



Introduction and previous work
Since the first determinations of the crystal structures of variscite (orthorhombic AlPO4⋅2H2O, Pbca) and topologically related metavariscite (monoclinic dimorph, P21/n) by Kniep et al. (Reference Kniep, Mootz and Vegas1977) and Kniep and Mootz (Reference Kniep and Mootz1973) (redetermination, correcting a previous study of Fayos and Salvador-Salvador, Reference Fayos and Salvador-Salvador1971), respectively, a large number of both naturally occurring and synthetic members of these two groups have been reported. These members comprise predominantly phosphates and arsenates, but also vanadates (only one representative) and selenates (three representatives). An up-to-date compilation of the known minerals and synthetic compounds and their crystal data is provided in Tables 1 and 2 (variscite- and metavariscite-types, respectively), along with footnotes pointing out some errors and inconsistencies in the published data. This compilation reveals an interesting observation: while there are six variscite-type arsenate members, the metavariscite-type members include only one arsenate member (ScAsO4⋅2H2O). The topology of the framework-based variscite and metavariscite structure types was elucidated recently by Ilyushin and Blatov (Reference Ilyushin and Blatov2017), who also discussed the functional role of the hydrogen bonds within the frameworks (see also the discussion of topological features below).
Table 1. Overview on variscite-type minerals and synthetic compounds (five phosphates, five arsenates, three selenates).
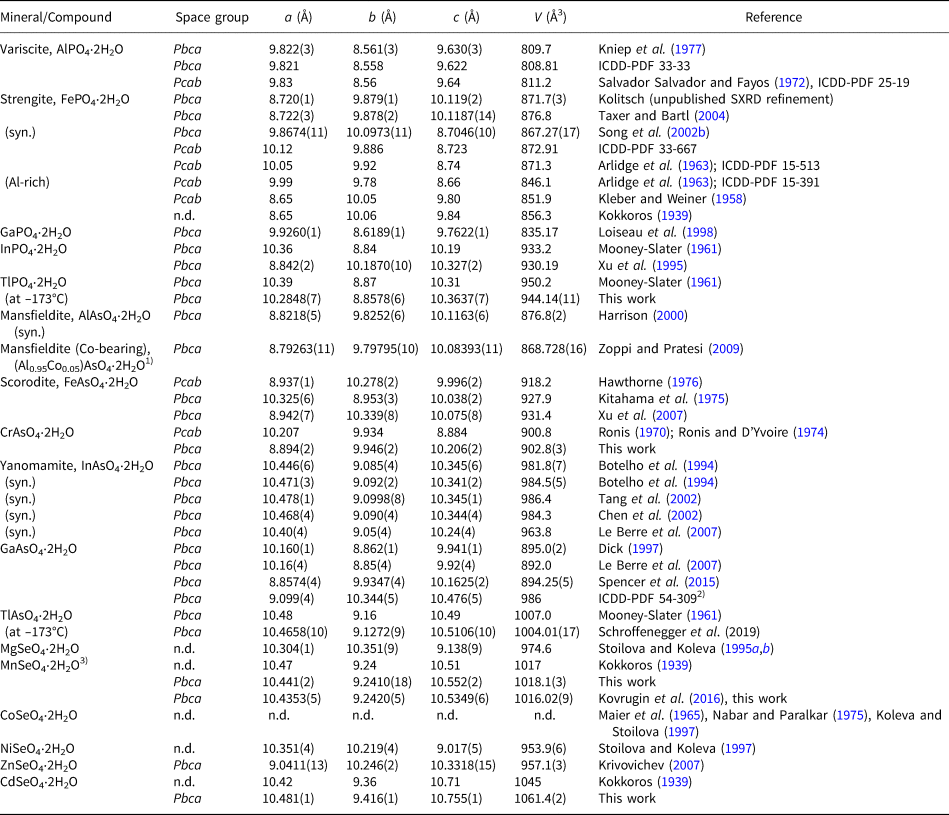
Notes: n.d. = not determined. Space groups are those given in the cited literature, some settings appear doubtful, although two different Pbca settings exist. A closely related, metastable species is parascorodite, trigonal FeAsO4⋅2H2O (space group P  $\bar{3}$c1; Perchiazzi et al., Reference Perchiazzi, Ondruš and Skála2004).
$\bar{3}$c1; Perchiazzi et al., Reference Perchiazzi, Ondruš and Skála2004).
1)Simplified; formula given by Zoppi and Pratesi (Reference Zoppi and Pratesi2009) is: (Al0.944Co3+0.046Cu2+0.005Fe3+0.003Zn2+0.002)Σ=1(As0.972Al0.022P0.006)Σ=1O3.975⋅2H2O (sic!).
2)The unit-cell parameters given in ICDD-PDF 54-309 (Wang, S.-L., Grant-in-Aid, 2002) appears to be those of InAsO4⋅2H2O, not of GaAsO4⋅2H2O; the sample was “prepared at 160°C for 72 h”.
3)MnSeO4·2H2O is also reported by Koleva and Stoilova (Reference Koleva and Stoilova1999), but without unit-cell parameters.
Table 2. Overview on metavariscite-type minerals and synthetic compounds (seven phosphates and one arsenate).

Note: Some unit-cell volumes were not given in the original publication and were therefore calculated from the published unit-cell parameters.
1) For GaPO4⋅2H2O, Mooney-Slater (Reference Mooney-Slater1966) gives an incorrect cell with a = 9.77, b = 9.64, c = 9.68 Å and β = 102.7°, and a doubtful interpretation of the crystal structure.
2) n.d. – not determined. Komissarova et al. (Reference Komissarova, Pushkina and Khrameeva1971) state that synthetic ScAsO4⋅2H2O has a “monoclinically distorted rhombic lattice”; see also Carron et al. (Reference Carron, Mrose and Murata1958), Ivanov-Emin et al. (Reference Ivanov-Emin, Korotaeva, Moskalenko and Ezhov1971) and Komissarova et al. (Reference Komissarova, Pushkina, Khrameeva and Teterin1973).
The aim of the present article is threefold. Firstly, we present the hydrothermal synthesis and crystal structures of four members of the variscite group, viz. the arsenate CrAsO4⋅2H2O, the phosphate Tl3+PO4⋅2H2O and the two selenates Mn2+SeO4⋅2H2O and CdSeO4⋅2H2O. In addition, the thermal behaviours of the two selenates are reported. For all compounds, only unit-cell parameters were previously available (Table 1).
The existence of CrAsO4⋅2H2O was first reported by Lukaszewski et al. (Reference Lukaszewski, Redfern and Salmon1961) in a study of the aqueous system Cr(III)–arsenic acid. The dihydrate, obtained from dehydration of the hexahydrate (in two steps at 0°C and 50°C, with the intermediate formation of a tetrahydrate), was found to be stable up to 120°C. Subsequently, CrAsO4⋅2H2O was also reported by Ronis (Reference Ronis1970) and Ronis and D'Yvoire (Reference Ronis and D'Yvoire1974) who synthesised the compound by heating a solution of Cr(H2AsO4)3⋅5H2O and H3AsO4 at 270°C in a sealed container and by double decomposition of the appropriate metal salts with H3AsO4 or by hydrolysis of Cr(H2AsO4)3, respectively. On the basis of an indexed powder X-ray diffraction (PXRD) pattern, these authors suggested that CrAsO4⋅2H2O is isotypic with variscite, and provided the following unit-cell parameters for space group Pcab (non-standard setting of the space group no. 61): a = 10.207, b = 9.934, c = 8.884 Å and V = 900.8 Å3. The two selenates, first reported by Kokkoros (Reference Kokkoros1939), were synthesised independently by two groups among the present authors. A brief, preliminary report on MnSeO4⋅2H2O has already been presented at a conference (Kovrugin et al., Reference Kovrugin, Aliev, Colmont, Mentré and Krivovichev2016).
