


Introduction
Many model systems for the study of host specialization and ecological speciation can be found for plant-feeding insects and spider mites (e.g. Jaenike, Reference Jaenike1990; Navajas et al., Reference Navajas, Tsagkarakou, Lagnel and Perrot-Minnot2000; Charaabi et al., Reference Charaabi, Carletto, Chavigny, Marrakchi, Makni and Vanlerberghe-Masutti2008; Ohshima, Reference Ohshima2008). The best documented models include two tephritid flies, Rhagoletis pomonella (Walsh) and Eurosta solidaginis (Fitch) (Bush, Reference Bush1969; McPheron et al., Reference McPheron, Smith and Berlocher1988; Abrahamson et al., Reference Abrahamson, Eubanks, Blair and Whiipple2001), the lepidopteran Zeiraphera diniana Guenée (Emelianov et al., Reference Emelianov, Marec and Mallet2004) and the aphid Acyrthosiphon pisum (Harris) (Ferrari et al., Reference Ferrari, Godfray, Faulconbridge, Prior and Via2006). It has been shown that due to adaptive differentiation to ecological conditions certain plant-feeding arthropods may represent different stages of the speciation process (from polymorphism through host races to full species), based on the current extent of reproductive barriers between them (Diehl & Bush, Reference Diehl and Bush1984; Drés & Mallet, Reference Drés and Mallet2002). Discrimination between different host-adapted biotypes is not only important for understanding the process of speciation and species diversity but also includes applications in agriculture. The question of whether a given animal is composed of one species or several host-specific races or species has significant practical implications for pest monitoring and biological control programs (Clarke & Walter, Reference Clarke and Walter1995). Host-plant related selection differences (i.e. host fidelity, preference and performance) and genetic differentiation should be investigated to determine where a given animal resides along a continuum between a set of panmictic populations and full species. Genetic differentiation may be measured by the direct observation of gene flow during hybridization and backcrossing or by the analysis of genetic markers (Drés & Mallet, Reference Drés and Mallet2002). In recent years, the number of studies using molecular markers to assess host-associated genetic differentiation in phytophagous arthropods has increased significantly (e.g. Price et al., Reference Price, Barker and Villet2007; Scheffer & Hawthorne, Reference Scheffer and Hawthorne2007). Some analyses have revealed that presumed polyphagous taxa are in fact complexes of cryptic species, each utilizing fewer hosts than the nominal species (e.g. Hebert et al., Reference Hebert, Ratnasingham and deWaard2003b; Blair et al., Reference Blair, Abrahamson, Jackman and Tyrrell2005; Stireman et al., Reference Stireman, Nason and Heard2005; Rach et al., Reference Rach, DeSalle, Sarkar, Schierwater and Hadrys2008). The results of these molecular tests have often been in contradiction with traditional taxonomy (Hebert et al., Reference Hebert, Penton, Burns, Janzen and Hallwachs2004).
Traditional taxonomy, based on key traits and ecological features, recognizes the cereal rust mite Abacarus hystrix (Nalepa) as a single species with a wide range of host plants. It belongs to the super-family Eriophyoidea, consisting of mites that are dependent on and intimately associated with their host plants. Most eriophyoid species are highly host-specific, being restricted to a single host species or a few hosts within a single genus (Oldfield, Reference Oldfield, Lindquist, Sabelis and Bruin1996). Thus, A. hystrix is regarded as one of a few exceptions among eriophyoids with reference to the pattern of host-plant utilization. The mite is widely distributed, and at least 60 grass species (including cereals, pasture grasses and weeds) have been recorded as its hosts (Amrine & Stasny, Reference Amrine and Stasny1994). Drifting on air currents was shown as the mode of dispersal between host plants for the cereal rust mite (Nault & Styer, Reference Nault and Styer1969). That is why the mite has long been regarded as a host generalist for whom host specialization would not be likely to evolve and as a species possessing a large adaptive potential for a wide variety of different grass species (Sabelis & Bruin, Reference Sabelis, Bruin, Lindquist, Sabelis and Bruin1996). The cereal rust mite's feeding causes direct damage of leaves, and also the mite is known as a vector of ryegrass and agropyron mosaic viruses (RgMV and AgMV) (Oldfield & Proeseler, Reference Oldfield, Proeseler, Lindquist, Sabelis and Bruin1996).
There are 52 species in the genus Abacarus and, aside from A. hystrix, all are known to be associated with only one or a few host plant species (Amrine & Stasny, Reference Amrine and Stasny1994; J. Amrine, personal communication). Thus, the cereal rust mite appears to be a suitable subject of study for host-associated genetic variation, particularly considering the results of recent studies focusing on differentiation of host populations of this mite, as outlined below.
