Introduction
Classical biological control entails the introduction of a co-evolved natural enemy meant to control an inadvertently introduced invasive pest. The objective of a novel introduction is the re-establishment of a natural balance, hence, control of the introduced pest in its new range. However, although about 30% of introductions result in establishment of the natural enemy, only 10% lead to economically significant control (Bellows et al., Reference Bellows, Fisher and Cattairone1999). Introductions of the pest and/or the natural enemy are both often followed by a rapid population expansion in the new geographical range, usually leading to loss of genetic diversity associated with the establishment constraints and determined by the diversity of the original founding population (Grevstad, Reference Grevstad1999; Sakai et al., Reference Sakai, Allendorf, Holt, Lodge, Molofsky, With, Baughman, Cabin, Cohen, Ellstrand, McCauley, O'Neil, Parker, Thompson and Weller2001; Memmott et al., Reference Memmott, Craze, Harman, Syrett and Fowler2005). For natural enemies, this effect can be reduced by large releases and multiple field recoveries of the natural enemy across its natural ecological range (Grevstad, Reference Grevstad1999; Roderick & Navajas, Reference Roderick and Navajas2003; Lloyd et al., Reference Lloyd, Hufbauer, Jackson, Nissen and Norton2005). However, this requires some knowledge of the underlying natural variation, which could be exploited to increase diversity of the population to be released in biocontrol programmes (Lockwood et al., Reference Lockwood, Cassey and Blackburn2005; Bacigalupe, Reference Bacigalupe2009). Also, the adaptability and genetic diversity may be lost during laboratory culturing if strong selection pressure occurs that enable the organism to survive under such conditions, thereby lowering the chance of establishment (Bigler, Reference Bigler and Ochieng-Odero1992). This may result in lower vigour of the natural enemy in the wild (Bellows et al., Reference Bellows, Fisher and Cattairone1999). Molecular markers are therefore important for assessing the underlying field diversity and monitoring its maintenance through the culturing and establishment periods.
The invasive larger grain borer (LGB), Prostephanus truncatus (Horn) (Coleoptera: Bostrichidae), is the most devastating pest of farm-stored maize in Africa (Schneider et al., Reference Schneider, Borgemeister, Setamou, Affognon, Bell, Zweigert, Poehling and Schulthess2004). It was accidentally introduced into the continent during the 1970s and has since spread to at least 18 countries (Schneider et al., Reference Schneider, Borgemeister, Setamou, Affognon, Bell, Zweigert, Poehling and Schulthess2004; Gueye et al., Reference Gueye, Goergen, Badiane, Hell and Lamboni2008). The LGB may cause grain weight losses of up to 34% within the first six months of grain storage, five times of that resulting from indigenous storage pests (Hodges et al., Reference Hodges, Dunstan, Magazini and Golob1983; Giles et al., Reference Giles, Hill, Nang'ayo, Farrell and Kibata1996). Storage loss is particularly costly as it occurs when all the production costs have been incurred and when plant compensatory growth is not possible.
Being an invasive species causing devastating outbreaks in its new areas, LGB appeared to be a prime candidate for classical biological control (Markham et al., Reference Markham, Wright and Ríos-Ibarra1991). Teretrius (formerly Teretriosoma) nigrescens Lewis (Coleoptera: Histeridae), found to be consistently associated with P. truncatus in Meso-America, has been introduced into several African countries (Meikle et al., Reference Meikle, Rees and Markham2002). Teretrius nigrescens is a specialist predator that is strongly attracted to the pheromone of P. truncatus, using it as a kairomone to locate its prey (Böye, Reference Böye1988). The low pest status of LGB in Central America and the occurrence of T. nigrescens under a wide range of environmental conditions suggested that the pest was under natural control in the area of origin. In West Africa, the release of a Costa Rican population of the predator considerably reduced trap catches of LGB in the warm humid coastal areas of Togo and Benin (Schneider et al., Reference Schneider, Borgemeister, Setamou, Affognon, Bell, Zweigert, Poehling and Schulthess2004), but the predator appeared not to be efficient in the dry-hot savannahs, and it did not establish in the cool highlands of Guinea Conakry (Borgemeister, ICIPE, Nairobi, Kenya unpublished data). A Mexican T. nigrescens population released in the semi-arid areas of Wundanyi and Makueni in eastern Kenya purportedly resulted in an 80% reduction in pest flight activity and predator dispersal of over 70 km within the first three years of its release (Nang'ayo, Reference Nang'ayo1996; Hill et al., Reference Hill, Nang'ayo and Wright2003). However, recent surveys indicated that T. nigrescens got extinct in the original release area. The failure of the populations of the predator introduced into Africa to establish in or spread to certain regions suggests the existence of ecotypes adapted to different climatic conditions. However, a review of the literature does not reveal a distinct biological discontinuity among geographical populations, making it difficult to estimate the level of differentiation that would be expected within and among the populations. Knowledge of the genetic structure of T. nigrescens populations has potential use in guiding the exploration for new populations, monitoring released populations and their interactions with the environment, other populations and the pest.