The second aim of the present paper is to present the crystal structure of bonacinaite (IMA2018-056; Cámara et al., Reference Cámara, Ciriotti, Kolitsch, Vignola, Hatert, Bittarello, Bracco and Bortolozzi2018), natural metavariscite-type (Sc,Al)(As,P)O4⋅2H2O from the abandoned Varenche mine, Saint-Barthélemy, Nus, Valle d'Aosta, Italy, from which it was briefly described by Barresi et al. (Reference Barresi, Kolitsch, Ciriotti, Ambrino, Bracco and Bonacina2005), including the reporting of the unit-cell parameters and space-group symmetry, based on a crystal-structure determination of U.K., the second author of the mentioned paper. The Varenche mine worked on a metamorphic manganese deposit containing a number of rare As and V minerals, as well as the scandium silicate thortveitite (Barresi et al., Reference Barresi, Kolitsch, Ciriotti, Ambrino, Bracco and Bonacina2005). Bonacinaite forms tiny colourless (partly pale violet due to a violet ring-like zone in the centre), tabular crystals that show a pseudohexagonal outline, have glassy lustre and are transparent. They sit in a small void of a matrix composed of granular braunite, quartz and manganese oxides, and are associated with corroded arseniopleite nearby the void.
Synthetic ScAsO4⋅2H2O is known and was reported for the first time by Carron et al. (Reference Carron, Mrose and Murata1958) who, in a study on REEXO4 (X = P, As or V) compounds, also prepared a “hydrated scandium arsenate from solutions of ScCl3 and dilute arsenic acid which was isostructural with the metavariscite group of minerals.” No crystal data or indexed powder diffraction pattern were given. Subsequently, ScAsO4⋅2H2O was also observed in a study of the system ScCl3–Na2HAsO4–H2O at 200°C by Ivanov-Emin et al. (Reference Ivanov-Emin, Korotaeva, Moskalenko and Ezhov1971). These authors synthesised the compound by reaction of a freshly prepared Sc(OH)3 solution with H3AsO4 at 25°C. Komissarova et al. (Reference Komissarova, Pushkina and Khrameeva1971) prepared ScAsO4⋅2H2O as a white crystalline powder from aqueous solutions at the As/Sc ratios of 0.45–3.9 and pH 2.0–7.1. They did not determine the compound's space-group symmetry, but suggested a “monoclinically distorted rhombic lattice”, with a = 5.64(5), b = 10.47(1), c = 9.36(1) Å, β ≈ 90 and V ≈ 553 Å3. Komissarova et al. (Reference Komissarova, Pushkina, Khrameeva and Teterin1973) reported infrared and nuclear magnetic resonance spectra of ScAsO4⋅2H2O and its solubility in some acids and bases.
The third aim of our study is to discuss the crystal chemistry and structural topology of the variscite and metavariscite groups on the basis of a comparison of standardised structure data, an attempt which has never been made so far. It is worth pointing out that many of the data on the members of these groups are reported with respect to the non-standard space-group setting Pcab, as the a and c unit-cell parameters in the variscite group are very similar and the convention a>b>c was usually followed. Furthermore, the relative thermodynamic stabilities of the members of both groups and their structural and topological complexities are evaluated. We also provide an insight into the possible application of selected members as electrode materials.
Experimental
Syntheses
CrAsO4⋅2H2O was synthesised as part of a larger project with the aim of an extensive study of the insufficiently known system M 1+–M 3+–As–O–H (M 1+ = Li, Na, K, Rb, Cs, Ag, Tl or NH4; M 3+ = Al, Ga, In, Sc, Cr or Fe) (e.g. Schwendtner and Kolitsch, Reference Schwendtner and Kolitsch2007a,Reference Schwendtner and Kolitschb, Reference Schwendtner and Kolitsch2017, Reference Schwendtner and Kolitsch2018, and references cited therein). CrAsO4⋅2H2O was prepared by mild hydrothermal methods in Teflon-lined stainless steel autoclaves (T = 220°C, duration 7 days with slow furnace cooling) from a mixture of reagent-grade Sr(OH)2⋅8H2O, Cr(NO3)3⋅9H2O, H3AsO4⋅0.5H2O (volume ratio ~1:1:3) and distilled water. The Teflon container was filled up with distilled water to ~70–80% of its inner volume. Initial and final pH values were ~0.5 and <~0.5, respectively. The reaction products were filtered and washed with distilled water. They contained deep green, tabular crystals with a maximum dimension of ~0.07 mm and a habit equivalent to that of strengite (i.e. with sword-shaped crystal terminations). CrAsO4⋅2H2O was also obtained as tiny green crystals from a hydrothermal run containing a mixture of reagent-grade PbO, Cr(NO3)3⋅9H2O, H3AsO4⋅0.5H2O and GeO2. Initial pH was <~0.5; after the run, the reaction products were nearly dry. These CrAsO4⋅2H2O crystals were associated with an uninvestigated greyish grainy material. The observation of Lukaszewski et al. (Reference Lukaszewski, Redfern and Salmon1961) that CrAsO4⋅2H2O is only stable below 120°C, suggests that in both hydrothermal runs the compound crystallised during furnace-cooling.
Synthesis and crystal growth of Tl3+PO4⋅2H2O was performed according to the method of Mooney-Slater (Reference Mooney-Slater1961): 1 g Tl2O3 was dissolved in boiling concentrated nitric acid (5 ml). The slightly opaque solution was then filtered warm through a glass frit and cooled down to room temperature. Concentrated phosphoric acid was added dropwise (altogether 2 ml) to the colourless solution. Then water was added to the still clear solution until clouding. The reaction mixture was subsequently heated to ~80°C for two hours. In that time, a cloudy greyish precipitate with an amorphous appearance formed. The mixture was then stored at a warm place (40°C) for three weeks and was allowed to slowly evaporate. Colourless crystals had formed from the precipitate with the typical form as snubbed bipyramids with maximum edge lengths of 0.3 mm.
The preparation of MnSeO4⋅2H2O was achieved by two different methods. In method 1, an excess of MnCO3 was dissolved in hot diluted H2SeO4 (Merck, p.A.). The resulting solution was then filtered from the remaining solid and concentrated on a water bath (90°C). After evaporation at ~60°C, pale pink single crystals of MnSeO4⋅2H2O with an unspecific form were picked out of the hot solution and enclosed in “magic oil” (low-viscous perfluoropolyether 216, Riedel de Haën). This was necessary because the crystals were found to be sensitive towards moisture and to decompose under inclusion of water. When the solution was allowed to cool down to room temperature, crystals exclusively of MnSeO4⋅5H2O (Euler et al., Reference Euler, Meents, Barbier and Kirfel2003) had formed.
In method 2, an aqueous solution of hydrated manganese(II) chloride (2.4 mmol), 40% selenic acid (4.7 mmol) and distilled water (10 ml) was used. The solution was stirred with a magnetic stirrer at 80°C for three hours until it became fully homogeneous. Then the solution was poured onto a watch glass and left in a fume hood to evaporate at room temperature. Single crystals of MnSeO4⋅2H2O suitable for single-crystal X-ray diffraction analysis appeared at the bottom of the watch glass after two days.
Preparation of CdSeO4⋅2H2O was as follows: CdO (Merck, p.A.) was dissolved in a diluted H2SeO4 (Merck, p.A.) solution in excess. After evaporation at room temperature, the resulting CdSeO4⋅2H2O crystals were recrystallised in demineralised water. Colourless translucent crystals with mostly plate-like forms and an edge-length of up to 1 mm were then obtained from the cooled solution. The crystals are non-sensitive towards moisture and are stable at room temperature. We note, however, that in an old study of the system CdSeO4–H2O (Klein, Reference Klein1940), CdSeO4⋅2H2O is considered a metastable phase above –11.0°C.