Morphometric analysis of three host-associated populations of A. hystrix (from quackgrass Elymus repens (L.) P. Beauv., ryegrass Lolium perenne L. and smooth brome Bromus inermis Leyss.) has revealed that these three populations are morphologically distinguishable by quantitative differences in body shape, length of setae and overall body size (Skoracka et al., Reference Skoracka, Kuczyński and Magowski2002). The morphological variation observed among populations of insects or mites living on different hosts may represent phenotypic adaptation to the local microenvironment on a particular host species (e.g. Downes, Reference Downes1990; Pegler et al., Reference Pegler, Evans, Stevens and Wall2005) or indicate some genetic separation (partial in the case of host races or complete in the case of separate species) (e.g. Edwards & Labhart, Reference Edwards and Labhart2000; Drés & Mallet, Reference Drés and Mallet2002). The latter phenomenon seems to be more likely in respect to the cereal rust mite since it has been demonstrated that two populations of this mite (quackgrass- and ryegrass-associated) differ in host preferences and performance (Skoracka & Kuczyński, Reference Skoracka and Kuczyński2006; Skoracka et al., Reference Skoracka, Kuczyński and Rector2007) and have different physiological host ranges (Skoracka, Reference Skoracka2009). Thus, these two populations of A. hystrix were concluded to be highly host-adapted.
Next, laboratory experiments have revealed the existence of reproductive barriers between these two populations. It has been shown that in addition to host fidelity, which acted as a pre-zygotic barrier, there were male-based sex ratio and low adult emergence, which acted as post-zygotic barriers to gene flow (Skoracka, Reference Skoracka2008). In this way, direct tests on the role of adaptation to host plants have shown that the cereal rust mite comprises at least two host-adapted races or even more genetically differentiated biotypes. Adaptation to the host plant is expected to translate into some degree of genetic differentiation between differentially adapted populations (e.g. Jaenike, Reference Jaenike1981; Mopper & Strauss, Reference Mopper and Strauss1998). Thus, it can be supposed that a high level of genetic divergence between ryegrass- and quackgrass-specialized races of A. hystrix exists. As yet, no studies have addressed this question. The significant morphometric differences among quackgrass-, ryegrass- and brome-associated mites (as described above: Skoracka et al., Reference Skoracka, Kuczyński and Magowski2002) may indicate that brome-associated mites also represent a specialized biotype.
In this study, we test the hypothesis that the cereal rust mite represents in fact a species complex consisting of host-associated genetically differentiated taxa. We discriminate between the three host-associated strains of A. hystrix (originating from quackgrass, ryegrass and smooth brome) and investigate species boundaries within the group using common DNA barcoding markers.
Material and methods
Mite sampling
A total of 25 eriophyoid mite populations were included in the present analyses (table 1). Twenty-two samples were collected directly from plants at several sites located in western Poland during the year 2007. Three samples (AH10, AH20 and AH30) originated from females that were reared as laboratory colonies previously used in cross-breeding and colonization experiments (e.g. Skoracka, Reference Skoracka2008, Reference Skoracka2009; where the details regarding rearing mites are described). The distances between collecting sites ranged from 1.5 to 105.5 km. Specimens of the cereal rust mite were collected from three grass species: quackgrass (Elymus repens), ryegrass (Lolium perenne) and smooth brome (Bromus inermis). Representative populations of additional eriophyoid species, Aculodes mckenziei (Keif.) from smooth brome and quackgrass, Aceria tulipae (Keif.) from garlic (Allium sativum L.) and onion (Allium cepa L.), Aceria tosichella Keif. from smooth brome, and Aceria eximia Sukh. from wood small reed (Calamagrostis epigejos (L.) Roth), were chosen as distant outgroups, while Abacarus acutatus Sukh. from wood small reed served as a close outgroup.
Table 1. Characteristics of the eriophyoid mites used in this study.

Mites obtained from one specific host plant species and locality were regarded as a single population for analysis, hereafter referred to as a ‘sample’ (see sample codes in table 1). Samples of A. hystrix obtained from one specific host plant species (i.e. host-associated populations) are hereafter referred as a ‘strain’ (see strain codes in table 1). One shoot (in respect to grasses) or one bulb (in respect to onion or garlic) from each sample was randomly selected and examined under an Olympus SZX12 stereomicroscope.
