, pairwise population ΦST, analogous to FST, was calculated with 999 random permutations to assess genetic differentiation. For nuclear markers, pairwise population Wright’s FST, G’ST and an estimate of Jost’s D, Dest, were calculated with 999 random permutations. P-values were corrected by either Holm-Bonferroni’s adjustment (Holm Reference Holm1979) or Benjamini and Hochberg’s correction (Benjamini & Hochberg Reference Benjamini and Hochberg1995). In order to evaluate the effect of sampling sizes, pairwise analyses were run on a random subsample of each population with 30 individuals with mitochondrial and nuclear markers. A Principal Coordinate Analysis and a Mantel test for the relationships between geographical distance and genetic diversity were also conducted (Diniz-Filho et al. Reference Diniz-Filho, Soares, Lima, Dobrovolski, Landeiro, Telles, Rangel and Bini2013).
Results
Nucleic acid extraction
Otolith extractions ranged between 1–5 ng.ul-1, with low 260/230 and 260/280 ratios. This indicates that a low quantity and poor quality DNA was extracted. However, the test PCR amplifications were successful, indicating that the otolith DNA was sufficient for recovery of markers.
Sequences analyses
Sequences were obtained for close to 400 individuals for all mitochondrial and nuclear markers, with the exception of the mtDNA marker CR4 with 265 individuals successfully sequenced (Table III). Between four and nine SNPs, including two to three common SNPs, were identified within the markers Ctr1, COI, LDHA and Mb. In contrast, CR4 was characterized by three rare SNPs present in a low number of individuals and many singleton substitutions were observed in CytB3. Dyst6a and Sec61a are also less variable with only one common SNP. Several SNPs were reported in the sequences from GenBank (SNPs underlined in Table III).
Table III Dissostichus eleginoides DNA polymorphisms with the number of individuals sequenced. Major SNPs are in bold (high frequencies). SNPs that were already reported in GenBank are underlined. S=singleton substitution, C=coding region.

Most of the SNPs were found in the third position of codons. All COI substitutions were synonymous, while two non-synonymous changes occurred in CytB3. In Sec61a, the coding SNP was in first position of the codon with the other SNP being non-synonymous. Other SNPs, nuclear or mitochondrial, were located in non-coding regions.
Among the mitochondrial markers, Ctr1 and CytB3 had the highest number of haplotypes (Table IV). Haplotype and nucleotide diversities varied across markers, respectively from 0.092 to 0.49 and from 0.00090 to 0.0044; Ctr1 presented the highest haplotype and nucleotide diversities, as would be expected for this non-transcribed mitochondrial region. The unbiased diversity (Hd) also varied across populations without any clear pattern (e.g. MACCA COI Hd=0.55, SSI COI Hd=0.088, SG CytB3 Hd=0.38, Crozet COI Hd=0.000, HIMI CR4 Hd=0.000). When sequences of three mtDNA markers (Ctr1, COI and CytB3) were combined, a dominant haplotype was shared in all populations; however, some haplotypes were specific to geographical locations. Most notably, MACCA had a unique haplotype shared by 31 individuals (Fig. 2). Tajima’s D test was negative for all markers but not significant, except for CytB3 (D=-2.02, P<0.05). This could be due to the fact that CytB3 presented several rare mitochondrial haplotypes with very low frequencies. Genotyping error rate was estimated at 5.49% for mitochondrial markers by comparing haplotypes and 7.04% for nuclear markers by comparing genotypes.

Fig. 2 Network showing relationships between mitochondrial sequences computed (Ctr1, CytB3 and COI) for six locations: South Sandwich Islands (SSI; n=42), South Georgia (SG; n=65), Macquarie Island (MACCA; n=105), Kerguelen (n=17), Heard and McDonald islands (HIMI; n=97) and Crozet (n=36). Each haplotype is represented by a circle, and coloured circles with a number indicate the presence of this haplotype in the population and its frequency. A non-coloured circle indicates that this haplotype is not present in the population concerned. A line separating two haplotypes represents a mutation that differentiates these haplotypes, any additional mutation is represented by a black dot. Lines that connect different layers indicate shared haplotypes between populations.