Single-crystal X-ray diffraction
Selected crystals of CrAsO4⋅2H2O and bonacinaite (ideally ScAsO4⋅2H2O) were studied with a Nonius KappaCCD diffractometer equipped with a 300 μm diameter capillary-optics collimator to provide increased resolution. Intensity data collections were carried out using the parameters listed in Tables 3 and 4, respectively. The measured intensity data were processed with the Nonius program suite DENZO-SMN and corrected for Lorentz polarisation, background and absorption effects (see Table 3).
Table 3. Crystal data, data collection information and refinement details for variscite-type synthetic CrAsO4⋅2H2O, TlPO4⋅2H2O, MnSeO4⋅2H2O and CdSeO4⋅2H2O.

Table 4. Crystal data, data collection information and refinement details for metavariscite-type bonacinaite (ideally ScAsO4⋅2H2O).

The crystal structure of CrAsO4⋅2H2O was solved in space group Pbca by direct methods (SHELXS-97; Sheldrick, Reference Sheldrick2008) and subsequent Fourier and difference Fourier syntheses, followed by full-matrix least-squares anisotropic refinement on F 2 (SHELXL-2018/3; Sheldrick, Reference Sheldrick2015). The structure model obtained confirmed the isotypy with variscite. All H atoms were located, and the O−H bond lengths restrained to 0.90(1) Å. Subsequently, the structure model of variscite (Kniep et al., Reference Kniep, Mootz and Vegas1977) was adopted for a final refinement step which led to R 1 = 2.14%.
The crystal structure of bonacinaite was solved in space group P21/n by direct methods (SHELXS-97; Sheldrick, Reference Sheldrick2008). Details on data collection and refinement are given in Table 4. A full-matrix least-squares anisotropic refinement on F 2 (SHELXL-2018/3; Sheldrick, Reference Sheldrick2015) provided a structure model in full agreement with a metavariscite-type atomic arrangement. Based on subsequent scanning electron microscopy using energy-dispersive spectroscopic analyses of the studied crystal, which showed minor Al (and traces of Fe) and P (and traces of Si) as impurity constituents substituting for Sc and As, respectively, the occupancies of the Sc and As sites were refined taking into account these substitutions. The refinement converged at R 1 = 3.66% and showed that ~19% of the Sc site contained Al, and ~23% of the As site contained P (the trace amounts of Fe and Si were ignored).
Intensity datasets of crystals of MnSeO4⋅2H2O (prepared by method 1) and CdSeO4⋅2H2O were measured with a Bruker SMART (CCD) diffractometer, while a crystal of MnSeO4⋅2H2O (prepared by method 2) was measured with a Bruker APEX DUO diffractometer. The measurement of a TlPO4⋅2H2O crystal was done with a Bruker APEX-II diffractometer at −173°C. Using Bruker software (Bruker AXS, 1997, 1998a,b), the datasets were processed and corrected for Lorentz polarisation, background and absorption effects (for details see Table 3). For all synthetic crystals, systematic extinctions and structure factor statistics unambiguously indicated the centrosymmetric space group Pbca. Using the published coordinate set for variscite (Kniep et al., Reference Kniep, Mootz and Vegas1977) as a starting model, the structures were refined with SHELXL-2018/3 (Sheldrick, Reference Sheldrick2015). The positions of the hydrogen atoms were clearly discernible from difference-Fourier maps and included in the model, while restraining the O−H bond length to 0.90(1) Å. No significant deviation from unit occupancy was observed for any of the atoms in any of the structures of the synthetic variscite-group representatives (including CrAsO4⋅2H2O).
The final positional and displacement parameters of all measured crystals are given in the deposited crystallographic information files deposited with the Principal Editor of Mineralogical Magazine and available as Supplementary material (see below). The latter include lists of observed and calculated structure factors. Selected bond lengths (including hydrogen bonds) and calculated bond-valence sums (BVSs) are presented in Tables 5, 6 and 7.
Table 5. Selected interatomic distances (Å) and calculated bond valences (vu) for CrAsO4⋅2H2O and Tl3+PO4⋅2H2O.

1) There is an additional, long hydrogen bond: Ow2–H21⋅⋅⋅O3 at 3.225(3) Å, ∠DHA = 135(5)°.
2) There is an additional, long hydrogen bond: Ow2–H22⋅⋅⋅O2 at 2.996(3) Å, ∠DHA = 117(3)°.
3) There is an additional, long hydrogen bond: Ow2–H21⋅⋅⋅O3 at 3.079(3) Å, ∠DHA = 120(5)°.
Notes: Bond-valence calculations were done with the program VALENCE (Brown, Reference Brown1996) and bond-valence parameters from Gagné and Hawthorne (Reference Gagné and Hawthorne2015); sum values are derived from unrounded bond-valence contributions. Bond-valence sums (vu) for the O atoms for CrAsO4⋅2H2O are O1: 1.75; O2: 1.78; O3: 1.86; O4: 1.78; Ow1: 0.43; Ow2: 0.48, and for Tl3+PO4⋅2H2O are O1: 1.74; O2: 1.75; O3: 1.72; O4: 1.76; Ow1: 0.42; Ow2: 0.50.
Table 6. Selected interatomic distances (Å) and calculated bond valences (vu) for MnSeO4.2H2O (crystal method 1) and CdSeO4.2H2O.

1) There is an additional, long hydrogen bond: Ow2–H11⋅⋅⋅Ow2 at 3.121(3) Å, ∠DHA = 115(3)°.
2) There is an additional, long hydrogen bond: Ow2–H21⋅⋅⋅O3 at 3.206(3) Å, ∠DHA = 126(3)°.
3) There is an additional, long hydrogen bond: Ow2–H11⋅⋅⋅Ow2 at 3.027(3) Å, ∠DHA = 173(4)°.
4) There is an additional, long hydrogen bond: Ow2–H21⋅⋅⋅O3 at 3.137(2) Å, ∠DHA = 125(3)°.
Notes: Bond-valence calculations were done with the program VALENCE (Brown, Reference Brown1996) and bond-valence parameters from Gagné and Hawthorne (Reference Gagné and Hawthorne2015); sum values are derived from unrounded bond-valence contributions. Bond-valence sums (vu) for the O atoms for MnSeO4⋅2H2O are O1: 1.82; O2: 1.84; O3: 1.85; O4: 1.87; Ow1: 0.33; Ow2: 0.38, and for CdSeO4⋅2H2O are O1: 1.82; O2: 1.81; O3: 1.83; O4: 1.86; Ow1: 0.33; and Ow2: 0.38.
Table 7. Selected interatomic distances (Å) and calculated bond valences (vu) for bonacinaite (ideally ScAsO4⋅2H2O) with the refined structural formula (Sc0.807(1)Al0.193)(As0.767(7)P0.233)O4⋅2H2O.
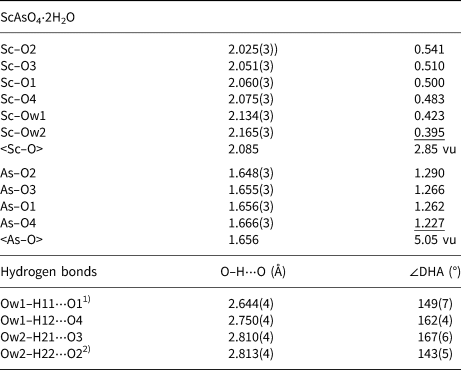
1) There is an additional, long hydrogen bond: Ow1–H11⋅⋅⋅O3 at 3.258(4) Å, ∠DHA = 123(5)°.
2) There is an additional, long hydrogen bond: Ow2–H22⋅⋅⋅O1 at 3.152(4) Å, ∠DHA =122(5)°.
Notes: Bond-valence calculations were done with the program VALENCE (Brown, Reference Brown1996) and bond-valence parameters from Gagné and Hawthorne (Reference Gagné and Hawthorne2015), taking into account the refined occupancies of the mixed Sc and As sites. Sum values are derived from unrounded bond-valence contributions. Bond-valence sums (vu) for the O atoms are O1: 1.76; O2: 1.83; O3: 1.78; O4: 1.71; Ow1: 0.42; and Ow2: 0.40.