DNA extraction, amplification and sequencing
Mites for DNA extraction were preserved in 96% ethyl alcohol or directly in the ATL buffer (Qiagen GmbH, Hilden, Germany). DNA was isolated from ca. 30 adult females collected from a single shoot or bulb using a non-destructive method as described by Dabert et al. (Reference Dabert, Ehrnsberger and Dabert2008). Later, mite exoskeletons were mounted on slides and deposited in the collection of the Department of Animal Taxonomy and Ecology, AMU, Poznan, Poland (specimen vouchers are described in table 1).
We used sequence data from the cytochrome oxidase subunit I (COI) gene fragment from the mitochondrial genome and D2 region of 28S rDNA as a representative of the nuclear DNA. Both markers have been applied with success in phylogenetic studies at the species level and can serve as DNA barcodes for the identification of species in animals (Hebert et al., Reference Hebert, Cywinska, Ball and deWaard2003a; Sonnenberg et al., Reference Sonnenberg, Nolte and Tautz2007).
The cytochrome oxidase subunit I (COI) gene fragment (covering ca. 670 bp of the 5′-terminus of COI gene) was amplified by PCR with the degenerate primers: bcdF04 (5′- TTTTCTACHAAYCAYAAAGATAT-3′) and bcdR04 (5′-TATAAACYTCDGGATGNCCAAAAAA-3′). PCRs were carried out in 25 μl reaction volumes containing reaction buffer (5 mM Tris-HCl pH 8.8, 25 mM KCl, 0.04% Nonidet P40), 1.5 mM MgCl2, 0.1 mM dNTPs, 1 μM each primer, 1.25 U Taq polymerase (Allegro, Novazym, Poznan, Poland) and 5 μl of DNA template using a thermocycling profile of one cycle of 3 min at 96°C followed by 35 steps of 10 s at 95°C, 30 s at 50°C, and 1 min at 72°C, with a final step of 5 min at 72°C.
Amplification of D2 region in 28S rDNA with primers D1D2fw2 and D1D2rev4 (Sonnenberg et al., Reference Sonnenberg, Nolte and Tautz2007) yielded single bands of amplified DNA, but they were heterogeneous in direct sequencing almost in all analyzed taxa. Therefore, we decided to amplify rDNA fragments, spanning the 3′-end of the 18S rDNA, ITS1-5.8S-ITS2 rDNA and D1D2 regions of 28S rDNA (ca. 2500 bp). Amplification was done with the primer developed in this study f1230 (5′-TGAAACTTAAAGGAATTGACG-3′) and D1D2rev4. The primers defined in the 18S and 28S regions corresponded, respectively, to nucleotides 1220-1250 and 4060-4079 of the Drosophila melanogaster ribosomal RNA gene cluster (GenBank accession no. M21017). PCRs were carried out in 25 μl reaction volumes containing reaction buffer, 1.5 mM MgCl2, 0.1 mM dNTPs, 0.25 μM each primer, 1.25 U Taq polymerase (HiFi, Novazym) and 5 μl of DNA template using a thermocycling profile of one cycle of 3 min at 96°C followed by 35 steps of 10 s at 95°C, 15 s at 50°C, and 2 min at 72°C, with a final step of 5 min at 72°C. The amplicons were used for direct sequencing of the D2 region using primers D1D2fw2 and D1D2rev4.
After amplification, 5 μl of the PCR reaction was analyzed by electrophoresis on a 1.5% agarose gel. Samples containing visible bands were directly sequenced in both directions using 1 μl of the PCR reaction and 50 pmoles of PCR primer. Sequencing was performed with BigDye Terminator v3.1 on an ABI Prism 3130XL Analyzer (Applied Biosystems, Foster City, CA, USA). Trace files were checked and edited using FinchTV 1.3.1 (Geospiza Inc.).
