



Introduction
Malaria is one of the most serious infectious diseases of humans with 200 million clinical malaria cases and around 429 000 deaths in 2015 (World Health Organization, World Malaria Report, 2016). Children under the age of 5 are particularly susceptible to malaria, accounting for 70% of the fatalities (WHO, World Malaria Report, 2016). Although there are five species of Plasmodium parasites that infect humans, most cases of severe disease and fatalities are caused by P. falciparum. Humans with no previous experience of malaria almost invariably become ill on their first exposure to the parasite. They develop a febrile illness, which may become severe and, in a proportion of cases, life threatening. Clinical cases are associated with a spectrum of disease syndromes including respiratory distress, metabolic acidosis, hypoglycaemia, renal failure, pulmonary oedema and cerebral involvement (reviewed by Cowman et al. Reference Cowman, Healer, Marapana and Marsh2016). The blood stage of Plasmodium is entirely responsible for malaria-associated pathology. Although the precise mechanisms of pathogenesis of malaria are not completely understood, it is thought to arise from the combined effects of host and parasite factors including the sequestration of infected erythrocytes in the microvasculature, as well as local and systemic host inflammatory responses to infection (Schofield and Grau, Reference Schofield and Grau2005).
The acquisition of immunity to P. falciparum appears to develop at slower rates than immunity to viral or bacterial pathogens (Maple et al. Reference Maple, Jones, Wall, Vyseb, Edmunds, Andrews and Miller2000; Hammarlund et al. Reference Hammarlund, Lewis, Hansen, Strelow, Nelson, Sexton, Hanifin and Slifka2003; Amanna et al. Reference Amanna, Carlson and Slifka2007). While children become immune to the most severe aspects of disease after a few symptomatic infections (Gupta et al. Reference Gupta, Snow, Donnelly, Marsh and Newbold1999), individuals living in malaria endemic areas develop immunity to clinical symptoms only after years of repeated infections (Marsh and Kinyanjui, Reference Marsh and Kinyanjui2006). With cumulative parasite exposure over time, immunity to clinical disease is acquired, conferring the ability to maintain parasitaemia below a clinical threshold and preventing the onset of disease symptoms (Schofield and Mueller, Reference Schofield and Mueller2006).
Landmark studies conducted in the 1960s showed that passive transfer of purified immunoglobulin G (IgG) from malaria immune individuals reduced parasitaemia in non-immune children by 99% within 4 days (Cohen et al. Reference Cohen, McGregor and Carrington1961), demonstrating a key role for antibodies in protection from symptomatic malaria. Subsequent work demonstrated that these purified IgG had an inhibitory effect on parasite growth in vitro (Cohen et al. Reference Cohen, Butcher and Crandall1969). These seminal observations provided a strong rationale supporting the idea that an effective anti-malaria vaccine targeting blood-stage parasites is achievable. Thus the identification of antigenic targets of naturally acquired immunity is an important step towards the identification of novel vaccine candidates prior to testing their efficacy in clinical trials. The contribution of cell-mediated immunity to protection from malaria in the context of vaccine development has recently been reviewed elsewhere (Stanisic and Good, Reference Stanisic and Good2016) so this review will focus on approaches to identify antigenic targets of naturally-acquired antibodies.
Methodological approaches to investigate antibody responses
Serological investigation of responses to malaria antigens has historically relied on the enzyme-linked immunosorbent assay (ELISA), the most commonly used quantification technique for measuring antibody. This assay generally measures antibody binding to individual recombinant proteins. More recently, new techniques using bead array have been developed that allow antibodies to multiple antigens to be measured at the same time. In this assay, differentially fluorescent microbeads are conjugated with a selection of antigens to capture antibodies via flow cytometric methods allowing multiplexing capability (Elshal and McCoy, Reference Elshal and McCoy2006). Although amenable to high throughput formats, traditional serological approaches do not provide estimates of real antibody content in circulation, which precludes estimations on the efficacy of the antibody response to infection. To overcome these issues, cytometric bead arrays (CBA) for assessment of antigen-specific antibody concentration have been recently developed, to evaluate associations between immune response and protection from symptomatic malaria (Chiu et al. Reference Chiu, White, Healer, Thompson, Siba, Mueller, Cowman and Hansen2016). The development of protein microarray technology provides an opportunity to measure antibody responses against large numbers of proteins representing a sizeable proportion of the P. falciparum genome (Davies et al. Reference Davies, Liang, Hernandez, Randall, Hirst, Mu, Romero, Nguyen, Kalantari-Dehaghi, Crotty, Baldi, Villarreal and Felgner2005). Table 1 provides an overview of the relative advantages of each of these platforms for serological assessment. However measurement of antibody reactivity alone provides no evidence of functional immunity, and additional investigations must be performed to differentiate whether those antibodies are markers of parasite exposure or are anti-parasite effectors (Dent et al. Reference Dent, Bergmann-Leitner, Wilson, Tisch, Kimmel, Vulule, Sumba, Beeson, Angov, Moormann and Kazura2008).
Table 1. Relative pros and cons of different methodologies for anti-malarial serological analyses
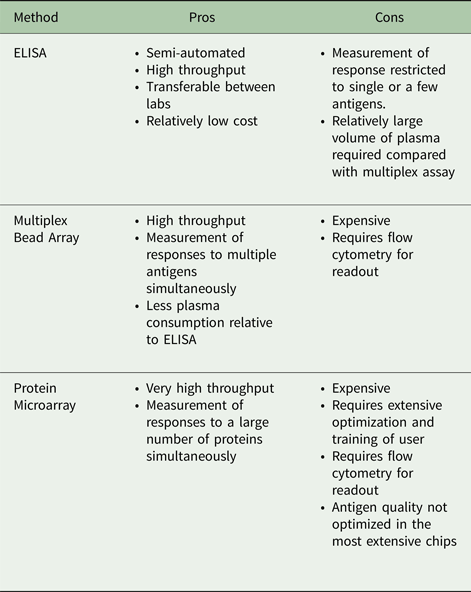
ELISA, enzyme-linked immunosorbent assay.