Thermal analysis
Thermoanalytical measurements of CdSeO4⋅2H2O and MnSeO4⋅2H2O were performed in an open system under a flowing N2 atmosphere and a heating rate of 5°C/min on a Mettler–Toledo TG50 (35–800°C, corundum crucibles) and a Mettler–Toledo DSC-25 system (35–550°C, aluminium capsules). The remaining solids obtained after heating were identified by PXRD.
Results
Thermal behaviour of CdSeO4·2H2O and MnSeO4·2H2O
The thermal behaviour on heating of the two synthetic variscite-type selenates is shown in Fig. 1 (TG and DSC curves). For the Cd compound the thermal dehydration follows a well-resolved two-step mechanism. In the first step, one mole H2O is released at ~112°C (reaction 1a, see below) and in the second step at ~178°C the other mole H2O is liberated and anhydrous CdSeO4 is formed (1b). The theoretical mass loss of 12.36% is in good agreement with the experimental value of 12.1%. Above 600°C, CdSeO4 starts to decompose under release of SeO2 and O2 into CdO (1c; PXRD-confirmed). The decomposition is completed at temperatures above 750°C.
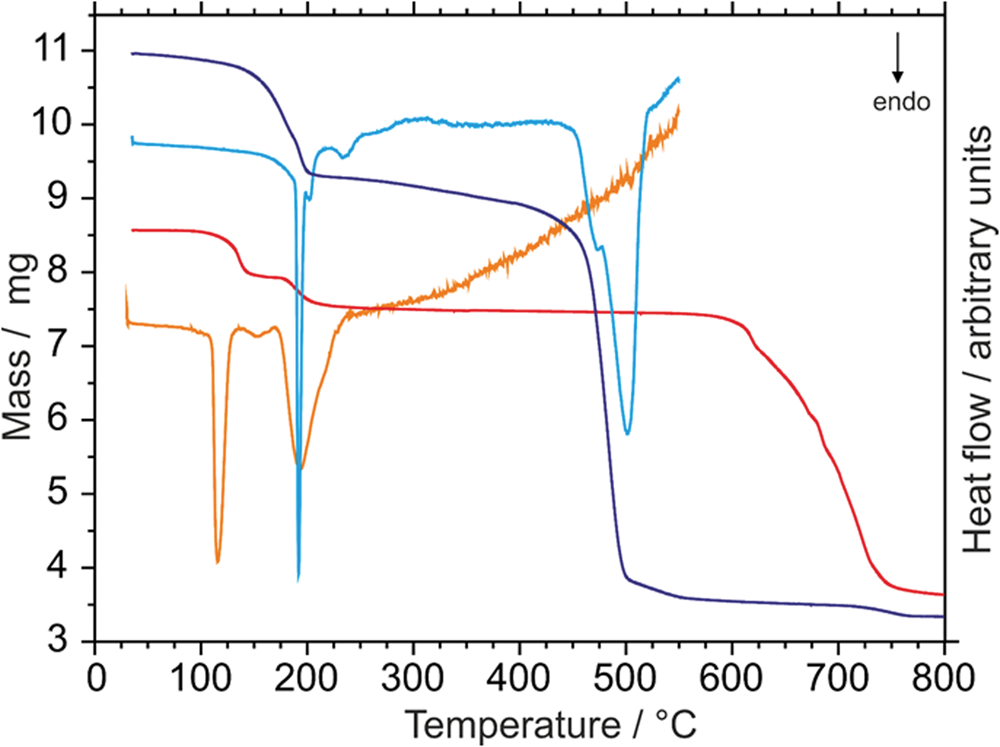
Fig. 1. Thermal analysis of CdSeO4⋅2H2O (TG curve: red and DSC curve: orange) and MnSeO4⋅2H2O (TG curve: dark violet and DSC curve: blue).
For the Mn compound the course of the thermal dehydration is different and the corresponding reaction steps are not well resolved. Above ~140°C the selenate starts to dehydrate and both water molecules are more or less released simultaneously (2a,b). The small hump in the DSC curve at ~205°C actually indicates a second step, but this is not clearly visible in the corresponding TG curve. The theoretical mass loss of 15.40% for the complete dehydration is also in good agreement with the experimental value of 15.1%. It should be noted that the previously reported thermal decomposition reactions of the pentahydrate, MnSeO4⋅5H2O, and the formed intermediate dihydrate MnSeO4⋅2H2O (Nabar and Paralkar, Reference Nabar and Paralkar1975), are different from the results of the present investigation. These authors observed a well-resolved two-step dehydration mechanism of the dihydrate with maxima at ~160°C and ~260°C, respectively. However, these observations could not be reproduced with our experimental set-up, possibly because of a different heating rate (10°C/min) and heating atmosphere (air flow) used by the authors of the 1975 paper. In agreement with the results of Nabar and Paralkar (Reference Nabar and Paralkar1975), MnSeO4 partly decomposes into MnSeO3 above 300°C, which is accompanied by a small mass loss but no DSC signal. At temperatures above 440°C, the anhydrous selenate decomposes completely and at temperatures >700°C Mn3O4 (synthetic hausmannite) is formed (2c) according to PXRD.
 $${\rm CdSe}{\rm O}_4\!\!\cdot\!\! 2{\rm H}_2{\rm O}\comma_{\rm s}\rightarrow {\rm CdSe}{\rm O}_4\!\!\cdot\!\! {\rm H}_2{\rm O}\comma_{\rm s} + {\rm H}_2{\rm O}\comma_{\rm g}$$
$${\rm CdSe}{\rm O}_4\!\!\cdot\!\! 2{\rm H}_2{\rm O}\comma_{\rm s}\rightarrow {\rm CdSe}{\rm O}_4\!\!\cdot\!\! {\rm H}_2{\rm O}\comma_{\rm s} + {\rm H}_2{\rm O}\comma_{\rm g}$$ $${\rm CdSe}{\rm O}_4\!\!\cdot\!\! {\rm H}_2{\rm O}\comma_{\rm s}\rightarrow {\rm CdSe}{\rm O}_4\comma_{\rm s} + {\rm H}_2{\rm O}\comma_{\rm g}$$
$${\rm CdSe}{\rm O}_4\!\!\cdot\!\! {\rm H}_2{\rm O}\comma_{\rm s}\rightarrow {\rm CdSe}{\rm O}_4\comma_{\rm s} + {\rm H}_2{\rm O}\comma_{\rm g}$$ $${\rm CdSe}{\rm O}_4,_{\rm s}\rightarrow {\rm CdO},_{\rm s} + {\rm Se}{\rm O}_2,_{\rm g} + {\rm }\raise.5ex\hbox{$\scriptstyle 1$}\kern-.1em/ \kern-.15em\lower.25ex\hbox{$\scriptstyle 2$} {\rm O}_2,_{\rm g}$$
$${\rm CdSe}{\rm O}_4,_{\rm s}\rightarrow {\rm CdO},_{\rm s} + {\rm Se}{\rm O}_2,_{\rm g} + {\rm }\raise.5ex\hbox{$\scriptstyle 1$}\kern-.1em/ \kern-.15em\lower.25ex\hbox{$\scriptstyle 2$} {\rm O}_2,_{\rm g}$$ $${\rm MnSe}{\rm O}_4\!\!\cdot\!\! 2{\rm H}_2{\rm O}\comma_{\rm s}\rightarrow {\rm MnSe}{\rm O}_4\!\!\cdot\!\! {\rm H}_2{\rm O}\comma_{\rm s} + {\rm H}_2{\rm O}\comma_{\rm g}$$
$${\rm MnSe}{\rm O}_4\!\!\cdot\!\! 2{\rm H}_2{\rm O}\comma_{\rm s}\rightarrow {\rm MnSe}{\rm O}_4\!\!\cdot\!\! {\rm H}_2{\rm O}\comma_{\rm s} + {\rm H}_2{\rm O}\comma_{\rm g}$$ $${\rm MnSe}{\rm O}_4\!\!\cdot\!\! {\rm H}_2{\rm O}\comma_{\rm s}\rightarrow {\rm MnSe}{\rm O}_4\comma_{\rm s} + {\rm H}_2{\rm O}\comma_{\rm g}$$
$${\rm MnSe}{\rm O}_4\!\!\cdot\!\! {\rm H}_2{\rm O}\comma_{\rm s}\rightarrow {\rm MnSe}{\rm O}_4\comma_{\rm s} + {\rm H}_2{\rm O}\comma_{\rm g}$$ $$3{\rm MnSe}{\rm O}_4\comma_{\rm s}\rightarrow {\rm M}{\rm n}_3{\rm O}_4\comma_{\rm s} + 3{\rm Se}{\rm O}_2\comma_{\rm g} + {\rm O}_2\comma_{\rm g}$$
$$3{\rm MnSe}{\rm O}_4\comma_{\rm s}\rightarrow {\rm M}{\rm n}_3{\rm O}_4\comma_{\rm s} + 3{\rm Se}{\rm O}_2\comma_{\rm g} + {\rm O}_2\comma_{\rm g}$$Descriptions of the structures
As the structure connectivity of the variscite and metavariscite structure types is well established, we provide only the most relevant aspects of the crystal structures. For further details, the reader is referred to dedicated previous publications (e.g. Moore, Reference Moore1966; Bennett et al., Reference Bennett, Dytrych, Pluth, Richardson and Smith1986; Taxer and Bartl, Reference Taxer and Bartl2004; Ilyushin and Blatov, Reference Ilyushin and Blatov2017).