The separate introduction of two populations of T. nigrescens recovered from Mexico and Costa Rica, over 2000 km apart, raises the possibility that distinct strains were involved. Both strains, however, were not effective in controlling LGB in warm dry and cool climates (Giles et al., Reference Giles, Hill, Nang'ayo, Farrell and Kibata1996; Hill et al., Reference Hill, Nang'ayo and Wright2003; Hodges et al., Reference Hodges, Addo and Birkinshaw2003; Schneider et al., Reference Schneider, Borgemeister, Setamou, Affognon, Bell, Zweigert, Poehling and Schulthess2004). Using data from extensive pheromone trapping in various climatic zones, Tigar et al. (Reference Tigar, Osborne, Key, FloresS and Vazquez-Arista1994) modelled the conditions responsible for the survival of both the pest and predator in Mexico and inferred that they could exist in a wide range of regions. Rees et al. (Reference Rees, Rivera and Rodriguez1990) recovered both LGB and T. nigrescens under hot-dry conditions in the Yucatan Peninsula. It is, thus, possible that the limitations of the T. nigrescens populations introduced into Africa are partially due to their inherent genetic limitations. To address this problem, four populations of the predator from Mexico and Honduras have been imported into the quarantine facilities of the International Centre of Insect Physiology and Ecology (ICIPE), Nairobi, for eventual release. Pertinent questions still to be answered are: What is the extent of genetic variation in T. nigrescens? Do geographical populations of T. nigrescens represent distinct ecotypes? Can simple genetic markers be used to distinguish between these ecotypes? How can this genetic diversity be incorporated into a biological control program? To investigate these questions, the sequence variations of the mitcochondrial cytrochome oxidase 1 (mtCOI) gene and the internal subscribed spacers, ITS1 and ITS2, spacer regions among the populations of the predator, were studied.
Materials and methods
Study materials
Insects
Starter colonies of T. nigrescens were obtained as outlined in table 1 and fig. 1. The strain from the Kiboko Field Station of the Kenya Agricultural Research Institute (KARI), which was released in Kenya in 1992, originally stemmed from Mexico (Giles et al., Reference Giles, Hill, Nang'ayo, Farrell and Kibata1996). The Benin strain was obtained from the International Institute of Tropical Agriculture (IITA), Abomey-Calavi, Benin. This population had been established from adults recovered from granaries in Benin, three years after field releases in Togo (C. Atcha, Africa Rice Centre, Benin, personal communication). The Togo releases had originated from Sardinal, Guanacaste, north-western Costa Rica (Böye, Reference Böye, Markham and Herren1990). Three geographical strains from colonies established using insects recovered from granaries at El Batan (hereafter referred to as the Batan strain), Oaxaca and Tlatizaspan in Mexico were imported from CIMMYT, Mexico. Three other populations were collected from Teupasenti, Gualaso and Yoro in Honduras between 2006 and 2007, and used to found laboratory cultures. The Kiboko colony was established using insects recovered in 2007 from the Kiboko Range, eastern Kenya within the original outbreak and release area (Giles et al., Reference Giles, Hill, Nang'ayo, Farrell and Kibata1996). The ‘Store’ colony was accidentally discovered in LGB-infested wooden pegs from a store at ICIPE (Nairobi). These pegs had reportedly been used for field sampling cereal stem borers and their parasitoids at the Kenyan coast a year earlier. The origin of this population was not known. All T. nigrescens strains were reared on P. truncatus raised on maize at the Animal Rearing and Quarantine Unit (ARQU) at ICIPE, Nairobi, Kenya. The LGB and T. nigrescens were reared using a modification of protocols described by Giles et al. (Reference Giles, Hill, Nang'ayo, Farrell and Kibata1996) and C. Atcha & C. Borgemeister (unpublished IITA manual).