Table IV Dissostichus eleginoides genetic diversity indices for mitochondrial markers. Significant values are given in bold.
In the nuclear markers, only one rare allele of Dyst6a was found specific to HIMI and it had a very low frequency (0.005). The number of effective alleles in comparison with the number of alleles demonstrated that the occurrence of rare alleles did not influence the genetic structure. There was a deviation from Hardy-Weinberg equilibrium with almost all populations and all loci (Table V). This was confirmed by the inbreeding coefficient FIS (between -0.661 to -0.191) for all markers and all populations, thereby indicating an excess of heterozygotes.
Table V Genetic diversity nuclear marker indices for each population and results for Hardy-Weinberg equilibrium test.

ns=non-significant, *P<0.05, ***P<0.001.
HIMI=Heard and McDonald islands, MACCA=Macquarie Island, SG=South Georgia, SSI=South Sandwich Islands.
Genetic structure results
The AMOVA mitochondrial analysis revealed significant overall differentiation between populations (ΦST=0.069, P=0.001) with 93% of the variation within populations (Table VI). CytB3 did not present any variation among populations while CR4 and Ctr1 showed a low proportion of genetic variability among regions. COI presented a high proportion of genetic variability among populations (39.92%) and revealed a significant ΦST value (0.40, P=0.001). This high variability among populations is explained by different haplotypes of COI found in MACCA. One haplotype is common and shared between HIMI, SSI, SG, Crozet and Kerguelen. This haplotype is also found in MACCA but other haplotypes are represented and differentiate MACCA from other locations. For nuclear markers, the variation was mostly within populations (99.42%, Table VI). However, a significant proportion of genetic variability was observed among populations with LDHA (FST=0.016, P=0.002) and when all markers were considered together (FST=0.008, P=0.003). Sec61a, Mb and Dyst6a presented low proportions of genetic variability among populations and did not present significant overall differentiation.
Table VI Analysis of molecular variance within and among Dissostichus eleginoides populations. Significant values are given in bold.

df=degrees of freedom, SS=sum of squares, MS=mean squares, pops=populations.
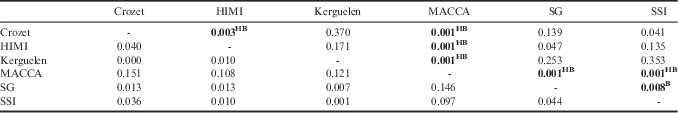
Pairwise ΦST across all mtDNA markers indicated that MACCA was highly differentiated from all other populations and that Kerguelen was not differentiated from Crozet or HIMI (Table VII). These two results were supported even when corrected for multiple comparisons and also by nuclear analysis (except between MACCA and Kerguelen) (Table VIII). The differentiation between MACCA and SSI was not supported after Holm-Bonferroni correction.
Table VII Pairwise ΦST estimates of mtDNA differentiation between Dissostichus eleginoides populations. ΦST values are below the diagonal and associated P-values above the diagonal (999 permutations). H=P-value significant after Holm-Bonferroni’s adjustment. B=P-value significant after Benjamini and Hochberg’s correction. G’ST, FST or Dest values are slightly different, but P-values are the same so only G’ST results are presented. Significant values are given in bold.
HIMI=Heard and McDonald islands, MACCA=Macquarie Island, SG=South Georgia, SSI=South Sandwich Islands.
Table VIII Pairwise G’ST (male or female together) estimates of nuclear DNA differentiation between Dissostichus eleginoides populations. G’ST values are below the diagonal and associated P-values above the diagonal (999 permutations). H=P-value significant after Holm-Bonferroni’s adjustment. B=P-value significant after Benjamini and Hochberg’s correction.

HIMI=Heard and McDonald islands, MACCA=Macquarie Island, SG=South Georgia, SSI=South Sandwich Islands.