Variscite-type synthetic compounds
The asymmetric unit of the four synthetic compounds with a variscite-type structure contains one M (M = Cr, Tl, Mn or Cd) atom, one T (T = As, P or Se) atom, six O atoms (two of which belong to water molecules) and four H atoms. The M atom is octahedrally coordinated by four O and two Ow ligands. The later are in cis configuration. The T atom is tetrahedrally coordinated by four O ligands. The TO4 tetrahedra share each of their four corners with the MO4(H2O)2 octahedra, thus forming a dense three-dimensional network. The H atoms of the two H2O molecules point into a channel running parallel to the a axis, and all H atoms participate in strong to weak, partly bifurcated hydrogen bonds. The latter are stronger for the arsenate and phosphate (2.581 to 3.226 Å, Table 5) and comparatively weaker for the two selenates (2.695 to 3.217 Å, Table 6). As examples, Fig. 2 shows polyhedral representations of the structures of CrAsO4⋅2H2O and CdSeO4⋅2H2O. Average M–O bond lengths are: <Cr3+–O> = 1.981 Å, <Tl3+–O> = 2.224 Å, <Mn–O> = 2.179 Å and <Cd–O> = 2.283 Å. The Cr value is slightly shorter than the grand mean value of 1.999 Å in inorganic compounds reported by Baur (Reference Baur, O'Keeffe and Navrotsky1981). The Tl3+ value is very similar to that observed in the isotypic arsenate analogue Tl3+AsO4⋅2H2O (2.23 Å, Schroffenegger et al., Reference Schroffenegger, Eder, Weil, Stöger, Schwendtner and Kolitsch2020), while the Mn value is slightly lower than the mean value of 2.205 Å given in Baur (Reference Baur, O'Keeffe and Navrotsky1981). The value for Cd is comparable to those in other Cd oxysalt compounds (to our knowledge, no review on Cd–O bonds in inorganic compounds exist).

Fig. 2. Polyhedral representation of the framework crystal structures of CrAsO4⋅2H2O (left) and CdSeO4⋅2H2O (right) in a view along [001]. Hydrogen bonds are shown with dashed lines. The unit cells are outlined. All structure drawings were done with ATOMS V. 6.3 (Dowty, Reference Dowty2011).
The tetrahedral anions show the following average T–O bond lengths: <As–O> = 1.685 Å, <P–O> = 1.545 Å, <Se–O> = 1.637 Å (Mn member) and 1.639 Å (Cd member). These values compare favourably with the corresponding grand mean bond lengths of 1.687(8) Å (Gagné and Hawthorne, Reference Gagné and Hawthorne2018), 1.537 Å (Huminicki and Hawthorne, Reference Huminicki, Hawthorne, Kohn, Rakovan and Hughes2002) and 1.638 Å (Krivovichev, Reference Krivovichev2009), respectively. The slightly elevated value observed for TlPO4⋅2H2O may be explained by a ‘relaxation’ of the structure due to the large Tl3+ cation and its relatively weak Tl3+–O bonds, allowing the P atom to compete less strongly for the common O ligands. The O ligands O1–4 are all somewhat underbonded and, accordingly, acceptors of hydrogen bonds (see below).
The flexibility of the variscite-type framework topology is predominantly due to the corner-linkage of the three-dimensionally linked octahedral and tetrahedral building units, allowing for considerable variation of the M–O–T angles. This is also reflected in the individual distortions of the MO4(H2O)2 octahedra, in which the sequence of O ligands with shortest vs. longest M–O bond varies from member to member, even when the two selenates are compared (Tables 5, 6). Moreover, the length of individual, topologically more or less equivalent hydrogen bonds, also shows distinct variation (Tables 5, 6). Bifurcation of hydrogen bonds may be present or absent. The comparatively shortest hydrogen bonds (O⋅⋅⋅O), 2.574(3) and 2.584(3) Å, are present in CrAsO4⋅2H2O, although TlPO4⋅2H2O has also one similarly short bond, 2.581(3) Å. In scorodite, FeAsO4⋅2H2O, the two shortest hydrogen bonds are 2.603(9) and 2.639(9) Å according to Hawthorne (Reference Hawthorne1976), somewhat different from those given for synthetic scorodite with a notably larger unit cell (cf. Table 1), 2.622(4) and 2.645(5) Å. In synthetic mansfieldite, AlAsO4⋅2H2O, the shortest O⋅⋅⋅O donor–acceptor distances are 2.578(4) and 2.592(3) Å (Harrison, Reference Harrison2000), i.e. very similar to those in CrAsO4⋅2H2O.
The four representatives characterised herein show that the stability range of the variscite structure type is apparently limited by the following ranges of the [6]M2+/3+ and [4]T5+/6+ cationic radii (values of Shannon, Reference Shannon1976): 0.535 Å (Al3+) to 0.95 Å (Cd2+) for M2+/3+ ions and 0.17 Å (P5+) to 0.28 Å (Se6+) for T5+/6+ ions. Therefore, we predict that the following hypothetical members may also be stable, possibly even in natural environments:
(1) The arsenates ScAsO4⋅2H2O (orthorhombic dimorph of bonacinaite, see below) and possibly LuAsO4⋅2H2O. Note that hypothetical V3+AsO4⋅2H2O is not considered to be stable because V3+ would be oxidised by the relatively strong oxidising agent As5+ (and, conversely, As5+ would be reduced by the relatively strong reducing agent V3+). No compound containing both V3+ and As5+ cations is present in the current version of the Inorganic Crystal Structure Database (Belsky et al., Reference Belsky, Hellenbrandt, Karen and Luksch2002), and no entry on anhydrous V3+AsO4 is found in any of the major literature databases.