Fig. 1. Map of Central America showing the field origin of Teretrius nigrescens populations used in this study.
Table 1. Field sources and culturing history of the sample populations of T. nigrescens used in this study.

1 Ethanol-preserved samples only used for molecular studies. The size of the original field-recovered population of the CIMMYT samples was about 200 beetles. The original size number of traps used and sampling duration for the recovery of KARI and Benin populations are not known.
Alcohol-preserved samples from three further populations were also used. The Ghana strain was obtained from a colony maintained at the Natural Resources Institute (NRI), UK, established with insects initially recovered from the field in Ghana in 1998 following releases in Togo, Benin and the Volta Region, in Ghana (R. Hodges, Natural Resource Institute, Greenwich, UK, personal communication). The Malawi samples originated from a colony maintained at the Crop Protection Agency, Ministry of Agriculture, Malawi. This colony was a subculture of the Benin colony (IITA, 1999). The Mombasa population was comprised of five individuals caught in a pheromone-baited sticky trap in the North Coast, Mombasa, Kenya. These traps were set after the discovery of the ‘Store’ population since the substrate material it was obtained from had been reportedly used in this region. Teretrius americanus (=T. latebricola) was used as the outgroup species. Specimens were obtained by J. Grubber (Russell Labs, Madison, WI, USA) from fallen tree barks in WI, USA, immediately preserved in 70% ethanol and shipped to ICIPE.
DNA extraction
The DNA was extracted from the head and thorax of adult insects using the phenol-chloroform-isoamyl alcohol protocol (Sambrook et al., Reference Sambrook, Fritsch and Maniatis1989). Briefly, insect tissue was homogenised in 100 μl of tissue-grinding buffer (10 mM Tris-Cl, 10 mM EDTA, 150 mM Sucrose, 60 mM NaCl, 0.5% SDS, 25 U ml−1 Proteinase, pH 7.5) and incubated at 55°C for one hour. An equal volume of the lysis buffer (0.3 M Tris-Cl, 0.1 M EDTA, 0.15 M Sucrose, 60 mM NaCl, 0.75% SDS, pH 7.5) was added to the homogenate, mixed gently and incubated on ice for 10 min. One volume of equilibrated phenol was added to the lysate, mixed by gentle inversion and centrifuged at 5000 g for 10 min. An equal volume (200 μl) of chloroform isoamyl alcohol (24:1) was added to the supernatant in a fresh tube, mixed gently and centrifuged at 13,000 g for 5 min. The DNA was precipitated by mixing the supernatant with a tenth volume of 5 M sodium acetate (pH 5.2) and an equal volume of cold isopropanol and incubating it overnight at −20°C. DNA was pelleted by centrifuging at 13,000 g for 25 min and tilting off the alcohol gently. The pellet was washed twice with 70% ethanol, each time centrifuging at 13,000 g for 5 min before decanting off the liquid phase. It was dried in a flow hood and resuspended in 300 μl of TE buffer (10 mM Tris pH 8.0, 1 mM EDTA).
Polymerase chain reaction
Cytochrome Oxidase 1
A 1200 bp region of the mtCOI gene was amplified by PCR using the universal insect mtCOI primers: C1J-17363 (5′ TATAGCATTCCCACTAATAAATAA 3′) forward and TL2-N-3014 (5′ TCCAATGACTAATCTGCCATATTA 3′) reverse, previously described by Zhang & Hewitt (Reference Zhang and Hewitt1997). The PCR amplifications were carried out in 30 μl reaction volumes containing 1×Genscript Taq polymerase buffer with 1.5 mM MgCl2, 400 μM dNTPs, 150 nM each of the primers, 1 U Taq polymerase and ∼20 ng genomic DNA (Genscript, Piscataway, NJ) and 10 ng of genomic DNA template. The thermal cycling programme was 94°C 3 min, followed by 35 cycles of denaturation at 94°C 1 min, annealing at 52°C for 40 s and extension at 72°C for 90 s, and then a final extension step of 72°C for 10 min. The PCR product was separated on a 1.2% agarose gel. The amplification product of 1200 bp size was excised from the gel and purified using Qiagen Min Elute Gel purification kit (Qiagen Inc., Valencia, CA, USA) according to manufacturer's instructions. The eluted product was sequenced in both directions using the PCR primers on an ABI chain termination sequencing technology at a commercial facility (Macrogen Inc., Korea).