There were different results between mitochondrial and nuclear markers. According to the mitochondrial results, HIMI was significantly different from Crozet (ΦST=0.040, P=0.003; both corrections) and SG was genetically different from SSI after Benjamini and Hochberg’s correction (ΦST=0.044, P=0.008; but not significant after the more conservative Holm-Bonferroni adjustment). However, these results were not confirmed by nuclear data (respectively, G’ST=-0.001, P=0.623; G’ST=-0.002, P=0.830). Conversely, some populations that were not differentiated with mtDNA markers showed some differentiation with nuclear markers. Thus, according to the mitochondrial analysis, there was no differentiation between SG and Crozet, between SSI and HIMI, and between Kerguelen and SG or SSI. However, all these locations were differentiated with nuclear markers. While SG and HIMI and SSI and Crozet were not significantly different according to the mtDNA analyses, these populations were differentiated according to nuclear analysis (respectively, G’ST=0.016, P=0.001; G’ST=0.009, P=0.006).
Overall, differentiation of the Pacific sector with the two other sectors was supported by nuclear and mitochondrial markers. The Atlantic and the Indian sectors are differentiated with nuclear markers but not with mitochondrial markers. When the mitochondrial analysis was re-tested with subsamples of 30 individuals for each population (as n=30 was the smallest population at Iles Kerguelen), MACCA was still differentiated from other populations, but after Holm-Bonferroni correction, there was no differentiation detected between populations with mitochondrial markers (MACCA: 0.007<P<0.050). With nuclear markers and a subset of 30 samples, Kerguelen was no longer differentiated with SSI (Table VIII).
With mitochondrial markers, the differentiation of MACCA from other populations was also shown by the Principal Coordinate Analysis. HIMI was opposite to Crozet and the same pattern was observed between SSI and SG which reflected the differentiations previously assessed (Fig. 3a). The Mantel test (comparing genetic distance between populations with the geographical distance) was not significant for mtDNA (Rxy=0.422, P=0.119). With nuclear markers, the differentiation between the three ocean sectors was confirmed by the Principal Coordinate Analysis as three groups were separated and followed the geographical pattern (Fig. 3b). The Mantel test was significant and supported the hypothesis of a genetic differentiation due to geographical distance (Rxy=0.829, P=0.004).

Fig. 3 Principal Coordinate Analysis results for a. mitochondrial markers and b. nuclear markers. Red diamond=Indian Ocean sector, blue diamond=Atlantic Ocean sector, green diamond=Pacific Ocean sector. SSI=South Sandwich Islands, SG=South Georgia, MACCA=Macquarie Island, HIMI=Heard and McDonald islands.
Discussion
Population structure
The mitochondrial and nuclear markers outlined in this study indicated high variation within populations, although overall ΦST and FST values indicate global differentiation among populations. Further quantification of differentiation within populations would require multiple temporal collections from the same area.
Concerning the south-west Pacific sector, MACCA is clearly genetically differentiated from all other regions in this study and it is the most geographically isolated toothfish population. It has a higher unbiased diversity, particularly for COI, and the highest number of specific haplotypes, including one that is shared by more than 30 individuals. This specific haplotype is due to the variability of COI in MACCA, through two SNPs, that differentiates the Pacific sector from the two other sectors. COI seems the most variable marker (mtDNA or nuclear) among populations but when all markers are examined together, differentiation of MACCA from other locations is still significant. The other markers are therefore contributing to this differentiation, although less strongly. MACCA is also genetically differentiated from all other locations with nuclear markers, aside from Kerguelen (P=0.059), possibly due to the relatively low number of individuals from Kerguelen (n=30) sequenced in this study. This differentiation of MACCA is consistent with previous studies such as Kalish & Timmiss (Reference Kalish and Timmiss2000; otolith’s core chemical composition), Appleyard et al. (Reference Appleyard, Ward and Williams2002; mitochondrial markers), Kuhn (Reference Kuhn2007; nuclear and mitochondrial markers) and Ward et al. (Reference Ward, Appleyard and Williams2002; mitochondrial and microsatellite markers). Differentiation was also supported by microsatellites in Smith & McVeagh (Reference Smith and McVeagh2000) but it was not consistent across loci and was not supported by allozymes. The Mantel test results suggest geographical distance is a major differentiating factor driving the significant nuclear marker outcomes. These test results were not significant with mitochondrial markers but the number of individuals successfully sequenced was lower and patterns of differentiation were not supported by geographical distances.