(2) The selenates MgSeO4⋅2H2O, FeSeO4⋅2H2O, CoSeO4⋅2H2O and NiSeO4⋅2H2O. A careful review of the literature showed that among these, FeSeO4⋅2H2O was never reported, but CoSeO4⋅2H2O was observed as an intermediate phase during the stepwise dehydration of CoSeO4⋅4H2O (Nabar and Paralkar, Reference Nabar and Paralkar1975) and CoSeO4⋅6H2O (Koleva and Stoilova, Reference Koleva and Stoilova1997). CoSeO4⋅2H2O was also reported earlier in a study of the thermodynamic properties of cobalt selenate hydrates (Maier et al., Reference Maier, Selivanova and Terent'eva1965). Similar to its Co analogue, NiSeO4⋅2H2O was observed as a result of dehydrating NiSeO4⋅6H2O (Klein, Reference Klein1940; Demassieux, Reference Demassieux1945), of heating NiSeO4⋅6H2O at 200°C (Snyman and Pistorius, Reference Snyman and Pistorius1963), and as an intermediate phase during the stepwise dehydration of NiSeO4⋅6H2O (Stoilova and Koleva, Reference Stoilova and Koleva1997). The latter authors found that NiSeO4⋅2H2O forms orthorhombic crystals with the unit-cell parameters a = 10.351(4), b = 10.219(4), c = 9.017(5) Å and V = 953.9(6) Å3. These values strongly suggest that the compound has in fact a variscite-type structure. MgSeO4⋅2H2O was detected on dehydrating MgSeO4⋅6H2O at 140°C (Klein, Reference Klein1940). “Orthorhombic” MgSeO4⋅2H2O was observed in a study of the dehydration of MgSeO4⋅6H2O, and the following unit-cell parameters were given: a = 10.304(1), b = 10.351(9) and c = 9.138(9) (Stoilova and Koleva, Reference Stoilova and Koleva1995a,Reference Stoilova and Kolevab). Again, these data serve as a strong indication that MgSeO4⋅2H2O is a variscite-type compound (Stoilova and Koleva, Reference Stoilova and Koleva1995a, already assumed that MgSeO4⋅2H2O is “isomorphous with MnSeO4⋅2H2O”, based on a comparison of PXRD data and cell parameters).
Metavariscite-type bonacinaite
The asymmetric unit of monoclinic bonacinaite contains one Sc site (with the refined occupancy Sc0.807(1)Al0.193), one As site (refined occupancy As0.767(7)P0.233), six O atoms (two of which belong to water molecules) and four H atoms, i.e. the same number of corresponding atoms as in the orthorhombic variscite-type compounds. The monoclinic angle β in bonacinaite, 91.94(3)°, is fairly close to 90°, reflecting pseudo-orthorhombic cell metrics. However, this angle β shows the largest deviation from 90° among all known metavariscite-type minerals and synthetic compounds. The structure of bonacinaite (Fig. 3) is very similar to that of the variscite-type compounds. The most notable structural difference is the orientation of the (As,P)O4 tetrahedron linked to the (Sc,Al)O4(H2O)2 octahedron and the environment of the H2O ligands. The latter results in different hydrogen-bonding schemes.

Fig. 3. Polyhedral representation of the framework crystal structure of metavariscite-type bonacinaite, (Sc,Al)(As,P)O4⋅2H2O, in a view along [100]. Hydrogen bonds are shown with dashed lines. The unit cell is outlined. Note the channels parallel to [100], which are relatively large by comparison to the denser variscite-type structures (Fig. 2).
Due to the partial substitution of Al for Sc in the (Sc,Al)O4(H2O)2 octahedron, the mean M–O bond length, 2.085 Å (Table 7), is slightly shifted from the value expected for pure Sc compounds, 2.105 Å (Baur, Reference Baur, O'Keeffe and Navrotsky1981) or 2.10(7) Å (Serezhkin et al., Reference Serezhkin, Kryuchkova and Kazakevich2003), towards the value of pure Al compounds, 1.909 Å (Baur, Reference Baur, O'Keeffe and Navrotsky1981). The mean T–O bond length in the (As,P)O4 tetrahedron, 1.656 Å, is between those of pure arsenates and phosphates (see above), and reflects the partial substitution of P for As. As in the four variscite-type compounds, the O ligands O1–4 in bonacinaite are all somewhat underbonded and, accordingly, acceptors of hydrogen bonds (Table 7). The latter are of medium strength and range between 2.644(4) and 2.813(4) Å (Table 7); two further, long hydrogen bonds to O3 and O1 at 3.258(4) and 3.152(4) Å, respectively, show that the bonds involving Ow1 and H11, and Ow2 and H22, are bifurcated.
Structure comparison
For a quantitative comparison of the isotypic crystal structures within the variscite and metavariscite groups, respectively, the program COMPSTRU (de la Flor et al., Reference de la Flor, Orobengoa, Tasci, Perez-Mato and Aroyo2016) available at the Bilbao Crystallographic Server (Aroyo et al., Reference Aroyo, Perez-Mato, Capillas, Kroumova, Ivantchev, Madariaga, Kirov and Wondratschek2006) was used. The criterion for this comparison is based on a full crystal-structure analysis from single-crystal diffraction data and a reliability factor R 1 < 0.05. Except for variscite-type TlPO4⋅2H2O and TlAsO4⋅2H2O (Schroffenegger et al., Reference Schroffenegger, Eder, Weil, Stöger, Schwendtner and Kolitsch2020), for which diffraction data were recorded at –173°C, all other crystals were measured at room temperature. The effect of the temperature on unit-cell parameters was neglected for the two cases. Due to different treatments of H atoms in the various refinements, e.g. by constrains/restraints regarding O–H bond lengths, our comparisons do not include hydrogen atoms.
Variscite (Kniep et al., Reference Kniep, Mootz and Vegas1977) and metavariscite (Kniep and Mootz, Reference Kniep and Mootz1973), respectively, were chosen as references, to which all other crystal structures in the corresponding groups were compared. For that purpose, literature data were standardised in terms of atom labelling and space-group setting and then were related to the reference structures. In the case of variscite, a cyclic transformation and a shift of the origin was necessary for some cases. In the case of metavariscite, all structures (including metavariscite itself) were transformed into the standard setting of space group type no. 14 (P21/c).
In Tables 8 and 9, numerical results of the comparisons are given, listing displacements of individual atoms relative to the standard. Also compiled are the degree of lattice distortion (S) which is the spontaneous strain, i.e. the sum of the squared eigenvalues of the strain tensor divided by 3, the arithmetic mean (d av) of the distances and the measure of similarity (Δ) (Bergerhoff et al., Reference Bergerhoff, Berndt, Brandenburg and Degen1999). The latter is a function of the differences in atomic positions (weighted by the multiplicities of the sites) and the ratios of the corresponding unit-cell parameters of the structures (de la Flor et al., Reference de la Flor, Orobengoa, Tasci, Perez-Mato and Aroyo2016).
Table 8. Numerical details from the comparisons of the crystal structure of variscite (AlPO4⋅2H2O) with isotypic structures1) of the variscite group using the COMPSTRU program (de la Flor et al., Reference de la Flor, Orobengoa, Tasci, Perez-Mato and Aroyo2016).

1) Atom labelling refers to the refinements in this study and corresponds to that originally given for variscite (Kniep et al., Reference Kniep, Mootz and Vegas1977).
2) P is the transformation and p is the shift of origin used for standardisation of crystal data relative to variscite.
Table 9. Numerical details from the comparisons of the crystal structure of metavariscite-type AlPO4⋅2H2O (Kniep and Mootz, Reference Kniep and Mootz1973) with isotypic structures1) of the metavariscite group using the COMPSTRU program (de la Flor et al., Reference de la Flor, Orobengoa, Tasci, Perez-Mato and Aroyo2016).

1) Atom labelling refers to the refinements in this study and corresponds to that originally given for metavariscite (Kniep and Mootz, Reference Kniep and Mootz1973).
In terms of atomic displacements, there is no clear trend recognisable for the variscite group, which may reflect the flexibility of the shared atomic arrangement. In the majority of cases (ten out of thirteen), atom O3 (which is one of the ligands linking MO4(H2O)2 octahedra to TO4 tetrahedra) shows the highest displacement in the crystal structure. The lowest displacements are shown by the O atom that belongs to water molecule Ow2 (six cases), the metal atom M (five cases) and the T atom in the TO4 tetrahedron (two cases). On the other hand, in the metavariscite group the trend regarding maximum and minimum atomic displacements is somewhat clearer: in four out of five cases, the maximum value is associated with the O atom of the water molecule Ow2 (Ow1 has the second highest values in all cases), and the minimum value pertains to the tetrahedrally coordinated T atoms. Thus, the TO4 groups behave, not surprisingly, as a rigid building unit.