Sequence and phylogenetic analyzes
The strand-bias in COI nucleotide composition was analyzed at fourfold degenerate sites by comparing the frequencies of complementary nucleotides at third positions of NNN codons and was described by skewness, which measures on one strand the relative number of As to Ts (AT skew=[A−T]/[A+T]) and Cs to Gs (CG skew=[C–G]/[C+G]) (Hassanin, Reference Hassanin2006). Contigs were aligned and assembled using ChromasPro v. 1.32 (Technelysium Pty Ltd., Halensvale, Australia). Low-quality base calls that are typically found near the 5′ and 3′ ends of the sequence were excluded from the final alignment. The final data set of aligned COI sequences comprised 605 bp of unambiguous sequence data for 25 samples. Nucleotide sequences were converted into amino acid sequences using GeneDoc v. 2.7.000 (Nicholas & Nicholas, Reference Nicholas and Nicholas1997). Pairwise distance calculations between nucleotide sequences were computed using Kimura's 2-parameter (K2P) distance model (Kimura, Reference Kimura1980) for all codon positions with MEGA 4 software (Tamura et al., Reference Tamura, Dudley, Nei and Kumar2007). Codon positions included were 1st+2nd+3rd. Nucleotide pair frequencies and transition/transversion ratios were calculated using MEGA 4. PAUP* 4.0b10 (Swofford, Reference Swofford2002) was used to perform a chi-square test of base frequency homogeneity across all taxa. Phylogenetic trees were estimated using both nucleotide and amino acid sequence data of the COI. For the nucleotide data set, Modeltest ver. 3.7 (Posada & Crandall, Reference Posada and Crandall1998) was performed; according to the Akaike information criterion (AIC), TrN+I+G was chosen as the model for the maximum likelihood (ML) analysis where the proportion of invariable sites (I)=0.5642 and the gamma distribution shape parameter (G)=0.7361. Estimated base frequencies were: A=0.2377, C=0.1786, G=0.1337 and T=0.4499. Amino acid alignment contained 25 sequences of length 201 and the observed number of invariant sites was 183. Model selection for the amino acid replacement was done with ProtTest ver. 1.4 (Abascal et al., Reference Abascal, Zardoya and Posada2005). Due to the AIC criterion model, MtArt+I performed best with our data set.
Contigs of the D2 28S rDNA were aligned and assembled manually in GenDoc. Nucleotide sequence divergence was computed using the K2P distance model with MEGA 4. All positions containing alignment gaps and missing data were eliminated only in pairwise sequence comparisons (pairwise deletion option). Identical sequences from the same strain were represented in the final data set by just a single sequence. For sequences with mixed bases, Y(C+T) and R(A+G), we created two pseudo-sequences with one having ‘C’ and the other having ‘T’ (or ‘A’ and ‘G’) for the unresolved site. In the analysis, they were treated as two separate sequences (marked as X and Y). The final data matrix for phylogenetic analysis consisted of 516 characters for 11 haplotypes representing 19 mite populations. The best-fit model selected by Modeltest 3.7 was HKY85+G where G=0.0881. Base frequencies were: A=0.19870, C=0.21510, G=0.28970 and T=0.29650.
ML analysis of the nucleotide alignments was done using PAUP* 4.0b10, while analysis of amino acid data set was performed in Treefinder (Jobb, Reference Jobb2008). Bootstrap support (BS) of the recovered ML trees was evaluated with non-parametric 100 bootstrap replicates. All sequences have been deposited in GenBank under accession nos indicated in table 1. The alignments are available upon request.
Results
COI sequence diversity
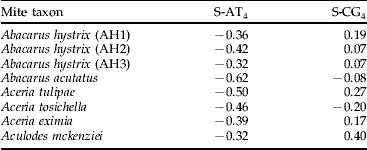
The final data set consisted of 25 aligned sequences representing 15 ingroup populations of A. hystrix strains, four populations of the close outgroup (A. acutatus), and six distant outgroups. No insertions/deletions were found. In total, 240/605 (39.7%) of the sites were variable. Among these sites, 194 (80.8%) variable sites were in the third codon position, 40 (16.7%) in the first codon position and six (2.5%) in the second codon position. COI sequences have a bias in nucleotide composition, with a majority of bases T (46.1%) and A (22.7%). For each of the species and A. hystrix strains examined, the strand bias in nucleotide composition was studied at synonymous sites (table 2). The results show that all AT skews were found negative, indicating a reverse strand-compositional bias of analyzed COI sequences. However, it was observed only in an excess of T relative to A nucleotides. The frequencies of G relative to C varied from −0.2 (in A. tosichella) to 0.4 (in A. mckenziei). Base frequencies were homogeneous across taxa (χ2=54.22, df=72, P=0.942). In A. hystrix, the average transition/transversion ratio was 2.7 for all codon positions and 1.1 calculated at fourfold degenerate sites. In the whole data set, the average transition/transversion ratio was similar and equaled 2.0 and 1.0, respectively. The translation of the nucleotide sequence resulted in 201 amino acid sequence, within which 18 (8.6%) amino acid positions were variable.
Table 2. Strand bias in COI nucleotide composition on synonymous sites. S-AT4 and S-CG4 are the skews calculated at fourfold degenerate sites.