Internal transcribed spacer regions (ITS)
Amplification of the two ITS regions and 5.8S rRNA coding site was done as above using standard forward primer ITS5 (5′GGAAGTAAAAGTCGTAACAAGG 3′) and reverse primer ITS4 (5′ TCCTCCGCTTATTGATATGC 3′) (White et al., Reference White, Bruns, Lee, Taylor, Innes, Gelfand, Sninsky and White1990). The thermal cycling conditions were similar to those of mtCOI (above) but with an annealing temperature of 54°C and a reduction to 25 cycles, done to limit polymerase errors. Fewer cycles were used to increase the integrity of the product for cloning. Preliminary direct sequencing of ITS genes produced equivocal results; hence, cloning was done to separate co-amplifying products.
Gel purified fragment was cloned into pGEMT-E vector (Promega, Madison, WI, USA) and used to transform DH5α chemically competent cells according to manufacturer's recommendations. Recombinant colonies were screened by PCR to confirm the presence of the insert. Five positive recombinant colonies per insect sample were selected for bacterial propagation and plasmid purification. Plasmid purification was done using Qiagen Miniprep Kit according to manufacturer's protocol. Plasmid and DNA samples were sequenced by the ABI terminator system using M13FpUC and M13RpUC universal primers (Promega) (Macrogen Inc., Seoul, South Korea).
PCR-RFLP analysis
Sequence alignment of the mtCOI fragment revealed parsimony informative sites associated with populations; thus, NEB cutter version 2 software (Vincze et al., Reference Vincze, Posfai and Roberts2003) was used to predict the diagnostic utility of the restriction endonucleases. The selected enzymes were then used to digest PCR products of the mtCOI from the five populations of T. nigrescens (Ghana, Oaxaca, Benin, KARI and Tlaltizapan) representing the main sub clusters in sequence analysis.
Data Analysis
DNA sequences were edited manually to confirm the positions of ambiguous nucleotides using BioEdit (Hall, Reference Hall1999). The reverse complement of the reverse sequence was contigged with the forward sequence after pairwise alignment of the termini produced at least 100 bp complete consensus overlap region. All complete sequences were aligned together and segregating sites reconfirmed by comparison with the trace diagrams and translation using the Invertebrate Mitochondrial codon table. Since mtCOI is an expressed gene, only fragments translating into a complete protein sequence were considered to minimise error from possible nuclear mitochondrial pseudogenes (Bensasson et al., Reference Bensasson, Zhang and Hewitt2000). Haplotype identification and frequencies were done manually by sequences alignment and grouping of sequences according to similarity in BioEdit (Hall, Reference Hall1999). Haplotype network was determined in TCS version 2.1 (Clement et al., Reference Clement, Posada and Crandall2000). Haplotypes were grouped into series of clades from the smallest group of individual haplotypes.
Multiple sequence alignment was performed using Clustal W version 1.8 as implemented in BioEdit. The numbers and types of segregating sites, Kimura 2-parameter distances (K2P) between individuals, populations and groups of populations were determined using MEGA 4.0 (Tamura et al., Reference Tamura, Dudley, Nei and Kumar2007). In this approach, the mean number of base substitutions per site from all 59 sequences and 1083 bases per sequence were conducted using the Maximum Composite Likelihood method in MEGA4 (Tamura et al., Reference Tamura, Nei and Kumar2004, Reference Tamura, Dudley, Nei and Kumar2007). Average within and between geographic population K2P distances and their standard deviation were calculated using Mega 4.0.