The most informative parameter from the comparisons is the measure of similarity (Δ). The smaller the number of Δ, the higher is the similarity of the two compared structures, with Δ = 0.114 for CdSeO4⋅2H2O being the maximum value for all structures. For both variscite and metavariscite groups, Δ appears not to depend on the variation of M1 and X1 or their formal charges in the dimorphic MXO4⋅2H2O compounds, but clearly on the crystal volume. Within a roughly linear correlation, Δ increases with increasing volume (Fig. 4).

Fig. 4. Dependence of the measure of similarity on the crystal volume for the variscite group (a) and the metavariscite group (b).
From the viewpoint of topology, both variscite and metavariscite are based upon four-connected 3D frameworks based upon simple hexagonal 2D nets. Note that, despite the fact that both variscite and metavariscite are based upon octahedral–tetrahedral frameworks, only four vertices of each AlO6 octahedron are bridging, whereas the two remaining ones are occupied by H2O molecules and do not participate in the intraframework linkage. Wells (Reference Wells1954) and Smith (Reference Smith1977) derived a number of 3D nets based upon hexagonal nets linked in the direction perpendicular to the net plane. Figure 5a and c shows the hexagonal net in the metavariscite and variscite frameworks, respectively, with black and white nodes pointed upward and downward, respectively. According to Smith (Reference Smith1977), two adjacent nodes in a hexagon can have the additional linkages pointing in either the same (S) or changed (C) direction, respectively. Thus, each edge of the net is associated with either the S or C symbol. The metavariscite 3D net is based upon the 2D net with the cyclic symbol SCCSCC (Fig. 5a), whereas that in variscite has the symbol SCSCCC (Fig. 5c). The linkage of the hexagonal 2D nets in both metavariscite and variscite results in the formation of four-membered rings oriented perpendicular to the 2D nets. The idealised topologies of the metavariscite and variscite frameworks are shown in Fig. 5b and d, respectively.

Fig. 5. Idealised topologies of frameworks in metavariscite (a, b) and variscite (c, d). The black and white vertices in the simple hexagonal nets in (a, c) correspond to the up and down linkages, respectively. The grey squares in (b, d) highlight the location of the four-membered rings. See text for details.
The metavariscite topology corresponds to the BCT type of zeolite topologies, which was observed in the framework alkali silicates with Si4+ replaced by Mg2+, Zn2+ or Fe2+ (type material: Dollase and Ross, Reference Dollase and Ross1993) and in svyatoslavite, a metastable polymorph of anorthite, CaAl2Si2O8 (Chesnokov et al., Reference Chesnokov, Lotova, Pavlyuchenko, Nigmatulina, Usova, Bushmakin and Nishanbaev1989; Krivovichev et al., Reference Krivovichev, Shcherbakova and Nishanbaev2012). The ideal symmetry of the BCT framework is described by the tetragonal space group I4/mmm. In metavariscite, the symmetry is reduced to P21/n, which is a subgroup of I4/mmm. The symmetry reduction can be viewed as consisting of two steps: the I4/mmm → P42/mnm transition due to the Al–P ordering of the framework nodes and the P42/mnm → P21/n transition due to the framework distortion. The topology is based upon four-membered rings of tetrahedra stacked in columns along the c axis and interlinked with similar rings in the adjacent columns (Fig. 5b).
The variscite topology was first recognised by Smith (Reference Smith1977) as the topology of the gallate framework in monoclinic CaGa2O4 (Deiseroth and Müller-Buschbaum, Reference Deiseroth and Müller-Buschbaum1973). Its ideal symmetry is described by the space group Cmca, which, due to the Al–P ordering, is reduced to its subgroup Pbca as observed in variscite. Whereas four-membered rings in the metavariscite topology are parallel to each other (Fig. 5b), they are inclined relative to each other in the variscite topology (Fig. 5d).
Relative stabilities of the two structure types
From our survey of the literature and the study of the title compounds and minerals, it appears not fully clear whether one of the two atomic arrangements can be considered thermodynamically more stable at ambient temperature, and how large the influence of kinetics and other parameters (e.g. pH) is. From hydrothermal syntheses of metavariscite and variscite, Sergeeva (Reference Sergeeva2016) concluded that metavariscite is the high-temperature dimorph and variscite the low-temperature one; both were found to coexist at a temperature of ~63°C. In a detailed study on natural strengite and phosphosiderite, Wilk (Reference Wilk1959) observed that phosphosiderite shows a reversible transformation at 95°C, but the nature of the high-temperature phase was not identified (the high-temperature Guinier powder pattern does not resemble that of strengite); this transformation may be related to a partial dehydration. A mixture of synthetic strengite and its dimorph phosphosiderite from hydrolysis of Fe(H2PO4)3⋅H2O at pH 2.8 was obtained by Eshchenko et al. (Reference Eshchenko, Shchegrov, Pechkovskii and Ustimovich1973). Strengite and phosphosiderite, as well as variscite and metavariscite, can also coexist in natural assemblages (e.g. phosphate pegmatites), suggesting very small differences in their Gibbs energy of formation. To our knowledge, only values for strengite and variscite have been determined (Robie et al., Reference Robie, Hemingway and Fisher1979, and Woods and Garrels, Reference Woods and Garrels1987, respectively), but none for phosphosiderite or metavariscite. The X-ray density values of dimorphic pairs are distinctly different: for strengite, D x = 2.85 mg m–3, while for phosphosiderite, D x = 2.72 mg m–3 (Taxer and Bartl, Reference Taxer and Bartl2004); for variscite, D x = 2.59 mg m–3 (Kniep et al., Reference Kniep, Mootz and Vegas1977), whereas metavariscite has D x = 2.535 mg m–3 (Kniep and Mootz, Reference Kniep and Mootz1973). This clearly demonstrates that the orthorhombic modification in the phosphate subgroups is denser and therefore can be considered more stable. The case is less clear-cut when synthetic InPO4⋅2H2O is considered: The orthorhombic dimorph has D x = 3.294 mg m–3 (Tang et al., Reference Tang, Gentiletti and Lachgar2002), while the monoclinic dimorph has D x = 3.300 (Sugiyama et al., Reference Sugiyama, Yu, Hiraga and Terasaki1999).
The influence of pH also plays a role: in the case of synthetic dimorphic InPO4⋅2H2O (Sugiyama et al., Reference Sugiyama, Yu, Hiraga and Terasaki1999), crystallisation of the variscite-type dimorph was favoured when mixtures with a higher H3PO4 content were used. The resulting lower pH may lead to a change in speciation in the hydrothermal fluid, and to the formation of polynuclear complexes that may then template specific crystalline solids. This observation suggests that (metastable?) metavariscite-type InAsO4⋅2H2O might be prepared under certain pH conditions and/or at elevated temperatures.
It is worth noting that Strunz and von Sztrokay (Reference Strunz and von Sztrokay1939) stated that the existence of a monoclinic, metavariscite-type “clinoscorodite” in nature is probable. However, such a dimorph was never confirmed in the numerous studies on the preparation and stability of scorodite, although it might crystallise from high(er)-temperature hydrothermal solutions if strongly stabilised in some way.