Pairwise comparison of the COI distances within host-associated clusters of A. hystrix, between clusters and congeneric species (A. acutatus), and interspecies distances in an outgroup Aceria is presented in fig. 1. Intraspecific nucleotide sequence divergences for outgroup taxa and host-associated strains (AH1, AH2 and AH3) averaged 5.7% (SE=5.3) and ranged from 0% to 13.1%. The average interspecific divergence calculated only for the outgroup taxa was 20.4% (SE=4.5) and ranged from 12.1% (between Aceria spp.) to 25% (between different genera). Fifteen COI sequences of host-associated strains of A. hystrix varied significantly and represented 12 different haplotypes; however, the quackgrass-associated samples AH10 and AH14 were polymorphic with purine nucleotides at two synonymous sites in each sequence. The average divergence among AH1, AH2 and AH3 was 22.6% (SE=0.9). The average distance within quackgrass- (AH1) and smooth brome-associated (AH3) strains of the cereal rust mite was relatively high: 10.8% (SE=1.2) and 13.9% (SE=1.4), respectively. These distances reflect deep differences between two clusters formed in both analyzed groups (AH1a vs AH1b and AH3a vs AH3b) (fig. 2). The mean intrapopulation sequence divergence at COI in frames of the particular cluster was lower and averaged 0.32% (SE=0.3). The ryegrass-associated strain (AH2) was homogenous and represented an average distance of 0.2% (SE=0.01). Distances between five clusters of the cereal rust mite (AH1a, AH1b, AH2, AH3a and AH3b), as well as between all five clusters and congeneric species (A. acutatus), were higher than distances within the genus Aceria. In ryegrass- and quackgrass-associated populations (AH1 and AH2) all nucleotide substitutions were synonymous, while two amino acid substitutions (isoleucine into valine and methionine into leucine) were found in comparison with the brome-associated strain (AH3).

Fig. 1. Estimates of average evolutionary divergence (%) over mtDNA COI sequence pairs within and between groups: (A) within clusters of Abacarus hystrix; (B) between clusters of A. hystrix; (C) between A. acutatus and clusters of A. hystrix; (D) between Aceria species. The number of base substitutions per site from averaging over all sequence pairs within each group is shown. All results are based on the pairwise analysis of 15 sequences. Standard error estimates are included and were obtained by a bootstrap procedure (500 replicates). Analyses were conducted using the Kimura 2-parameter method in MEGA4.
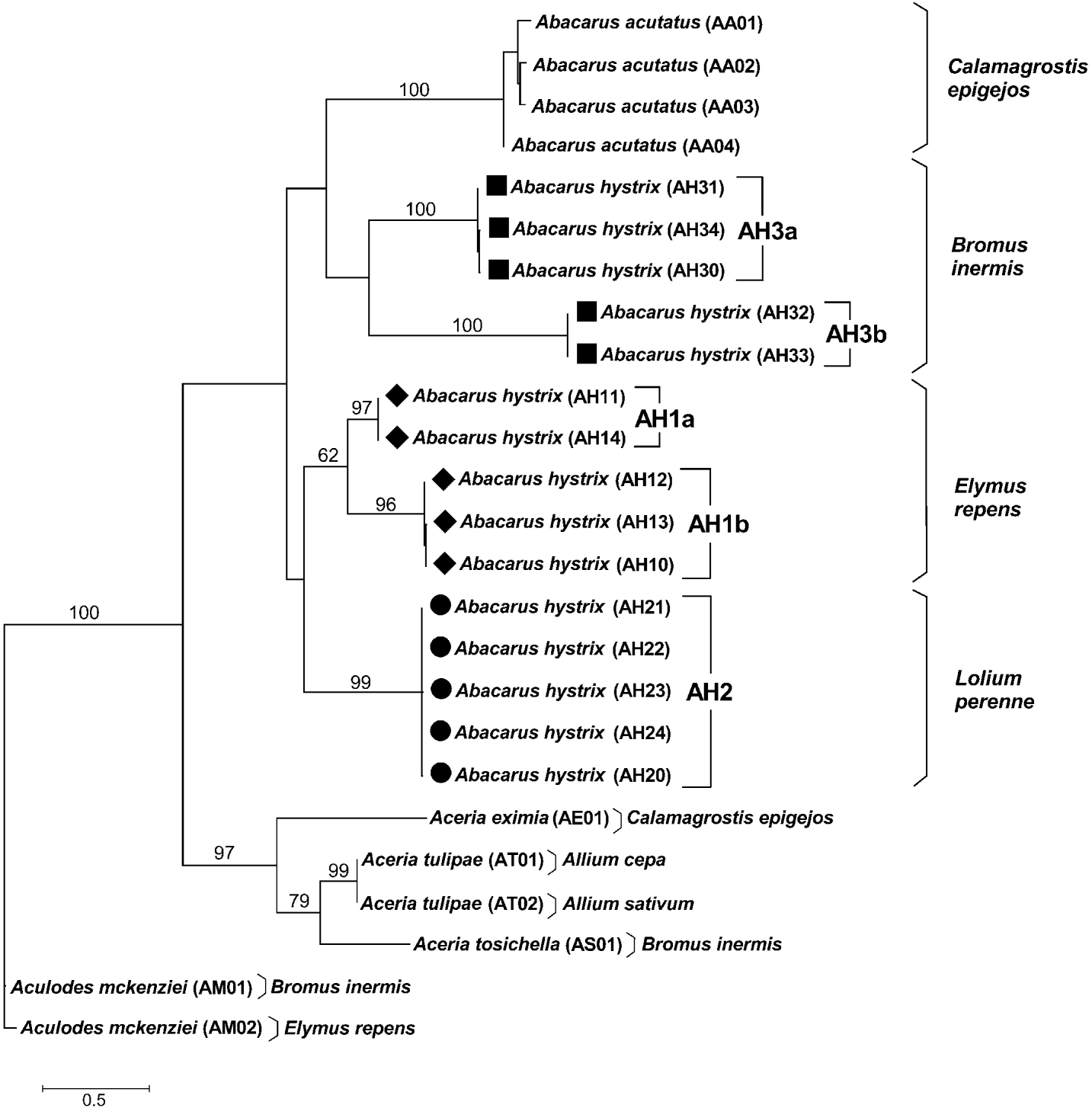
Fig. 2. Maximum likelihood tree for eriophyoids mites calculated from mitochondrial COI sequences. Bootstrap values are indicated at nodes >60% (100 replicates).
D2 sequence diversity