The phylogenetic relationships between the samples were inferred using a neighbour-joining tree on MEGA 4 (Saitou & Nei, Reference Saitou and Nei1987). Sequence alignment was done in Clustal X to create an alignment file for MEGA. The evolutionary relationships were inferred with the neighbour-joining tree method, with 1000 bootstrap replicates. The evolutionary distances used to compute tree-branch length were determined based on the K2P parameter (Kimura, Reference Kimura1980). A similar length mtCOI sequence from Teretrius americanus LeConte (=T. latebricola Lewis) was used as an outgroup species.
Isolation by distance
Mantel tests were done between pairwise genetic and geographic orthodromic distances, in kilometers, (raw and log-transformed distances). A partial mantel test (reduced major axis regression) was subsequently done using associated indicator matrices as conditional matrices. Two associated matrices were used, one based on splitting between populations (introduction matrix) and the other linked to geographical obstacles. Mantel test for significance was employed with 30,000 randomisations. The 99% confidence intervals were also estimated with 30,000 bootstrap replicates over independent population pairs. All regression analyses were performed using the web-based IBDWS Version 3.15 software (Jensen et al., Reference Jensen, Bohonak and Kelley2005).
The ITS1 and ITS2 sequences were aligned manually on BioEdit, since, due to numerous indels, automatic alignment was not satisfactory. The repeat regions were identified manually from the BioEdit alignment and using SSR Finder (Benson, Reference Benson1999). The boundaries of the 18S, 5.8S and 21S rRNA genes were delimited by comparisons with sequences of Timarcha and Tetranynchus sequences (Navajas et al., Reference Navajas, Lagnel, Gutiérrez and Boursot1998; Gomez-Zurita et al., Reference Gomez-Zurita, Juan and Petitpierre2000).
Results
MtCOI Sequence variation
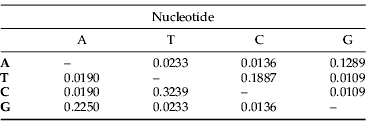
A 1200 bp region was amplified with the mtCOI primers. After editing, a 1083 bp fragment was used in the analyses whose results are presented below. These sequences of the 22 unique haplotypes have been deposited in GenBank (accession numbers: GC920842–GQ920863). The nucleotide frequencies were 0.285 (A), 0.349 (T), 0.203 (C) and 0.163 (G), comparable to mitochondrial sequences in other Coleoptera (Cognato et al., Reference Cognato, Harlin and Fisher2003; Mynhardt et al., Reference Mynhardt, Harris and Cognato2007). There were 102 polymorphic sites, of which 72 were parsimony informative (fig. 2). This variation occurred mostly at third (83.3%), followed at the first (13.7%) and was lowest at the second (9%) codon positions. The rate and direction of nucleotide substitutions are summarised in table 2.

Fig. 2. Nucleotide variation among 22 mtCOI haplotypes (H1 to H22) on 1083 bases of the mtCOI T. nigrescens used in this study. Numbers at the top of each nucleotide position represent the segregating site position relative to the first nucleotide of this fragment. Haplotypes are arranged in order of relative similarity. Dots represent similarity with haplotype H8 while the nucleotide in each segregating site is labelled vertically (such that the first is 27 bp and the last is 1068 bp from the origin).
Table 2. Maximum composite likelihood estimate of the pattern of nucleotide substitution.
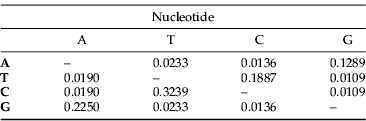
Each entry shows the probability of substitution from one base (row) to another base (column).
Genetic diversity
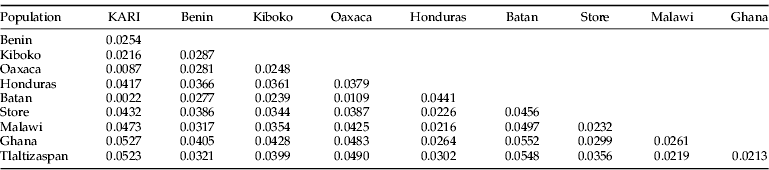
Pair-wise divergence between populations based on the K2P parameter are shown in tables 3 and 4. The evolutionary divergence among samples from Honduras/Costa Rica samples was greater than among samples from Mexico. However, the Mexican Tlaltizapan population was closest to individuals from Ghana and Malawi, both originating from the Costa Rica population reared and released in Benin.