Dispersal can occur through two mechanisms, egg and larval dispersal or adult movement. According to Shaw et al. (Reference Shaw, Arkhipkin and Al-Khairulla2004), deep waters may represent a barrier for adults and the Antarctic Polar Front could prevent larval transport. Yet, in areas with strong currents between two locations, the long pelagic larval stage could favour juvenile dispersal. According to tagging studies, this species does not usually move large distances (Welsford et al. Reference Welsford, McIvor, Candy and Nowara2012) and metapopulations are therefore a credible hypothesis. However, there are exceptions as proved by Rogers et al. (Reference Rogers, Morley, Fitzcharles, Jarvis and Belchier2006) that showed occasional movements of adults from SG to the Patagonian shelf, and Welsford et al. (Reference Welsford, Candy, Lamb, Nowara, Constable and Williams2011) has documented movements between island groups of at least 2500 km. As an extreme, Møller et al. (Reference Møller, Nielsen and Fossen2003) documented the catch of a Patagonian toothfish in Greenland. However, demographic movements of fish may not necessarily imply genetic contributions and the Patagonian toothfish is still generally considered to be a philopatric species (Kuhn Reference Kuhn2007, Welsford et al. Reference Welsford, Candy, Lamb, Nowara, Constable and Williams2011). The genetic differentiation observed at MACCA might be driven by oceanographic conditions including deeper waters and large currents (Collins et al. Reference Collins, Brickle, Brown and Belchier2010). Furthermore, MACCA is north of the Antarctic Polar Front which may be a major factor that limits larval movement from south of the Antarctic Polar Front. Tagging studies have not recorded individuals moving between MACCA and other locations, which is consistent with the genetic results presented here; toothfish at MACCA should be considered as a separate stock for fisheries management purposes.
In the western Indian sector, based on the mitochondrial sequences, HIMI was found to be significantly different from Crozet which indicates that genetic panmixia may not be present in the western Indian Ocean sector of the Southern Ocean. This could be explained by the specificity of some haplotypes to Crozet but they are mostly shared by only few individuals and not shared across all populations. This differentiation is not consistent with mitochondrial analysis from Appleyard et al. (Reference Appleyard, Williams and Ward2004) that reported homogeneity in the western Indian sector. This is despite some of the same individuals being used in this study and in the Appleyard et al. (Reference Appleyard, Williams and Ward2004) study. This conflicting result can be explained by the fact that different mitochondrial markers were used by Appleyard et al. (Reference Appleyard, Williams and Ward2004) (BCL, containing the D-loop, and ND2 containing the NADH dehydrogenase subunit 2 gene). This highlights the importance of the choice of mitochondrial markers in genetic differentiation assessment and that more markers (or whole mitochondrial genome sequencing) should be undertaken.
Unequal sample sizes may also have a sampling effect. Crozet was tested for 30 individuals whereas more than 100 individuals were sequenced at HIMI and approximately 60 individuals were sampled at Kerguelen. HIMI is south of the Antarctic Polar Front whereas Crozet is located north of it and this front could represent an obstacle to passive or active larval transport and limit genetic mixing (Shaw et al. Reference Shaw, Arkhipkin and Al-Khairulla2004). This result is consistent with consideration of Crozet and HIMI as different fisheries units (subarea 58.6 and division 58.5.2, respectively; CCAMLR 2014). However, Williams et al. (Reference Williams, Tuck, Constable and Lamb2002) and Welsford et al. (Reference Welsford, Candy, Lamb, Nowara, Constable and Williams2011) demonstrated that long distance travel by adult toothfish from HIMI to Crozet occurs, even if this phenomenon has not been found to be widespread so far. The fact that no differentiation was found with nuclear markers in this study can support the hypothesis that males disperse further than females between Crozet and HIMI. Indeed, tagging studies in the Kerguelen Plateau revealed that males represented a higher proportion of individuals travelling more than 500 km (Welsford et al. Reference Welsford, Candy, Lamb, Nowara, Constable and Williams2011). This could explain the difference in results between the two markers. It could also be explained by the fact that mtDNA is more sensitive to genetic drift and population bottlenecks than nuclear DNA. Therefore, if toothfish populations have ever experienced a decrease in the number of individuals, genetic drift could have accentuated mitochondrial differences.