The nuclear data, including 516 positions for the D2 region of 28S rDNA, were obtained for 15 A. hystrix populations, four populations of the close outgroup (A. acutatus) and one distant outgroup (A. eximia). The sequence obtained from the AH12 population was excluded from the analysis due to too many missing data. One sample in A. acutatus (AA03) and two samples in the A. hystrix brome-associated strain (AH32 and AH33) were found to be polymorphic, displaying one and two transversions in each sequence, respectively. No variation was found within the quackgrass-associated (AH1) and ryegrass-associated (AH2) strains. These strains differed from each other only in one nucleotide position in the analyzed D2 region (0.2%). The brome-associated strain (AH3) revealed two types of D2 sequences that differed at 22 nucleotide positions (2.45%, SE=2.39). These groups reflected AH3a and AH3b clusters on the COI tree (fig. 2) with the single exception of the AH34 sample, which displayed the same D2 sequence as the AH32 and AH33 variants. No variation in sequence was found in the AH30 and AH31. The average distances between groups are presented in fig. 3.

Fig. 3. The average K2P distance (%) between D2 28S rDNA sequences calculated between: (A) Aceria eximia (AE) and Abacarus species: A. acutatus (AA) and clusters of A. hystrix (AH); (B) Abacarus acutatus (AA) and clusters of A. hystrix (AH); (C) clusters of A. hystrix.
Phylogenetic analysis
The phylogenetic tree obtained with the ML analysis of nucleotide COI data sets showed that A. hystrix as a taxon was not monophyletic, and three host-adapted strains formed distinct clades (fig. 2). Furthermore, the quackgrass- (AH1) and brome-associated (AH3) strains were internally divided each into two well-supported monophyletic clusters (96–97% and 100% BS, respectively). Phylogenetic relationships that had moderate or high bootstrap support and that were pertinent to questions of monophyly of species were: A. acutatus (100% BS), A. hystrix ryegrass-associated populations AH2 (99% BS) and A. hystrix quackgrass-associated populations AH1 (62% BS). There was low bootstrap support (<50% BS) for the monophyly of the A. hystrix brome-associated strain AH3. Moreover, on the amino acid tree (not shown) the AH3 strain was represented by two separate, well-supported (65% and 81% BS) clades, which formed an unresolved polytomic clade (86% BS) with A. acutatus (91% BS) and AH1–AH2 clusters (67% BS). The separation of the quackgrass- (AH1) and ryegrass-associated (AH2) strains on the basis on nucleotide COI data sets was not supported on the amino acid tree. Both nucleotide and amino acid trees showed well-supported monophyly of genera used in the analyses (Abacarus (both 86% BS), Aceria (97% BS and 91% BS, respectively) and Aculodes (both 100% BS)).