Table 3. Estimates of evolutionary divergence over sequence pairs between populations.
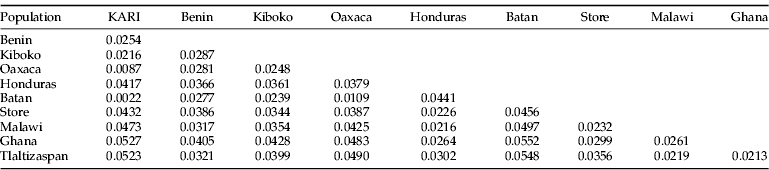
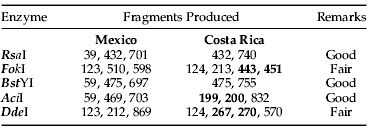
Table 4. Predicted products of the cleavage of 1200 bp mtCOI PCR product using four potentially diagnostic endonucleases1.
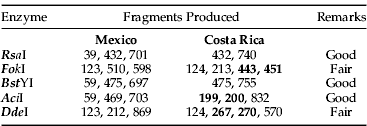
1 Fragments in bold, for each enzyme, are not distinguishable on the agarose gel.
The evolutionary relationships between the 57 T. nigrescens samples revealed two major clades supported by >98% bootstrap values (fig. 3). One cluster contained most samples originally recovered from Mexico, while the other grouped samples from Honduras, Costa Rica and Tlaltizapan (Mexico). Other minor clusters that were highly supported (e.g. all ‘Store’ samples) grouped samples from highly monomorphic populations. Twenty-two haplotypes were observed, of which 15 were unique (observed in only one individual each) (fig. 2). Haplotype diversity was highest among the Honduras populations (seven haplotypes) and lowest among the KARI, ‘Store’ and Malawi populations (two haplotypes each). Haplotype H19 was both the most geographically diverse and the most frequent, occurring in five of the populations and 23% of the individuals (fig. 4).

Fig. 3. Evolutionary relationships of 57 samples of T. nigrescens from seven populations analysed using the UPGMA method. The tree is rooted using a homologous sequence from T. americanus. This consensus tree inferred from 5000 replicates is shown, with the bootstrap values next to the branches.

Fig. 4. Network showing the most parsimonious relationship between 22 mtCOI haplotypes of T. nigreescens from ten geographical populations. Circles represent detected haplotypes, black dots signify intermediate (unsampled) haplotypes and lines between haplotypes denote single nucleotide change. Sample populations in which each haplotype ws sampled are shown: Bat, Batan; Fld (Kiboko, field); Str, store; Oax, Oaxaca; Mlw, Malawi; Yor,Yoro; Teu, Teupasenti; Ghn, Ghana/NRI; Tla, Tlaltizaspan; Ben, Benin; Kar, KARI.
Islation by distance
Figure 5 shows scatterplots of the pairwise corrected genetic distances of all populations versus geographic orthodromic distances (fig. 5a); and versus log geographic distances (fig. 5b). Measures of geographic and genetic distance exhibited a significant positive correlation in the RMA analysis, indicating that geographic distance explains a significant proportion of the variance in genetic distance between populations (r2=0.145, P<0.02, y=0.01129+1.933×10−6x). However, there was a higher correlation between the genetic distance and the indicator matrix defining geographical between source populations and split events between source populations and those released for biological control in Africa (r2=0.216, P<0.004). A partial correlation of genetic and geographical distances controlling for the indicator matrix was insignificant. A similar analysis excluding any secondary split laboratory samples (only field samples and the first introduction populations) revealed that log(geographical distance) explained a significant portion of the observed genetic diversity (r2=0.153, P<0.05, y=−0.08323+0.3951x, n=28). The partial correlation between genetic diversity and the geographical split events was not significant.

Fig. 5. Graph of RMA regression for pairwise geographic distances and mtCOI corrected genetic distances for all populations. Genetic distances are used as orthodromic distances between points in kilometres.
PCR-RFLP identification of populations
In silico restriction analysis showed that the restriction endonucleases AciI, BstYI, DdeI, FokI and RsaI, had their recognition sites flanking parsimony-informative sites and, thus, could resolve four populations into two major clades (table 4). The results of these digestions are shown in fig. 6.