Two main approaches have been pursued to investigate functional antibody responses to blood-stage malaria. In vitro assays measuring direct antibody-mediated regulation of parasite growth, such as parasite growth inhibitory assays (GIA) (Dent et al. Reference Dent, Bergmann-Leitner, Wilson, Tisch, Kimmel, Vulule, Sumba, Beeson, Angov, Moormann and Kazura2008; Miura et al. Reference Miura, Zhou, Moretz, Diouf, Thera, Dolo, Doumbo, Malkin, Diemert, Miller, Mullen and Long2008; Crompton et al. Reference Crompton, Miura, Traore, Kayentao, Ongoiba, Weiss, Doumbo, Doumtabe, Kone, Huang, Doumbo, Miller, Long and Pierce2010b) as well as indirect antibody-mediated mechanisms such as opsonization (Hill et al. Reference Hill, Eriksson, Li Wai Suen, Chiu, Ryg-Cornejo, Robinson, Siba, Mueller, Hansen and Schofield2013), antibody-dependent respiratory burst (Llewellyn et al. Reference Llewellyn, Miura, Fay, Williams, Murungi, Shi, Hodgson, Douglas, Osier, Fairhurst, Diakite, Pleass, Long and Draper2015) and complement deposition/complement-mediated lysis assays (Boyle et al. Reference Boyle, Reiling, Feng, Langer, Osier, Aspeling-Jones, Cheng, Stubbs, Tetteh, Conway, McCarthy, Muller, Marsh, Anders and Beeson2015) are useful tools to quantify the functional activity of antibodies to merozoite antigens by measuring parasite growth in the presence of immune compared with non-immune sera/plasma samples (Persson et al. Reference Persson, Lee, Marsh and Beeson2006; Murungi et al. Reference Murungi, Sonden, Llewellyn, Rono, Guleid, Williams, Ogada, Thairu, Farnert, Marsh, Draper and Osier2016). These in vitro functional assays have been complemented by field studies, which provide a solid platform to reveal associations between antibody responses and protection against symptomatic malaria. In particular, longitudinal cohort studies are informative to explore relationships between various immune responses (such as antibody levels and frequency of memory B cells) and disease/infection outcomes.
Acquisition of humoral immunity
In regions where malaria is stably endemic, the prevalence of severe malaria and death rises and declines rapidly within the first 5 years of life (Cowman et al. Reference Cowman, Healer, Marapana and Marsh2016). From then onwards, infected individuals may continue to experience bouts of non-severe malaria for 10–20 years, until they finally reach clinical immunity in early adulthood, where parasitaemia is often undetectable by microscopic examination but may be evident by polymerase chian reaction (PCR) (Okell et al. Reference Okell, Ghani, Lyons and Drakeley2009). This observation is often interpreted that immunity to malaria develops in distinct steps, such that immunity to severe disease (anti-disease immunity) precedes and is qualitatively different from that against malaria infection (anti-parasite immunity). However, an interesting feature of malaria infections is that in the vast majority of cases, the normal state is asymptomatic parasitaemia (Cowman et al. Reference Cowman, Healer, Marapana and Marsh2016).
Both host and parasite-based models have been proposed to explain the slow acquisition of immunity to malaria, and both parasite and host factors are at play in determining host disease outcomes. On one hand, acquisition of protective immunity can be explained in terms of additive or cumulative immunity against infecting parasite isolates that, over time, as the repertoire of strains survived increases, allows the breadth of response grow to a point at which clinical immunity is achieved. Whether this response targets poorly immunogenic conserved epitopes or strain-specific antigens is not yet clear, but this is an area of much focus and is particularly relevant to vaccine development. It is known that P. falciparum undergoes clonal antigenic variation and expresses highly polymorphic antigens in the blood-stage merozoite form and on the surface of infected erythrocytes (Cowman and Crabb, Reference Cowman and Crabb2006; Scherf et al. Reference Scherf, Lopez-Rubio and Riviere2008). Because of the multiple invasion pathways (Cowman and Crabb, Reference Cowman and Crabb2006) and functional redundancy of invasion ligands of the merozoites (Tham et al. Reference Tham, Healer and Cowman2012) as well as the diverse PfEMP1 on the infected erythrocytes that contribute to the diversity of parasite within the host (Roberts et al. Reference Roberts, Craig, Berendt, Pinches, Nash, Marsh and Newbold1992), repeated exposure to diverse strains are necessary for the acquisition of a repertoire of functional antibodies to the surface antigens on both the merozoite and infected erythrocyte (PfEMP1) to protect against heterologous parasite challenge.
In addition to parasite survival mechanisms, it has also been proposed that the development of naturally acquired immunity is dependent on age (Baird, Reference Baird1995). Cross-sectional studies on malaria-naive immigrants to endemic areas revealed that older people have better clinical outcomes than children (Baird et al. Reference Baird, Jones, Danudirgo, Annis, Bangs, Basri, Purnomo and Masbar1991) suggesting that immunity to malaria is more efficient in older individuals. Emerging evidence in human studies and infection models suggests that defects in the induction of B cell immunological memory might be also responsible for the compromised humoral responses to infection (reviewed in(Ryg-Cornejo et al. Reference Ryg-Cornejo, Ly and Hansen2016b; Hansen et al. Reference Hansen, Obeng-Adjei, Ly, Ioannidis and Crompton2017)). Several field studies demonstrated that P. falciparum-specific antibodies are inefficiently acquired and short-lived, particularly in children (Cavanagh et al. Reference Cavanagh, Elhassan, Roper, Robinson, Giha, Holder, Hviid, Theander, Arnot and McBride1998, Reference Cavanagh, Dodoo, Hviid, Kurtzhals, Theander, Akanmori, Polley, Conway, Koram and McBride2004; Kinyanjui et al. Reference Kinyanjui, Bull, Newbold and Marsh2003, Reference Kinyanjui, Conway, Lanar and Marsh2007). Children and young adults in areas of high seasonal transmission are characterized by a delayed development of memory B cells specific for P. falciparum despite repeated exposure to the parasite (Weiss et al. Reference Weiss, Traore, Kayentao, Ongoiba, Doumbo, Doumtabe, Kone, Dia, Guindo, Traore, Huang, Miura, Mircetic, Li, Baughman, Narum, Miller, Doumbo, Pierce and Crompton2010). In contrast, adults living in areas of low transmission that experience fewer clinical episodes appear to generate memory B cells specific for malarial antigens that are stable even in the absence of frequent boosting (Wipasa et al. Reference Wipasa, Suphavilai, Okell, Cook, Corran, Thaikla, Liewsaree, Riley and Hafalla2010). These studies and a growing body of evidence suggest that inflammatory responses contributing to clinical episodes might have a detrimental effect on the development of parasite-specific memory B cells able to produce high-affinity antibody, required for effective control of blood-stage parasite replication. In support of that concept, recent studies revealed acute malaria infections in children are characterized by T helper (TH)1-like T follicular helper (TFH) cells with impaired B cell helper function (Obeng-Adjei et al. Reference Obeng-Adjei, Portugal, Tran, Yazew, Skinner, Li, Jain, Felgner, Doumbo, Kayentao, Ongoiba, Traore and Crompton2015). Moreover, similar findings in a mouse model revealed that inflammatory cytokines responsible for the development of severe malaria infection also inhibit the differentiation of TFH cells required for germinal centre responses and the induction of long-lasting humoral immunity (Ryg-Cornejo et al. Reference Ryg-Cornejo, Ioannidis, Ly, Chiu, Tellier, Hill, Preston, Pellegrini, Yu, Nutt, Kallies and Hansen2016a).