The phylogenetic tree obtained with the ML analysis of the D2 region data set did not support monophyly of the cereal rust mite (fig. 4). Quackgrass- (AH1) and ryegrass-associated (AH2) populations formed two distinct clades. Monophyly of the brome-associated (AH3) strain was not recovered. The strain was internally divided into two clades; thus, AH3 was paraphyletic in origin. One of these lines, consisting of two identical sequences (AH30 and AH31), formed a well-supported clade with A. acutatus (97% BS), and the second one (AH32, AH33, AH34, 87% BS) was their sister group (64% BS).

Fig. 4. Maximum likelihood tree for eriophyoid mites calculated from nuclear D2 28S rDNA sequences. Bootstrap values are indicated at nodes >50% (100 replicates). Alternative haplotypes are marked as X and Y.
Discussion
Species status of the cereal rust mite
Our results did not confirm the placement of three host-associated strains of A. hystrix within one, ostensibly generalist, species. Therefore, they are not in accordance with the hitherto prevailing taxonomy and wide host-range data for this mite (Amrine & Stasny, Reference Amrine and Stasny1994; Sabelis & Bruin, Reference Sabelis, Bruin, Lindquist, Sabelis and Bruin1996). Populations of A. hystrix originating from three different host plants (quackgrass, ryegrass and smooth brome) clustered into different clades on the mitochondrial and nuclear trees. In addition, A. acutatus, a species chosen as the nearest outgroup, did not form a sister group for A. hystrix. In contrast, on the COI nucleotide tree, A. acutatus was found inside the Abacarus complex as a fourth host-associated cluster. Moreover, on the D2 28S rDNA tree, A. acutatus grouped in the same clade with two populations of brome-associated mites (AH30 and AH31). Thus, it could be hypothesized that A. acutatus and A. hystrix belong to the same complex of species.
Apart from host-associated differentiation within Abacarus complex, further delineation inside two host-adapted populations was observed. The mitochondrial COI nucleotide tree showed a well-supported split within the AH1 strain. This splitting was not supported by the COI amino acid tree, and this may suggest a very recent divergence. Similar splitting was also observed within the brome-associated AH3 strain. Two distinct clades of the AH3 strain were observed on the mitochondrial COI nucleotide tree, and both strains were confirmed on the COI amino acid tree. The clade AH3b consists of populations originating from the same region, which is geographically more distinct than regions of origin of populations of AH3a. This suggests the possibility of diversification within the brome strain influenced by geographical distance. However, on the D2 28S rDNA tree, such a relation between clusters and the origin of the population was not observed. Instead, two populations of AH3 strain clustered with A. acutatus, which may suggest that they are more closely related to A. acutatus than to other strains of A. hystrix. Undoubtedly, increased sampling throughout the entire range of Abacarus complex and consideration of the role of causes other than host plant are needed to explain the relationships between and within host strains.
Genetic divergences in Abacarus hystrix complex
Three host-associated strains of the cereal rust mite exhibited more than 22% nucleotide sequence divergence, which was higher than those within each host strain (0.2%–13.9%) and each cluster (0.32%). It should be noted that the threshold for diagnosis of eriophyoid species on the basis of the COI gene has not been presented up to now. Hence, this is the first report in which COI distances for eriophyoid mites has been interpreted.
Other studies of the same region in arthropods have documented different levels of COI divergence for species belonging to various taxa. Values of intraspecific distances have ranged from 0.1% to 3.6%, whereas interspecific distances have ranged from 2.3% to 29.9% in several species of insects, spiders and mites (e.g. Hebert et al., Reference Hebert, Penton, Burns, Janzen and Hallwachs2004; Barret & Hebert, Reference Barrett and Hebert2005; Monti et al., Reference Monti, Nappo and Giorgini2005; Anderson & Morgan, Reference Anderson and Morgan2007; Dabert et al., Reference Dabert, Ehrnsberger and Dabert2008; Tixier et al., Reference Tixier, Kreiter, Croft and Cheval2008). The observed COI distances between the three host-associated cereal rust mite strains seem to be large enough to represent differences between species. Hebert et al. (Reference Hebert, Ratnasingham and deWaard2003b, Reference Hebert, Penton, Burns, Janzen and Hallwachs2004) and Witt et al. (Reference Witt, Threloff and Hebert2006) suggested that a threshold level of intraspecies versus interspecies variance could be used as a proxy to identify groups of species. They proposed a standard sequence threshold as ten times the mean intraspecific variation for the group under study and termed it the species screening threshold (SST). The within-cluster divergences of the studied cereal mite populations resulted in an SST of 3.4%. This value is placed between threshold divergence values for birds (Hebert et al., Reference Hebert, Penton, Burns, Janzen and Hallwachs2004) and amphipods (Witt et al., Reference Witt, Threloff and Hebert2006).