Fig. 6. The ≈1200 bp amplicon and products of its single digestion with three restriction enzymes. Sample abbreviations are: K, KARI; Bt, Batan; Ox, Oaxaca; G, Ghana, Tl, Tlatizapan; ‘−ve’, negative control set up without DNA.
Internal transcribed spacer regions
Cloning enabled the sequencing of the whole of the ITS amplicons with flanking coding sequences and enabled comparison between samples and outgroup species. The length of the ITS sequences ranged between 1250 and 1350 bp. Intra-individual variation of up to ten bases was observed. Most of this variation involved insertions and deletions of tandem sequence repeat units. Differences were, therefore, in multiples of repeat lengths. Because of little variation in the non-repeat sequences and high polymorphism in the repeat variable regions, the ITS was not found useful for the phylogenetic analysis in this study.
Discussion
The mtCOI sequences were the most useful in the detection of population genetic variations in T. nigrescens. The ITS 1 and ITS 2 sequences showed comparable sizes and coding region similarity with the previously studied beetle species (Gomez-Zurita et al., Reference Gomez-Zurita, Juan and Petitpierre2000). Since the variation was predominantly due to two or three base pair tandem repeats, it might be interesting to investigate how the repeat-length fit with microsatellite models in population genetic analysis. However, one must first resolve the apparent polyploid genotype pattern created by the intragenomic variation of these markers and to estimate the most dominant variants.
The mtCOI gene sequence analysis revealed two major clades of T. nigrescens associated with defined geographical ranges. Apart from the ‘Store’ samples, whose origin was not clearly known, all insect samples from Africa clustered with their original source populations, confirming suitability of this marker for phylogeographic analysis (Otranto et al., Reference Otranto, Traversa, Guida, Tarasitano, Fiorente and Stevens2003). The 1200 bp region sequenced in this study covered two highly variable regions and a much more conserved portion of this gene (Lunt et al., Reference Lunt, Zhang, Szymura and Hewitt1996; Zhang & Hewitt, Reference Zhang and Hewitt1997). This strategy promised greater accuracy since the phylogenetic differentiation of the species was initially unknown. Several regions of the sequence with a greater density of parsimony informative sites have been identified in this study. Shorter fragments spanning these desirable regions would be more practical, to reduce the potential analysis challenges associated with a large number of single sequence haplotypes and potential error in sequencing large fragments. Although very few parsimony-informative segregating sites are within five bases of each other, careful primer design might reveal a single step multiplex PCR protocol for identifying these populations (Lindell & Murphy, Reference Lindell and Murphy2008).
The 22 haplotypes separated into two clusters revealed two distinct maternal lineages, suggesting historical geographical and hence genetic fragmentation. This could have impeded interaction between populations causing independent evolution of this gene in the two populations. The region between Oaxaca and Tlaltizapan is the possible contact zone, where both subtypes exist. Lindell & Murphy (Reference Lindell and Murphy2008) have identified the Baja peninsula in north-western Mexico as a possible hybrid zone for the mitochondrial forms of the fence lizard Uta stansburiana, Baird & Girard. This hybrid zone was explained to reflect a complex of geological history events since the Miocene era (Lindell et al., Reference Lindell, Ngo and Murphy2006). Although all samples of T. nigrescens were obtained south of the Baja peninsula, the extent of the historical geological disturbances would vary for individual species depending on their original distribution and the effect of geological events on their eventual dispersal. Thus, a study of the geological and ecological history of the natural range of T. nigrescens would clarify this possibility. This was, however, beyond the scope of the study. Alternatively, the T. nigrescens could have spread north and southwards from the zone around Oaxaca, founding the two different clades. This hypothesis is supported by the haplotype differentiation observed.
The observation of significant association between geographical distances and genetic distances further supports this hypothesis. Geographical obstacles seemed to increase this association, implying that active dispersal was limited by such factors. Taken together, the results of the isolation by distance analysis underscore the importance of artificial population subdivisions and depth of sampling coverage in defining the genetic diversity of introduced populations.