The role of antibodies in naturally acquired immunity
Despite the key role that antibodies play in protection, the key antigenic targets of naturally acquired immunity or the effector mechanisms responsible for these processes have not been fully elucidated. Field studies provide a platform to reveal associations between antibody responses and protection against clinical malaria. In particular, longitudinal cohort studies are informative in exploring relationships between various immune responses (such as antibody levels and frequency of memory B cells) and disease/infection outcomes. Various treatment-reinfection studies conducted in Papua New Guinea (Cole-Tobian et al. Reference Cole-Tobian, Michon, Biasor, Richards, Beeson, Mueller and King2009; Stanisic et al. Reference Stanisic, Richards, McCallum, Michon, King, Schoepflin, Gilson, Murphy, Anders, Mueller and Beeson2009; Reiling et al. Reference Reiling, Richards, Fowkes, Barry, Triglia, Chokejindachai, Michon, Tavul, Siba, Cowman, Mueller and Beeson2010; Richards et al. Reference Richards, Stanisic, Fowkes, Tavul, Dabod, Thompson, Kumar, Chitnis, Narum, Michon, Siba, Cowman, Mueller and Beeson2010; Wilson et al. Reference Wilson, Fowkes, Gilson, Elliott, Tavul, Michon, Dabod, Siba, Mueller, Crabb and Beeson2011; Reiling et al. Reference Reiling, Richards, Fowkes, Wilson, Chokejindachai, Barry, Tham, Stubbs, Langer, Donelson, Michon, Tavul, Crabb, Siba, Cowman, Mueller and Beeson2012; Hill et al. Reference Hill, Eriksson, Li Wai Suen, Chiu, Ryg-Cornejo, Robinson, Siba, Mueller, Hansen and Schofield2013; Chiu et al. Reference Chiu, Healer, Thompson, Chen, Kaul, Savergave, Raghuwanshi, Li Wai Suen, Siba, Schofield, Mueller, Cowman and Hansen2014; Reference Chiu, Hodder, Lin, Hill, Li Wai Suen, Schofield, Siba, Mueller, Cowman and Hansen2015; Reference Chiu, White, Healer, Thompson, Siba, Mueller, Cowman and Hansen2016) and Africa (Ogutu et al. Reference Ogutu, Apollo, McKinney, Okoth, Siangla, Dubovsky, Tucker, Waitumbi, Diggs, Wittes, Malkin, Leach, Soisson, Milman, Otieno, Holland, Polhemus, Remich, Ockenhouse, Cohen, Ballou, Martin, Angov, Stewart, Lyon, Heppner and Withers2009; Gomez-Escobar et al. Reference Gomez-Escobar, Amambua-Ngwa, Walther, Okebe, Ebonyi and Conway2010; McCarra et al. Reference McCarra, Ayodo, Sumba, Kazura, Moormann, Narum and John2011; Dent et al. Reference Dent, Moormann, Yohn, Kimmel, Sumba, Vulule, Long, Narum, Crabb, Kazura and Tisch2012; Mugyenyi et al. Reference Mugyenyi, Elliott, McCallum, Anders, Marsh and Beeson2013; Tran et al. Reference Tran, Ongoiba, Coursen, Crosnier, Diouf, Huang, Li, Doumbo, Doumtabe, Kone, Bathily, Dia, Niangaly, Dara, Sangala, Miller, Doumbo, Kayentao, Long, Miura, Wright, Traore and Crompton2014; Osier et al. Reference Osier, Mackinnon, Crosnier, Fegan, Kamuyu, Wanaguru, Ogada, McDade, Rayner, Wright and Marsh2014b; Boyle et al. Reference Boyle, Reiling, Feng, Langer, Osier, Aspeling-Jones, Cheng, Stubbs, Tetteh, Conway, McCarthy, Muller, Marsh, Anders and Beeson2015) demonstrated that increased antibody levels were associated with protective clinical outcomes, which highlights the importance of antigen-specific antibodies in the development of naturally acquired immunity. The immune effector mechanisms mediated by anti-merozoite antibodies are described below and summarized in Fig. 1.