The lack of overlap between intra-strain (⩽13.9%) and inter-strain (22.6%) COI divergence additionally argues for species delineation among strains of the cereal rust mite. However, Avise (Reference Avise2000) pointed out that intraspecific genetic divergences are rarely greater than 2% and most are less than 1%. Thus, it could be concluded that among the three strains of A. hystrix studied here, only the ryegrass-associated strain exhibits an appropriate level of intraspecific divergence with an intraclade divergence of only 0.2%. COI divergences within the quackgrass and brome strains seem to be much higher than a typical intraspecific level (10.8% and 13.9%, respectively). However, the divergences within clusters (AH1a, AH1b, AH3a and AH3b; see fig. 1) clearly suit the intraspecific rank.
The discrimination of genetically distinct entities within the cereal rust mite on the basis of COI sequences was validated here by D2 28S rDNA sequences, although with a few differences. In contrast to the observed delineation within the quackgrass strain on the basis of COI sequences, no variation was observed in D2 sequences within either the quackgrass or ryegrass strains. Moreover, all sequences within a particular strain were identical. Quackgrass- and ryegrass-associated populations each formed a separate clade and appeared to be each other's most closely related taxon. It is known that the nuclear ribosomal sequences are much more conserved than COI, so closely related species often possess 28S rDNA identical or nearly so (Lee & Ó Foighil, Reference Lee and Ó Foighil2004). The observed variation in D2 sequences between the quackgrass and ryegrass strains was less (0.2%) than that of other between-strain, between-cluster and between-Abacarus spp. comparisons (1.3–5.7%). So, it could be concluded that the divergence of the quackgrass and ryegrass strains is a relatively recent event. This presumption is supported also by COI data; when nucleotide COI sequences were translated into amino acids, mites from quackgrass and ryegrass clustered into one clade. This may suggest a shared origin and closer relationships to each other than to the smooth brome strain. Moreover, the very low intraclade COI divergence of the ryegrass-associated strain of the cereal rust mite (0.2%) may suggest that this strain could have shifted from the quackgrass strain. The host-shift model assumes that speciation via host shift should result in significantly lower levels of genetic variation in populations infesting the derived host because of the founder effect and that populations on the ancestral host have had a greater amount of time to differentiate than populations on the derived host (Harrison, Reference Harrison1991). Earlier results showed the existence of asymmetry in reproductive barriers between the quackgrass and ryegrass strains with the origin of the female nymph acting as a predictor of hybrid inviability (Skoracka, Reference Skoracka2008). Asymmetry in gene flow seems to be a very common isolating barrier between closely related species (Wu & Beckenbach, Reference Wu and Beckenbach1983). To clarify the relationships within different host strains of the cereal rust mite and the direction of their evolutionary changes would require more molecular data from other host-plant species and different geographical regions. At this moment, we can conclude that two reproductively incompatible strains of the cereal rust mite exhibit more than 20% sequence divergence in the COI gene and 0.2% sequence divergence in the D2 region.
High molecular divergence within the cereal rust mite A. hystrix revealed that A. hystrix does not exist as a monophyletic taxon. Over its huge host range (ca. 60 host species), the cereal rust mite may comprise more hidden species than shown in this study. The unresolved relationship between A. hystrix and A. acutatus suggests that the taxonomy of the whole grass-associated Abacarus complex may be more complicated than was thought. This study (and our continuous observations) indicates that DNA barcoding is a valuable tool in identifying morphologically similar eriophyoid species and should be widely applied in eriophyoid systematics. This is especially the case considering that the present classification of this group is thought to be artificial, and revisions of many taxonomically problematic species are urgently required (Lindquist, Reference Lindquist, Lindquist, Sabelis and Bruin1996; Lindquist & Amrine, Reference Lindquist, Amrine, Lindquist, Sabelis and Bruin1996).
Acknowledgements
We thank Jacek Dabert (AMU, Poznan, Poland) for help in phylogenetic analysis and remarks on the manuscript, David Orwin (University of Luton, England) and Brian Rector (USDA, Montpellier, France) for editorial corrections, two anonymous reviewers for helpful and valuable remarks on the manuscript, and Katarzyna Zawadzka (AMU, Poznan, Poland) for technical help. The study was supported by the Polish Ministry of Science and Higher Education (grant no. NN303089434).