Finally, no differentiation was found between Kerguelen/HIMI and Kerguelen/Crozet with mitochondrial or nuclear markers. This supports the conclusions of Appleyard et al. (Reference Appleyard, Williams and Ward2004) who proposed that these populations occasionally interbreed. This is also consistent with tagging studies, such as that of Williams et al. (Reference Williams, Tuck, Constable and Lamb2002) which reported the movement of an individual from HIMI to Kerguelen, but this observation is not enough itself to explain a possible homogeneity between these two locations. Moreover, these two locations are separated by the Antarctic Polar Front, but it is not clear how significant this is to reducing toothfish movement. It is thought that adults move from HIMI to Kerguelen to spawn, and then juveniles move from Kerguelen to HIMI, as no larvae or one-year-old juveniles have been recorded in HIMI (Welsford et al. Reference Welsford, McIvor, Candy and Nowara2012). This supports the hypothesis of a single stock across the Kerguelen Plateau and genetic homogeneity between these two sites. If all recruits come from the Iles Kerguelen that would represent a spawning site. However, this homogeneity is not consistent with Kuhn (Reference Kuhn2007) who identified differentiation between Kerguelen and HIMI. Additional testing of the null hypothesis (i.e. no genetic structure among the sector) is required on a larger set of samples, especially for Crozet and Kerguelen given that these populations had a lower number of individuals sequenced in comparison to HIMI or MACCA and at different temporal scales (12 years between Crozet and HIMI sampling).
In the south-west Atlantic sector, a differentiation was found between SG and SSI after Benjamini and Hochberg’s correction. However, it is not confirmed after Holm-Bonferroni’s adjustment. Furthermore, this marginal evidence for differentiation was not confirmed by nuclear marker results. Moreover, one fish tagged near the SSI was recaptured near SG approximately 740 km from the point of release; it was suggested that the population in the north of the SSI might belong to the SG stock (Collins et al. Reference Collins, Brickle, Brown and Belchier2010). If both results are correct and not artificial results due to genotyping errors, it could be explained by a difference in behaviour between males and females. However, if SSI and SG are really differentiated it would be consistent with the current CCAMLR fisheries management that considers these two locations separately (subareas 48.4 and 48.3, respectively) (CCAMLR 2014). Only 50 SSI individuals were sequenced; an increased sample size may be required to inform management decisions.
With mitochondrial markers, no differentiation was detected between SG/SSI and the western Indian sector. Because of the distance and the multiple obstacles such as deep waters, gyres and frontal systems, a genetic structure could be expected between Crozet/Kerguelen/HIMI and SSI/SG. Significant differentiations were detected with nuclear markers between the two ocean sectors. Moreover, Appleyard et al. (Reference Appleyard, Ward and Williams2002) and Ward et al. (Reference Ward, Appleyard and Williams2002) reported a differentiation between HIMI and Shag Rocks/SG which is consistent with our nuclear analysis. The lower sample size for SG, especially with mitochondrial markers, could underestimate differentiation. When each area was sub-sampled to 30 individuals to be equivalent with Kerguelen, no differentiation was detected. However, with mitochondrial markers, some variation was detected for MACCA and HIMI, which correspond to the two biggest sample sizes; thereby underlining the importance of power analyses for differentiation studies.