There is a possibility of population fragmentation, as suggested by a high frequency of unique haplotypes only shared among a few individuals within specific populations. Local eco-geographic features may also be responsible for the variation and lineage divisions observed in this study. It is recognized that the adaptability of an invader, rather than sheer genetic diversity or numbers, are responsible for the establishment of a species in the new range (Lloyd et al., Reference Lloyd, Hufbauer, Jackson, Nissen and Norton2005; Zayed et al., Reference Zayed, Constantin and Packer2007; Bacigalupe, Reference Bacigalupe2009). However, adaptability is also a factor of the diversity of the gene pool introduced, upon which selection occurs to produce an adaptable population (Roderick & Navajas, Reference Roderick and Navajas2003). The chances of establishment and range expansion, therefore, may be higher with an increased genetic base (Kolar & Lodge, Reference Kolar and Lodge2001; Allendorf & Lunquist, Reference Allendorf and Lundquist2003; Phillips et al., Reference Phillips, Baird, Iline, McNeill, Proffitt, Goldson and Kean2008). Adequate sampling for a greater genetic pool is important in recovery of natural enemies (Omwega & Overholt, Reference Omwega and Overholt1996; Phillips et al., Reference Phillips, Baird, Iline, McNeill, Proffitt, Goldson and Kean2008) and should guide future sampling plans for a more genetically inclusive approach to the biological control of the LGB.
Genetic diversity in laboratory samples was generally lower than that of field samples. Similarly, both the Malawi and NRI populations showed a much lower level of mitochondrial genetic diversity than their parental Benin population. This suggests the disproportionate contribution of a few related females to the genetic pool of these populations, population expansion from a few haplotypes or problems with genetic bottlenecks during recovery, quarantine or rearing (Lloyd et al., Reference Lloyd, Hufbauer, Jackson, Nissen and Norton2005; Zayed et al., Reference Zayed, Constantin and Packer2007). The demographic history of the laboratory populations used in this study is not certain on a few important events. For instance, insects isolated from Mexico were studied in the IITA Benin laboratory, but their possible genetic contribution to the Benin populations is not clear (W. Meikle, USDA, Weslaco, TX, USA, personal communication). In Kenya, mite infestation and possible mycoplasma infections were reported leading to culling of a part of the colony, though the size of the eventual founding populations are not documented nor is the extent of this possible bottleneck known (R. Hodges, NRI, Greenwich, UK; P. Likhayo and G. Kibata, KARI, Nairobi, Kenya, personal communications). Recent population genetic shifts and admixture events could also have contributed significantly to the genetics of the populations we used and eventually to the performance of those released for biological control. These hypotheses may be tested more efficiently in recent population time scales using hypervariable multilocus markers, such as microsatellites and gene expression profiles.
The variability of establishment and success of T. nigrescens in the control of the larger grain borer in Africa is possibly associated with the genetic variation within the populations used. Here, we show the existence of two clades of the predator dominant at the two areas of earlier field recovery of the species and sympatric in southern Mexico. This difference still is largely maintained in the laboratory samples from the two control programmes, even after many cycles of rearing. The association of this diversity with definite adaptability and potential survival in different regions in Africa could be tested by ecological experiments and gene expression assays, especially of stress related factors. It would be interesting to explore the full range of genetic diversity of this predator in a new effort to the biological control of the LGB in Africa.
Acknowledgements
We are grateful to the following colleagues for kindly donating Teretrius nigrescens samples used in this study: Richard Hodges (NRI, Greenwich, UK), Paddy Likhayo and Josephine Songa (KARI, Kiboko, Kenya), Rhoderick Ndawala (NARO, Malawi), Cyprian Atcha (IITA, Abomey Calavi, Benin), David Bergvinson (CIMMYT), Lucia Orantes and Alfredo Rueda (Zamorano University, Tegucigalpa, Honduras). We thank Jeffrey Grubber (Russel Laboratories, WI, USA) for his kind gift of T. americanus samples; Pierpaolo Vienna (Lido, Italy) and Riaan Stals for identifying samples. We also thank Sarine Adhiambo, Ogola Gerphas and Robert Mutweti for technical assistance. We are indebted to Richard Hodges, William Meikle, FLO Nang'ayo, P. Likhyayo and Garry Hill for important consultations and advice and to Anne-Emmanuelle Felix for suggestions on the manuscript. This study was supported Kirkhouse Trust, UK, through a PhD Fellowship to BOA. Field sampling in Honduras was funded by the Swiss Development Cooperation.