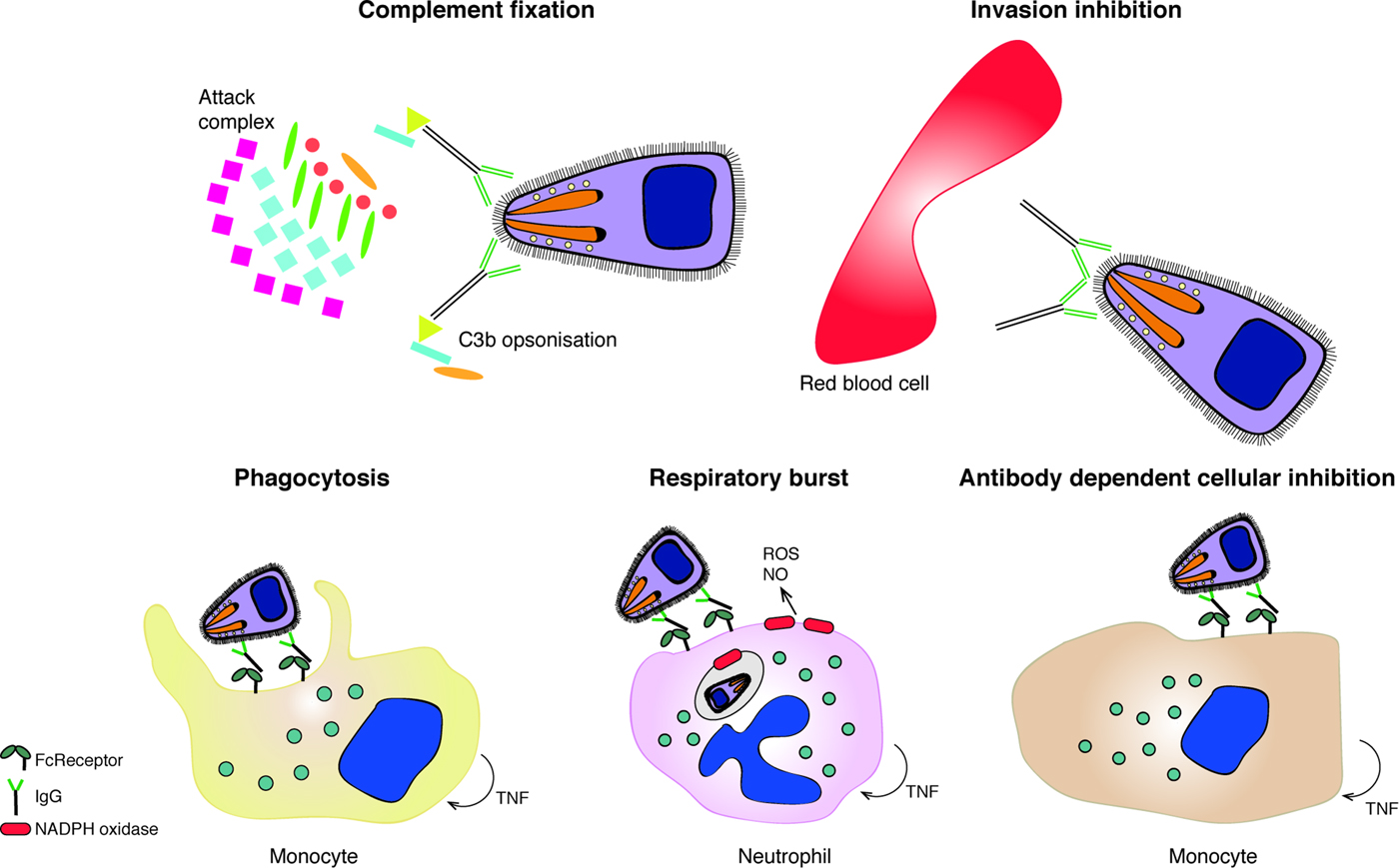
Fig. 1. The functional mechanism of anti-merozoite antibodies. Antibodies to merozoite surface proteins can mediate several effector mechanisms, including complement fixation due to cytophillic antibodies that result in merozoite lysis of C3b opsonization; inhibition of merozoite invasion into the RBC; phagocytosis of IgG-opsonized merozoites; production of reactive oxygen species (ROS) or Nitric oxide (NO) in response to opsonized parasites and antibody dependent cellular inhibition (ADCI) triggered by IgG-opsonized merozoites.
Antibody-mediated effector mechanisms
Inhibition of parasite replication and development
Early microscopic observations revealed that immune sera could agglutinate free merozoites upon schizogony (Cohen and Butcher, Reference Cohen and Butcher1970). It has been shown that parasite-specific antibodies can access the interior of infected erythrocytes at early stages of schizont rupture. Antibodies recognizing the major serine repeat antigen of P. falciparum, SERA 5, mediate agglutination of merozoites, thus preventing the dispersal of daughter merozoites (Pang et al. Reference Pang, Mitamura and Horii1999). Agglutination has also been shown to occur through the binding of antibodies to surface proteins on the free merozoites such as MSP-119 (Gilson et al. Reference Gilson, O'Donnell, Nebl, Sanders, Wickham, McElwain, de Koning-Ward and Crabb2008) MSP-142 (Bergmann-Leitner et al. Reference Bergmann-Leitner, Duncan and Angov2009) proteolytic cleavage products of the major merozoite surface protein, MSP1, MSP-2 (Ramasamy et al. Reference Ramasamy, Yasawardena, Kanagaratnam, Buratti, Baralle and Ramasamy1999), as well as to PfEMP1 on the infected erythrocyte surface (Biggs et al. Reference Biggs, Goozé, Wycherley, Wollish, Southwell, Leech and Brown1991; Joergensen et al. Reference Joergensen, Salanti, Dobrilovic, Barfod, Hassenkam, Theander, Hviid and Arnot2010). What role parasite agglutination per se plays in naturally acquired protective immunity, however, is unclear. It was apparent in the early studies that parasite agglutination was not necessarily associated with reductions in parasite growth, since agglutination of infected RBCs in vitro occurs at antibody concentrations much lower than that responsible for parasite growth inhibition; moreover when agglutinating serum was absorbed against mature schizont-infected RBCs, agglutination was reduced to below 10% of the level seen prior to absorption, but was still capable of inhibiting parasite growth in vitro (Cohen and Butcher, Reference Cohen and Butcher1971).
Antibodies that recognize merozoite surface proteins (MSPs) involved in different steps of the erythrocyte invasion process have been found to block parasite growth. The inhibitory effect of these antibodies is usually evaluated in vitro by comparing the end-point parasitaemia after co-incubation of uninfected erythrocytes and merozoites with and without antibody of interest. GIA can measure the effect of antibodies on the invasion of uninfected erythrocytes, intraerythrocytic arrest, schizont rupture or parasite growth over multiple cycles. It is well established that antibodies specific to the MSP-1 complex (Kauth et al. Reference Kauth, Woehlbier, Kern, Mekonnen, Lutz, Mucke, Langowski and Bujard2006; Lin et al. Reference Lin, Uboldi, Marapana, Czabotar, Epp, Bujard, Taylor, Perugini, Hodder and Cowman2014, Reference Lin, Uboldi, Epp, Bujard, Tsuboi, Czabotar and Cowman2016), members of the EBL and PfRh protein families (reviewed in (Tham et al. Reference Tham, Healer and Cowman2012)), (Baum et al. Reference Baum, Chen, Healer, Lopaticki, Boyle, Triglia, Ehlgen, Ralph, Beeson and Cowman2009; Chen et al. Reference Chen, Lopaticki, Riglar, Dekiwadia, Uboldi, Tham, O'Neill, Richard, Baum, Ralph and Cowman2011; Reddy et al. Reference Reddy, Amlabu, Pandey, Mitra, Chauhan and Gaur2015; Volz et al. Reference Volz, Yap, Sisquella, Thompson, Lim, Whitehead, Chen, Lampe, Tham, Wilson, Nebl, Marapana, Triglia, Wong, Rogers and Cowman2016) and the components of tight junction between the parasite and the RBC (Hodder et al. Reference Hodder, Crewther and Anders2001; Srinivasan et al. Reference Srinivasan, Beatty, Diouf, Herrera, Ambroggio, Moch, Tyler, Narum, Pierce, Boothroyd, Haynes and Miller2011) can mediate effective parasite growth inhibition.