Biasing factors in genetic studies
Sex bias is an important factor to consider in population genetics studies. Males and females can disperse differently and influence genetic structure. In this study, differences between the two types of markers can be due to sample size, but could also be due to a sex-specific gene flow (e.g. males dispersing further than females between Crozet and HIMI). Ideally, to estimate population genetic structure, individuals should belong to the same generation. Indeed, allele frequencies vary over time and are influenced by genetic drift or bottlenecks. However, in real populations, generation-specific samples are generally not possible to obtain. By using otoliths for both genetic and ageing studies, sampled individuals can be allocated to different year classes. This possible overlap between generations could be one of the reasons why most of the allele frequencies of the nuclear markers are not in Hardy-Weinberg equilibrium. Other explanations could be that allele frequencies are not equal in the sexes, non-random mating is present in toothfish populations, outbreeding, selection of certain alleles (although this was not tested in this study) or sampling effects (i.e. if many alleles/haplotypes then sampling was not large enough to adequately represent the genetic variability).
The most likely explanation for the Hardy-Weinberg disequilibrium is genotyping error. A common problem working with biparental genetic markers is the rate of genotyping errors (coupled with sequencing technology). Genotyping errors reported in this study were high (5% and 7% for mtDNA and nuclear markers, respectively) and there was an excess of heterozygotes with nuclear markers. From low quality samples, such as otolith DNA, per-genome error rates can reach 50% and it must be noted that error rates reported in studies are always different from zero, no matter the sample quality (Morin et al. Reference Morin, Leduc, Archer, Martien, Huebinger, Bickham and Taylor2009). Morin et al. (Reference Morin, Leduc, Archer, Martien, Huebinger, Bickham and Taylor2009) demonstrated that even low error rates can lead to significant deviations from Hardy-Weinberg equilibrium.
The excess of heterozygotes was unexpected and probably results from the sequencing coverage used here for scoring genotypes with this next-generation sequencing approach. The number of individuals missing in the dataset increases when the minimum coverage increases but the number of genotyping errors decreases when the sequencing depth increases. In order to limit errors, only clearly defined nuclear SNPs found in a large number of individuals were scored. Multiplexing primers decreases the number of sequencing runs needed and thus the price of experiments; however, sequencing coverage per individual is lower. High throughput sequencing is a quite recent method, and it can be expensive and challenging to optimize protocols, especially when several primers are multiplexed. While we have been able to effectively genotype some nuclear SNPs using otolith DNA, further work to optimize the method will contribute to decrease genotyping errors and allow larger numbers of nuclear SNPs to be genotyped. This work provides some guidance on sequencing depth for future studies using this experimental approach.
Usefulness of otoliths
Publications on fish otolith applications have grown exponentially in the last 30 years. Otoliths were mostly used to determine annual age and growth (80% of the publications surveyed between 1999 and 2004) or sometimes to study chemistry or the microstructure of toothfish (Ashford et al. Reference Ashford, Jones, Hofmann, Everson, Moreno, Duhamel and Williams2008). However, despite the reported interest in using otoliths for genetic purposes, suitable DNA methods are limited (e.g. Cuveliers et al. Reference Cuveliers, Bolle, Volckaert and Maes2009). We believe the full potential of information available from otolith collections remains underutilized. Extracting DNA from otoliths is one of the main challenges as DNA is often degraded, in low yields and susceptible to contamination. These factors make DNA from otoliths more difficult to amplify and analyse than DNA purified from skin or muscle. Otoliths are acellular, so DNA is extracted from tissue or dried blood that has adhered to the rough surface of otoliths. The degraded DNA means that appropriate genetic markers must be chosen with PCR amplicons as small as possible, preferably less than 200 bp. It is also necessary to design species-specific primers to avoid contamination issues associated with working with degraded DNA.
As otoliths are relatively easy to collect and store, large collections often exist in museums or scientific research institutes. Consequently, the ability to extract DNA from otolith collections enables researchers to work with larger sample sizes than may be possible with tissue samples. This is particularly valuable for Antarctic fish that are especially hard to regularly sample due to access and resourcing issues. It would be useful to apply the same DNA extraction protocol on otoliths that have been stored for longer periods, to determine if storage time has an impact on the success of extraction. The otolith samples collected in this study were initially intended for age determination; thus as well as providing a source of population genetic information, age data is generally also available. The combination of age data and population genetic data could eventually permit comparisons of past and present levels of genetic diversity to estimate the effective population size and census population size. Effective population size is the key parameter to estimate for population management. As otoliths can be stored for a long period, they can be used to study long-term changes in demography and the potential adaptation of fish populations facing harvesting pressure.