Topological and structural complexity and stability
The relative complexity of the structures and topologies of variscite and metavariscite can be analysed using the information complexity measures proposed in Krivovichev (Reference Krivovichev2012, Reference Krivovichev2013a,Reference Krivovichevb, Reference Krivovichev2014). Within this approach, the crystal-structure complexity is numerically estimated as the amounts of structural Shannon information per atom (strI G) and per unit cell (strI G,total), calculated according to the following equations:
 $$^{str} I_G = - \sum\nolimits_{i = 1}^k {\,p_i\;{\rm lo}{\rm g}_2p_i\;\lpar {{\rm bits}/{\rm atom}} \rpar } $$
$$^{str} I_G = - \sum\nolimits_{i = 1}^k {\,p_i\;{\rm lo}{\rm g}_2p_i\;\lpar {{\rm bits}/{\rm atom}} \rpar } $$ $$^{str} I_{G\comma total} = - v I_G = - v\;\sum\nolimits_{i = 1}^k {\,p_i\;{\rm lo}{\rm g}_2p_i\,\,\lpar {{\rm bits}/{\rm cell}} \rpar } $$
$$^{str} I_{G\comma total} = - v I_G = - v\;\sum\nolimits_{i = 1}^k {\,p_i\;{\rm lo}{\rm g}_2p_i\,\,\lpar {{\rm bits}/{\rm cell}} \rpar } $$where k is the number of different crystallographic orbits in the structure and pi is the random choice probability for an atom from the ith crystallographic orbit, that is:
 $$p_i = m_i/ v$$
$$p_i = m_i/ v$$where mi is a multiplicity of a crystallographic orbit (i.e. the number of atoms of a specific Wyckoff site in the reduced unit cell), and v is the total number of atoms in the reduced unit cell.
The topological complexity is calculated by taking into account the complexity of the idealised framework topology, where each T atom (T = Al or P) is associated with a node, and each edge is associated with the O atom in the middle of the T–T link (Krivovichev, Reference Krivovichev2013b).
The topological and structural complexity parameters for variscite and metavariscite are given in Table 10. It can be seen that variscite is both structurally and topologically more complex than metavariscite. This kind of complexity relations is observed for stable and metastable polymorphs that form in an Ostwald sequence of phases in inorganic systems. According to the Goldsmith's simplexity principle (Goldsmith, Reference Goldsmith1953), metastable kinetically stabilised phases in the Ostwald cascades are usually structurally simpler than their thermodynamically stable polymorphs. Krivovichev (Reference Krivovichev2013a) confirmed the validity of this principle using information-based complexity measures, and several other examples have been accumulated recently (Cempírek et al., Reference Cempírek, Grew, Kampf, Ma, Novák, Gadas, Škoda, Vašinová-Galiová, Pezzotta, Groat and Krivovichev2016; Zaitsev et al., Reference Zaitsev, Zhitova, Spratt, Zolotarev and Krivovichev2017; Krivovichev et al., Reference Krivovichev, Hawthorne and Williams2017; Plášil et al., Reference Plášil, Petříček and Majzlan2017; Plášil, Reference Plášil2018; Majzlan et al., Reference Majzlan, Dachs, Benisek, Plášil and Sejkora2018; Huskić et al., Reference Huskić, Novendra, Lim, Topić, Titi, Pekov, Krivovichev, Navrotsky, Kitagawa and Friščić2019; Majzlan, Reference Majzlan2020). Thus, metavariscite and phosphosiderite can be considered as metastable phases, in contrast to their stable counterparts, variscite and strengite, respectively. This hypothesis is in agreement with the differences in the physical densities (see above): according to the Ostwald–Volmer rule, the polymorphs with the lowest density in Ostwald sequences are formed first (Bach et al., Reference Bach, Fischer and Jansen2013). The difference in complexity between variscite and metavariscite is also in agreement with the identification of the two phases as low- and high-temperature polymorphs, respectively (Sergeeva, Reference Sergeeva2016), as the empirical rule states that the high-temperature phase is usually less complex than the low-temperature phase (Krivovichev, Reference Krivovichev2013a).
Table 10. Information-based complexity parameters for variscite and metavariscite (v in atoms per cell, IG in bit per atom and IG,total in bits per cell).

Sp. gr. – space group.
Compounds of the variscite and metavariscite groups as potential electrode materials
Both FePO4⋅2H2O polymorphs, orthorhombic strengite and monoclinic phosphosiderite, have been the subject of several studies focused on evaluation of their electrochemical properties (Hong et al., Reference Hong, Ryu, Park, Kim, Lee and Chang2002; Masquelier et al., Reference Masquelier, Reale, Wurm, Morcrette, Dupont and Larcher2002; Song et al., Reference Song, Yang, Zavalij and Whittingham2002a; Delacourt et al., Reference Delacourt, Poizot, Bonnin and Masquelier2009). In particular, these compounds, composed of abundant and low-cost elements, look attractive as they provide 3D frameworks suitable for small-sized ion intercalation. Moreover, the presence of transition metal cations in the structure of strengite and phosphosiderite suggests that it would operate on the Fe3+/Fe2+ redox couple and might be used as an electrode material in a rechargeable Li ion battery with a promising theoretical specific capacity value of 143.4 mAh/g. Indeed, the crystalline and amorphous FePO4⋅2H2O phases were promptly patented by Masquelier et al. (Reference Masquelier, Morcrette, Reale and Wurm2003), and after a while Delacourt et al. (Reference Delacourt, Poizot, Bonnin and Masquelier2009) revealed a decent electrochemical behaviour of both variscite- and metavariscite-type crystalline phases with a good capacity retention delivering reversible capacities of ~140 and 120 mAh/g, respectively, at an average potential of ~3 V vs. Li+/Li. Delacourt et al. (Reference Delacourt, Poizot, Bonnin and Masquelier2009) also observed a beneficial role of constitutional water molecules promoting fast ionic conduction.
The better performance of variscite-type strengite agrees well with our findings about the structural stability of the polymorphs: the denser and more complex variscite-type framework provides appropriate stability essential for extensive electrochemical (de)intercalation of Li into its crystal structure.
In view of the foregoing, VPO4⋅2H2O (Schindler et al., Reference Schindler, Joswig and Baur1995) would be also of interest to investigate the formation of a theoretical LixVPO4⋅2H2O composition (theoretical specific capacity of 147.3 mAh/g) by electrochemical lithiation. It is important to note, however, that the reduction mechanism of V3+ to V2+ generally occurs through lithium insertion at a quite low potential of <2 V vs. Li+/Li (Masquelier and Croguennec, Reference Masquelier and Croguennec2013). Hence, a positive effect of higher theoretical capacity of the vanadium-based phase can be diminished by low operating voltages in practical tests.
In this regard, scorodite, FeAsO4⋅2H2O, is worthy of the attention as well as the inductive effect (Padhi et al., Reference Padhi, Nanjundaswamy and Goodenough1997) of As5+ cations on the Fe3+/Fe2+ couple is similar to that of P5+ due to close electronegativity values of As and P (Allen, Reference Allen1989). Thus, the Fe3+/Fe2+ redox process would probably occur at a similar potential value of ~3 V vs. Li+/Li. This phase exhibits a moderate theoretical capacity (116 mAh/g), which also promotes further electrochemical tests.
The summarising list of selected candidates for the role of electrode materials in Li ion batteries is given in Table 11.
Table 11. List of selected phases of the variscite and metavariscite groups as potential electrode materials for Li ion batteries.

1) Experimental values from Delacourt et al. (Reference Delacourt, Poizot, Bonnin and Masquelier2009)
2) Expected values, see details in text.
n.d. – not determined.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1180/mgm.2020.57
Acknowledgements
Part of this work was done while the first author was financially supported by the Austrian Science Foundation (FWF). S.V.K was supported in this work by the Russian Science Foundation (grant 19-17-00038). V.K. would like to thank Olivier Mentré, Marie Colmont and Almaz Aliev from the UCCS laboratory (University of Lille 1) for providing laboratory equipment for the synthesis of MnSeO4⋅2H2O (method 2). The article was improved by constructive reviews by Juraj Majzlan and two anonymous reviewers and editorial assistance by Principal Editor Stuart Mills.


