Antibodies may impair intra-erythrocytic growth and/or parasite egress (Jensen et al. Reference Jensen, Boland and Akood1982). Several mechanisms by which antibodies could bind to intracellular antigens have been suggested. Firstly, some MSPs such as MSP-119, MSP-2 and MSP-4 are not shed after invasion. Consequently, antibodies bound to these MSPs could be potentially carried into the erythrocyte during the invasion (Blackman et al. Reference Blackman, Scott-Finnigan, Shai and Holder1994; Dluzewski et al. Reference Dluzewski, Ling, Hopkins, Grainger, Margos, Mitchell, Holder and Bannister2008; Boyle et al. Reference Boyle, Langer, Chan, Hodder, Coppel, Anders and Beeson2014). Secondly, antibodies can access the parasitophorous vacuole membrane via a putative parasitophorous duct, which was found by the observation of extracellular macromolecules and IgG in the infected erythrocyte (Pouvelle and Gysin, Reference Pouvelle and Gysin1997). Finally, the permeability of the erythrocyte membrane is increased at late schizogony (Green et al. Reference Green, Morhardt, Brackett and Jacobs1981; Lyon et al. Reference Lyon, Thomas, Hall and Chulay1989), which could potentially allow access of antibodies to the erythrocyte cytosol. Antibodies that bind to intracellular antigens are thought to act on different intraerythrocytic developmental stages such as formation of the food vacuole (Dluzewski et al. Reference Dluzewski, Ling, Hopkins, Grainger, Margos, Mitchell, Holder and Bannister2008), developmental arrest at early trophozoite stage (Bergmann-Leitner et al. Reference Bergmann-Leitner, Duncan and Angov2009), schizont development (Ahlborg et al. Reference Ahlborg, Iqbal, Bjork, Stahl, Perlmann and Berzins1996) and merozoite dispersal upon egress (Pang et al. Reference Pang, Mitamura and Horii1999; Bergmann-Leitner et al. Reference Bergmann-Leitner, Duncan and Angov2009; Raj et al. Reference Raj, Nixon, Nixon, Dvorin, DiPetrillo, Pond-Tor, Wu, Jolly, Pischel, Lu, Michelow, Cheng, Conteh, McDonald, Absalon, Holte, Friedman, Fried, Duffy and Kurtis2014). How relevant these antigen–antibody interactions are in naturally acquired protection from malarial disease is unknown but deserve further investigation.
Antibody-mediated opsonization for cell and complement mediated-killing
It has been shown that complement is activated during human malaria infections (Silver et al. Reference Silver, Higgins, McDonald and Kain2010). Antibodies from immune sera specific for the abundant MSP-1 and MSP-2 antigens have been shown to mediate merozoite lysis in the presence of complement and this was associated with immune status (Boyle et al. Reference Boyle, Reiling, Feng, Langer, Osier, Aspeling-Jones, Cheng, Stubbs, Tetteh, Conway, McCarthy, Muller, Marsh, Anders and Beeson2015). Accordingly, merozoites have evolved strategies to evade complement activation. Pf92, a member of the six-cysteine family of malaria proteins (Gilson et al. Reference Gilson, Nebl, Vukcevic, Moritz, Sargeant, Speed, Schofield and Crabb2006) binds Factor H, a complement-regulatory protein present in human serum, protecting merozoites from complement-mediated lysis (Simon et al. Reference Simon, Lasonder, Scheuermayer, Kuehn, Tews, Fischer, Zipfel, Skerka and Pradel2013; Kennedy et al. Reference Kennedy, Schmidt, Thompson, Weiss, Taechalertpaisarn, Gilson, Barlow, Crabb, Cowman and Tham2016). In addition, blocking of complement has been shown to inhibit the phagocytosis of infected erythrocytes in vitro (Turrini et al. Reference Turrini, Ginsburg, Bussolino, Pescarmona, Serra and Arese1992).
Parasite-specific IgM and IgG1 can initiate the classical pathway of the complement cascade and the consequent deposition of C3b acts as a phagocyte receptor on parasites (Newman and Mikus, Reference Newman and Mikus1985). Both complement- and antibody-mediated opsonization of malaria merozoites have been found to trigger phagocytosis by effector cells including, neutrophils (Witko-Sarsat et al. Reference Witko-Sarsat, Rieu, Descamps-Latscha, Lesavre and Halbwachs-Mecarelli2000) and monocytes (Hill et al. Reference Hill, Eriksson, Li Wai Suen, Chiu, Ryg-Cornejo, Robinson, Siba, Mueller, Hansen and Schofield2013; Osier et al. Reference Osier, Feng, Boyle, Langer, Zhou, Richards, McCallum, Reiling, Jaworowski, Anders, Marsh and Beeson2014a). IgG1 and IgG3 are the cytophilic isotypes that can bind to the Fc receptors on phagocytes to signal the bound merozoites for phagocytosis. Monocyte-mediated phagocytosis of merozoites via opsonizing antibody (Hill et al. Reference Hill, Eriksson, Carmagnac, Wilson, Cowman, Hansen and Schofield2012) has been shown to correlate with clinical immunity to P. falciparum amongst children in PNG (Hill et al. Reference Hill, Eriksson, Li Wai Suen, Chiu, Ryg-Cornejo, Robinson, Siba, Mueller, Hansen and Schofield2013) and Africa (Osier et al. Reference Osier, Feng, Boyle, Langer, Zhou, Richards, McCallum, Reiling, Jaworowski, Anders, Marsh and Beeson2014a). In addition, opsonized merozoites can be phagocytosed by neutrophils in vitro (Celada et al. Reference Celada, Cruchaud and Perrin1982; Khusmith et al. Reference Khusmith, Druilhe and Gentilini1982) and in vivo (Sun and Chakrabarti, Reference Sun and Chakrabarti1985). It is generally accepted that the release of reactive oxygen species (ROS) occurs downstream of phagocytosis (Joos et al. Reference Joos, Marrama, Polson, Corre, Diatta, Diouf, Trape, Tall, Longacre and Perraut2010).