Ageing studies are often carried out on one otolith and sometimes only one otolith is available in collections. Appropriate methods for collecting all information needed (i.e. ageing, microchemistry and DNA data) on one otolith without destructive sampling is required. Indeed, otoliths represent a limited source of DNA since only two otoliths per fish can be collected; otolith collections are very valuable. According to Cuveliers et al. (Reference Cuveliers, Bolle, Volckaert and Maes2009), a trade-off must be found between DNA quality and otolith preservation. Contrasting conclusions were given for several species concerning the effect of DNA extraction on ageing success (e.g. Cuveliers et al. Reference Cuveliers, Bolle, Volckaert and Maes2009). It underlines the importance of minimizing the damage to the otolith, depending on the method used and different factors such as incubating time or concentration of EDTA or SDS. However, otoliths from different species are likely to present different sensitivities to degradation; therefore, a specific study on Patagonian toothfish otoliths is necessary. An ageing study on the otoliths used in this study would allow parallel improvement of the DNA and ageing protocols. The incubation time could perhaps be lowered or the concentration of reagents modified while maintaining a suitable DNA extraction protocol.
Conclusion
This study was successful in establishing a procedure for extracting DNA from toothfish otoliths and allowing large numbers of individual fish, from a wide variety of sites covering most of the species’ range, to be analysed for mtDNA and nuclear SNPs. The results of this study are generally consistent with previous work on Patagonian toothfish. Genetic differentiation between toothfish populations in the three ocean sectors of the Southern Ocean was observed with nuclear markers, while only the Pacific sector was differentiated from the two other sectors with mitochondrial markers. To further examine these differences, it is necessary to sequence more individuals and have larger datasets (at least 100 individuals per location).
These results contribute to the ongoing management of toothfish fisheries. It would be interesting to integrate additional locations (and temporal sampling) to have a more complete indication of the genetic structure of the Patagonian toothfish, notably samples from the Ross Sea and the Patagonian Shelf, as these regions are poorly characterized from a genetic perspective. It is also advisable, thanks to high throughput sequencing methods, to include more variable nuclear markers, microsatellites and sequence the whole mtDNA genome. The availability of more genetic markers would enable other tests such as close-kin recognition and parentage analysis that would enhance knowledge of the breeding biology of toothfish. Provenance assignment tests could also be developed which may enable the origin of toothfish catches to be established while helping to minimize illegal fishing.
Knowledge of the movements of larvae, juveniles and adults are also key to understanding the toothfish life-cycle and genetic structure. As these movements and the precise locations of spawning areas are not fully understood yet, more studies are necessary on these parameters to improve interpretation of genetic assessments. Finally, many factors can affect population structure, past historical events, such as separation of different oceans or, at smaller scales, the topography of the environment. As structure evolves through time, a long-term study should be realized to determine if the current population structure is stable or if it is evolving in the face of population harvesting and climate change.
Acknowledgements
We thank the observers who collected and documented the samples, the CCAMLR (Hobart), the Australian Antarctic Division (Kingston), the CSIRO National Research Collections and the British Antarctic Survey for providing samples. The authors would like to thank the reviewers for their comments which helped to improve the manuscript.
Author contributions
All authors contributed to writing this manuscript. Lola Toomey, Dirk Welsford, Sharon A. Appleyard and Mark Belchier were responsible for sample collection, archiving and preparation. Lola Toomey, Sharon A. Appleyard, Andrea Polanowski, Cassandra Faux, Bruce E. Deagle, Simon Jarman worked on molecular method development. Lola Toomey, Andrea Polanowski, Cassandra Faux and James Marthick performed the experiments. Lola Toomey, Bruce E. Deagle and Simon Jarman worked on the data analysis.