Respiratory burst refers to the process by which soluble mediators, such as ROS, are released by activated polymorphonuclear cells (PMN) such as neutrophils. The antibody-dependent respiratory burst assay (ADRB) can be used to quantify the ability of antibodies to induce neutrophil-mediated intracellular and extracellular release of ROS in response to blood-stage parasites (Llewellyn et al. Reference Llewellyn, Miura, Fay, Williams, Murungi, Shi, Hodgson, Douglas, Osier, Fairhurst, Diakite, Pleass, Long and Draper2015). The extracellular release of ROS has been shown to be associated with protection from clinical malaria and appears to require opsonizing antibodies to bind to FcγRI (Joos et al. Reference Joos, Marrama, Polson, Corre, Diatta, Diouf, Trape, Tall, Longacre and Perraut2010). On the other hand, the engagement of opsonized merozoites to FcγRII has been found to induce intracellular release of ROS as the main mode of anti-parasitic action for neutrophils (Kapelski et al. Reference Kapelski, Klockenbring, Fischer, Barth and Fendel2014).
It has been also shown that the binding of cytophilic antibodies to the Fc receptors on monocytes leads to the release of soluble mediators including TNF, which inhibit in vitro intraerythrocytic parasite growth (Bouharoun-Tayoun et al. Reference Bouharoun-Tayoun, Oeuvray, Lunel and Druilhe1995). Recently, antibody-dependent cytotoxic inhibition (ADCI) was found to increase with age and was associated with protection from malarial clinical episodes amongst Ghanaian children (Tiendrebeogo et al. Reference Tiendrebeogo, Adu, Singh, Dziegiel, Nebie, Sirima, Christiansen, Dodoo and Theisen2015). These effector mechanisms are not restricted to merozoite neutralization and may play a critical role in the clearance of parasitized erythrocytes via anti-PfEMP1 responses.
Merozoite targets of naturally acquired immunity
The erythrocyte is the only host cell type permissive to Plasmodium infection in the bloodstream phase of development and P. falciparum parasites must relocate from one spent host cell to a new one every 48 h. While inside the erythrocyte, the parasite is relatively sheltered from immune attack. The extracellular invasive merozoite has evolved various strategies to avoid immune destruction (Weiss et al. Reference Weiss, Crabb and Gilson2016). Merozoites are extracellular for only a few minutes at most, exposed antigens are highly polymorphic and essential ligands necessary for erythrocyte invasion are sequestered in organelles until specific signals authorize their release to the host cell–parasite interface and some of those exposed antigens subvert complement-mediated host defence mechanisms (Kennedy et al. Reference Kennedy, Schmidt, Thompson, Weiss, Taechalertpaisarn, Gilson, Barlow, Crabb, Cowman and Tham2016). It is worth reflecting on the findings of the early passive transfer studies in humans if we wish to evaluate the relative importance of infected RBC surface antigens (e.g. PfEMP1, stevor, rifin) and merozoite surface antigens in conferring protective immunity (McGregor, Reference McGregor1964). In these in vivo studies, the critical time for parasite survival was around schizogony. It was noted that after schizogony, parasitaemia was markedly reduced and that if the successful invasion had occurred, there was no growth inhibitory effects of the serum until the next round of schizont rupture. This finding implicates antibodies targeting the free merozoite as effective mediators of protective immunity. On the other hand, however, the evolution of the massive genetic variation in PfEMP1 implicates it as a key immune target. Identification of the antigen targets of protective immunity is a route to the development of an antimalarial vaccine and because of the great degree of diversity in PfEMP1, this is not seen as a feasible prospect for vaccine development, except in the case of pregnancy-related malaria, where the allelic variants that bind to placenta are relatively conserved (Fried et al. Reference Fried, Nosten, Brockman, Brabin and Duffy1998; Salanti et al. Reference Salanti, Dahlback, Turner, Nielsen, Barfod, Magistrado, Jensen, Lavstsen, Ofori, Marsh, Hviid and Theander2004).
We have made enormous strides, especially since the publication of the P. falciparum genome in 2002 (Gardner et al. Reference Gardner, Hall, Fung, White, Berriman, Hyman, Carlton, Pain, Nelson, Bowman, Paulsen, James, Eisen, Rutherford, Salzberg, Craig, Kyes, Chan, Nene, Shallom, Suh, Peterson, Angiuoli, Pertea, Allen, Selengut, Haft, Mather, Vaidya, Martin, Fairlamb, Fraunholz, Roos, Ralph, McFadden, Cummings, Subramanian, Mungall, Venter, Carucci, Hoffman, Newbold, Davis, Fraser and Barrell2002) towards identifying the proteins present on the merozoite (reviewed by (Cowman and Crabb, Reference Cowman and Crabb2006)); what many of their respective roles are in the process of erythrocyte invasion, and even revealing the molecular structure of some of the key proteins e.g. MSP-1 C-terminal region (Chitarra et al. Reference Chitarra, Holm, Bentley, Petres and Longacre1999), AMA1 (Bai et al. Reference Bai, Becker, Gupta, Strike, Murphy, Anders and Batchelor2005; Pizarro et al. Reference Pizarro, Vulliez-Le Normand, Chesne-Seck, Collins, Withers-Martinez, Hackett, Blackman, Faber, Remarque, Kocken, Thomas and Bentley2005), EBA-175 (Tolia et al. Reference Tolia, Enemark, Sim and Joshua-Tor2005), EBA-140 (Lin et al. Reference Lin, Malpede, Batchelor and Tolia2012) Rh5 (Chen et al. Reference Chen, Xu, Healer, Thompson, Smith, Lawrence and Cowman2014; Wright et al. Reference Wright, Hjerrild, Bartlett, Douglas, Jin, Brown, Illingworth, Ashfield, Clemmensen, de Jongh, Draper and Higgins2014), CyRPA, (Chen et al. Reference Chen, Xu, Wong, Thompson, Healer, Goddard-Borger, Lawrence and Cowman2017; Favuzza et al. Reference Favuzza, Guffart, Tamborrini, Scherer, Dreyer, Rufer, Erny, Hoernschemeyer, Thoma, Schmid, Gsell, Lamelas, Benz, Joseph, Matile, Pluschke and Rudolph2017), but definitive identification of antigens conferring protective immunity has been elusive.
Immunoepidemiological studies examining associations between antibody responses and protection from symptomatic malaria have been numerous but inconsistent in their findings, mainly due to differences in study protocols (Fowkes et al. Reference Fowkes, Richards, Simpson and Beeson2010). The most informative studies of the association between antibodies and naturally acquired immune protection are prospective cohorts, in which a relationship between antibody level and protection from clinical symptoms upon parasite re-exposure can be established. The quality, number and conformational state of the antigens used for immunological evaluations might have an impact on the study the outcome. Whilst early studies had mainly focussed on a few merozoite antigens identified as potential vaccine candidates, the availability of the genome (Gardner et al. Reference Gardner, Hall, Fung, White, Berriman, Hyman, Carlton, Pain, Nelson, Bowman, Paulsen, James, Eisen, Rutherford, Salzberg, Craig, Kyes, Chan, Nene, Shallom, Suh, Peterson, Angiuoli, Pertea, Allen, Selengut, Haft, Mather, Vaidya, Martin, Fairlamb, Fraunholz, Roos, Ralph, McFadden, Cummings, Subramanian, Mungall, Venter, Carucci, Hoffman, Newbold, Davis, Fraser and Barrell2002), transcriptome (Bozdech et al. Reference Bozdech, Llinas, Pulliam, Wong, Zhu and DeRisi2003; Le Roch et al. Reference Le Roch, Zhou, Blair, Grainger, Moch, Haynes, De La Vega, Holder, Batalov, Carucci and Winzeler2003) and proteome (Florens et al. Reference Florens, Washburn, Raine, Anthony, Grainger, Haynes, Moch, Muster, Sacci, Tabb, Witney, Wolters, Wu, Gardner, Holder, Sinden, Yates and Carucci2002) of P. falciparum provided the opportunity to take a systematic approach to immunoepidemiological profiling as a means to identify protective antigens in an unbiased way (Doolan et al. Reference Doolan, Mu, Unal, Sundaresh, Hirst, Valdez, Randall, Molina, Liang, Freilich, Oloo, Blair, Aguiar, Baldi, Davies and Felgner2008). The two main challenges of this approach are scale (the number of potential merozoite antigens is around 100, and the numbers of protective epitopes within this number is unknown) and antigen quality (production of conformationally intact proteins is difficult to achieve, even on a bespoke scale). Different in vitro systems used to generate the antigen targets for such immunoprofiling studies rely on either bacterial (Doolan et al. Reference Doolan, Mu, Unal, Sundaresh, Hirst, Valdez, Randall, Molina, Liang, Freilich, Oloo, Blair, Aguiar, Baldi, Davies and Felgner2008; Crompton et al. Reference Crompton, Kayala, Traore, Kayentao, Ongoiba, Weiss, Molina, Burk, Waisberg, Jasinskas, Tan, Doumbo, Doumtabe, Kone, Narum, Liang, Doumbo, Miller, Doolan, Baldi, Felgner and Pierce2010a; Dent et al. Reference Dent, Nakajima, Liang, Baum, Moormann, Sumba, Vulule, Babineau, Randall, Davies, Felgner and Kazura2015), wheatgerm (Tsuboi et al. Reference Tsuboi, Takeo, Arumugam, Otsuki and Torii2010; Richards et al. Reference Richards, Arumugam, Reiling, Healer, Hodder, Fowkes, Cross, Langer, Takeo, Uboldi, Thompson, Gilson, Coppel, Siba, King, Torii, Chitnis, Narum, Mueller, Crabb, Cowman, Tsuboi and Beeson2013) or mammalian cell expression (Crosnier et al. Reference Crosnier, Wanaguru, McDade, Osier, Marsh, Rayner and Wright2013; Osier et al. Reference Osier, Mackinnon, Crosnier, Fegan, Kamuyu, Wanaguru, Ogada, McDade, Rayner, Wright and Marsh2014b) or using synthetic peptide display (Agak et al. Reference Agak, Bejon, Fegan, Gicheru, Villard, Kajava, Marsh and Corradin2008) approaches have yielded different outcomes with limited overlap in the antigens that correlate with protection.
Determining immune correlates of protection with immunoprofiling studies using longitudinal cohort sera
The first proteome array produced for malaria immunoprofiling was a high throughput platform developed by Felgner and colleagues (Davies et al. Reference Davies, Liang, Hernandez, Randall, Hirst, Mu, Romero, Nguyen, Kalantari-Dehaghi, Crotty, Baldi, Villarreal and Felgner2005). This was a protein microarray consisting of 2320 P. falciparum proteins or fragments representing 1204 unique proteins. These individual proteins were printed onto glass slides that allow incubation with human serum and quantitative detection by scanning for fluorescently labelled secondary antibodies (Doolan et al. Reference Doolan, Mu, Unal, Sundaresh, Hirst, Valdez, Randall, Molina, Liang, Freilich, Oloo, Blair, Aguiar, Baldi, Davies and Felgner2008). A number of studies have taken advantage of this comprehensive array that represents proteins from all stages of the P. falciparum life cycle. A recent review covers the details of this platform and some of the studies that have used it to evaluate differences in antibody responses among malaria-exposed individuals (Davies et al. Reference Davies, Duffy, Bodmer, Felgner and Doolan2015).
A seminal prospective population study used this array to explore the kinetics of antibody responses and identify responses that are associated with protection from malaria in children and young adults in Mali (Crompton et al. Reference Crompton, Kayala, Traore, Kayentao, Ongoiba, Weiss, Molina, Burk, Waisberg, Jasinskas, Tan, Doumbo, Doumtabe, Kone, Narum, Liang, Doumbo, Miller, Doolan, Baldi, Felgner and Pierce2010a). Examination of plasma antibody titres measured before and after the malaria transmission season against over 1200 proteins was performed to identify P. falciparum-specific antibody responses that associate with protection from malaria symptoms. Hundreds of antigens were recognized by plasma from both age groups, highlighting one of the main challenges of using serological analyses to determine protective immune responses: which antibodies mediate protective responses to parasites in vivo and which reflect exposure without contributing to protection. In the Crompton study, individuals were followed up for 8 months and monitored for parasitaemia to determine exposure and for cases of symptomatic malaria. Those children experiencing one or more episodes of malaria were designated as ‘susceptible’ whereas those without clinical malaria were ‘protected’. The antibody profiles before the malaria season were compared between the two groups to determine whether specific responses were correlated with protection. Of the 491 antigens recognized, 49 were identified against which antibody responses were significantly higher in protected compared with susceptible children in the 8–10 age group. A total of 5·5% of the immunoreactive proteins were classified as merozoite-specific. Interestingly, leading malaria vaccine candidate antigens MSP-1, MSP-2 and AMA-1 were not among those proteins associated with protection (Crompton et al. Reference Crompton, Kayala, Traore, Kayentao, Ongoiba, Weiss, Molina, Burk, Waisberg, Jasinskas, Tan, Doumbo, Doumtabe, Kone, Narum, Liang, Doumbo, Miller, Doolan, Baldi, Felgner and Pierce2010a), while many of the proteins with the strongest association with protection from uncomplicated malaria were hypothetical (i.e. not yet assigned functionality). In another study examining serological associations with protection from malaria in different age groups in Western Kenya, using a similar microarray platform (a subset of 824 proteins of the larger 2320 protein array used in the former study), antibodies against MSP1 and MSP2 were correlated with protection, as well as those targeting other MSPs such as MSP7, MSP10 and MSP11 (Dent et al. Reference Dent, Nakajima, Liang, Baum, Moormann, Sumba, Vulule, Babineau, Randall, Davies, Felgner and Kazura2015). The differences in target antigens identified in these studies most likely reflect differences in study design rather than qualitative differences in parasite antigen expression between the study populations. While the collection and preparation of plasma for testing followed the same protocols, and the age groups of individuals were similar, there were some differences between the two studies. The main differences being the malaria endemicity in the two regions, with the result that protective immunity was acquired at a much younger age in the Kenyan cohort. Overall, 81% of children and 98% of adults were ‘protected’ in the Kenyan study, whereas in Mali, the percentage of ‘protected’ individuals was only 13·5% in the younger age groups, with a similar level of protection in adults (92%). Another difference was in the microarray platforms used – the Kenyan study platform contained a subset of proteins of the larger Malian study platform, however, this is probably less significant than the degree of malaria exposure between the two cohorts.
Other prospective cohort studies have used merozoite-stage restricted protein libraries curated to only include those proteins with known or predicted localization to the merozoite surface or invasion organelles in order to examine associations between antibody level and protection among a cohort of children acquiring immunity to malaria in Papua New Guinea (PNG) (Richards et al. Reference Richards, Arumugam, Reiling, Healer, Hodder, Fowkes, Cross, Langer, Takeo, Uboldi, Thompson, Gilson, Coppel, Siba, King, Torii, Chitnis, Narum, Mueller, Crabb, Cowman, Tsuboi and Beeson2013) or Kenya (Osier et al. Reference Osier, Mackinnon, Crosnier, Fegan, Kamuyu, Wanaguru, Ogada, McDade, Rayner, Wright and Marsh2014b). The PNG study used a wheatgerm cell-free expression system to produce proteins for analysis. Antigens produced in this system have been validated extensively for their conformation and function (Tsuboi et al. Reference Tsuboi, Takeo, Arumugam, Otsuki and Torii2010). The PNG study found strong associations with protection for antibodies recognizing individual invasion-related proteins Rh2b and Rh4, as well as EBA140, RhopH1 antigen family members, RON family proteins (RON2 & 6) and the MSP-DBL1; none of which are currently in the vaccine development pipeline. In addition, this study found that certain combinations of antibodies were more strongly associated with protection than responses to their single antigen components. The Kenyan study utilized a protein library expressed in a human (HEK) cell line that specifically contained full-length proteins (Crosnier et al. Reference Crosnier, Wanaguru, McDade, Osier, Marsh, Rayner and Wright2013) to increase the likelihood of correct folding and conformation of the antigens. As in the PNG study, strong associations with protective immunity were found with responses to combinations of antigens, consolidating the idea that to boost efficacy, vaccines may benefit from incorporating combinations rather than single antigen targets. Another consistent finding between serological studies is that the breadth and intensity of antibody responses increase with age and exposure and that this is important for immune protection. Interestingly, however, the antigen targets associated with protection from malaria in these two studies were quite distinct, and none of the top ten highest ranking antigens were identified in both studies. This could be due to qualitative differences between the proteins analysed in the two studies since different expression platforms were used. Different ELISA protocols or different statistical analyses could potentially explain the lack of correlation between the different studies. Alternatively, the parasite antigens that induce protection from malaria may be genetically variable in geographically distinct sites. Sharing of the antigen platforms between laboratories that test different serological cohorts is one way to unravel these apparent differences.
Concluding remarks
While the genomics era has provided the basic information for interrogating the immune response to P. falciparum, we still lack a clear understanding of what constitutes a protective immune response that confers resistance to malaria. Future studies will advance the development of in vitro assays that provide more accurate correlates of immunity and these could then be used to guide vaccine development. Whether vaccine targets should be validated targets of naturally acquired immunity, is however, another question. On the one hand, we can induce potent neutralizing immune responses using vaccines whose targets are not necessarily among those highly recognized by immune sera, for example with the merozoite invasion protein, PfRH5 (Douglas et al. Reference Douglas, Baldeviano, Lucas, Lugo-Roman, Crosnier, Bartholdson, Diouf, Miura, Lambert, Ventocilla, Leiva, Milne, Illingworth, Spencer, Hjerrild, Alanine, Turner, Moorhead, Edgel, Wu, Long, Wright, Lescano and Draper2015). It has yet to be determined whether current adjuvants, e.g. AS01 (Didierlaurent et al. Reference Didierlaurent, Laupeze, Di Pasquale, Hergli, Collignon and Garcon2017) are potent enough for induction of the high antibody titres required for sustainable reductions in parasite multiplication rate with this vaccine candidate, and whether the impressive preliminary results described in the Aotus challenge model can be achieved in human volunteers and a clinical trial with that goal is planned (SJ Draper, personal communication). On the other hand, an approach that more closely recapitulates human exposure conditions, such as a live, attenuated vaccine, where multiple antigens are processed and presented to the immune system, may be a more promising strategy. This, however, has its own challenges in manufacture, stability, delivery route and logistics for distribution; but in a controlled trial, these vaccines may provide crucial information regarding antigen targets and development of protective immune mechanisms that could be harnessed for the production of more efficacious subunit vaccines.
Financial support
JH is supported by a grant from the National Health and Medical Research Council of Australia (NHMRC 1092789), DH & CC are supported by NHMRC Independent Research Institute Infrastructure Support Scheme and Project Grants (1058665, 1107812).


