


Introduction
Mineral–organic interactions are of paramount relevance in the origins of life (Cleaves et al. Reference Cleaves, Michalkova Scott, Hill, Leszczynski, Sahai and Hazen2012). The interaction of organics with different minerals is a source of chemical complexity (Colin-Garcia et al. Reference Colin-Garcia, Heredia, Negron-Mendoza and Ramos-Bernal2012). Pyrite (FeS2) is one of the most common sulphide minerals on the Earth. Geologic records indicate that banded iron formations dating from 3.9 Ga, representing probably the earliest sedimentary rocks, contain pyrite (Hazen et al. Reference Hazen, Papineau, Bleeker, Downs, Ferry, McCoy, Sverjensky and Yang2008). On Mars, the assemblage of goethite–jarosite–opal–clay observed was probably derived from the oxidation of iron-containing minerals (Burns & Fisher Reference Burns and Fisher1990; Zolotov & Shock Reference Zolotov and Shock2005).
Specifically, the high-pressure reaction between FeS and H2S (Rickard Reference Rickard2015) is identified as a primitive path for fixing carbon, through a chain of redox processes (Russell et al. Reference Russell, Daniel, Hall and Sherringham1994). In this chemical process, the elemental composition and crystalline structure of pyrite mineral (FeS2) is of main significance (46.6% of Fe and 53.4% of S). For instance, the crystal system (cubic) of pyrite opens the way for coordination of organics in its sulphur-rich surface (Betejtin Reference Betejtin1970). Pyrite is involved in CO2 fixation in the presence of H2S and was a potential catalyst in the primitive Earth towards synthesis of more complex molecules associated with the origins of living matter (Tributsch et al. Reference Tributsch, Fiechter, Jokisch, Rojas-Chapana and Ellmer2003). Therefore, unravelling the potential chemical processes involved in small organics condensation and complexation will lead to a further understanding of the molecular mechanisms of prebiotic molecular evolution and how molecular and physicochemical inorganic aspects contributed to the origin of life (Cleaves et al. Reference Cleaves, Lazcano, Ledesma Mateos, Negrón-Mendoza, Peretó and Silva2014).
Glycine (NH3 +CH2COO − ) is a low-molecular-weight and structurally simple molecule (75 g mol−1) that might easily be synthesized in comet impact simulations (Goldman et al. Reference Goldman, Reed, Fried, William Kuo and Maiti2010) and is found in other interstellar bodies, such as meteorites (Glavin & Bada Reference Glavin and Bada2001). Moreover, those interstellar bodies contain, among other solids, pyrite mineral (Moriarty et al. Reference Moriarty, Hibbitts, Lisse, Dyar, Harlow, Ebel and Peale2010). In primitive Earth studies, special emphasis must be paid to inorganics interacting with organics (Degens Reference Degens1989) under different environmental conditions. Moreover, terrestrial planets such as Mars (Patel et al. Reference Patel, Bérces, Kerékgyárto, Rontó, Lammer and Zarnecki2004) and the primitive Earth, a lack of an ozone layer in the first stages was possible, having a strong ultraviolet (UV) irradiation as a potential driving force for chemical activation (Cockell Reference Cockell2000; Horneck Reference Horneck2007).
Many attempts have been made through different characterization methods in order to understand the molecular changes in glycine monomer, interacting with minerals of astrobiological relevance (Pilling et al. Reference Pilling, Mendes, Bordalo, Guaman, Ponciano and da Silveira2013) supporting the Oparin–Haldane hypothesis of chemical evolution (Okihana & Ponnamperuma Reference Okihana and Ponnamperuma1982). To understand the mechanism of chemical evolution, infrared (IR) experimental methods and theoretical characterization studies are of main relevance (Baran & Ratajczak Reference Baran and Ratajczak2005; Contreras-Torres & Basiuk Reference Contreras-Torres and Basiuk2005). Here, we perform an experimental and theoretical study of the interaction of glycine with the mineral pyrite when the system is exposed to UV radiation. The aim was to figure out the molecular changes promoted by the mineral in the self-assembly of the amino acid as an example of a process of chemical evolution.
Materials and methods
Pyrite (Figs 1 and 2) was obtained from the collection of the Institute of Geology, UNAM and Rominmex S.A. de C.V. (Atlixco, Puebla, México), and glycine was obtained from Sigma-Aldrich (CAS-56-40-6). The pyrite sample was ground in an agate mortar and subsequently mixed with glycine solution (0.01 M) and left in the overnight up to dehydration. Solid solutions were performed with 0.2 g of mineral and put to interact with 0.5 ml of glycine solution. To maximize reproducibility, the same initial stock of ground pyrite was used for all the experimental procedures after it had been cleaned thoroughly in a series of acetone–distilled water–acetone–distilled water rinses. At least two repetitions per sample were performed to confirm the presence and consistency of bands. The precise frequencies of the different chemical groups were determined by cross-referencing against previously reported group-specific frequencies (Table 1).

Fig. 1. Pyrite mineral showing its distinctive crystal habits (a) and a more detailed image (b) showing no furrows or sheets in the structure.
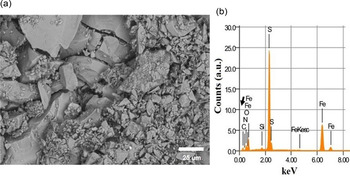
Fig. 2. SEM microphotograph of a characteristic sample of pyrite with self-assembled glycine (a) and the corresponding EDS analysis showing the presence of carbon (black arrow in b).
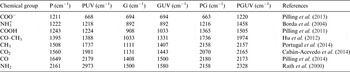
Table 1. Assignment of detected attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) frequencies of the different samples, pyrite (P), pyrite UV (PUV), glycine (G), glycine UV (GUV), pyrite–glycine (PG) and glycine–pyrite UV (PGUV). The assignment is based on the previously reported data
Scanning electron microscopy (SEM) and energy-dispersive spectroscopy (EDS)
The SEM analysis was performed at the Laboratory of Analysis and Characterization (Academic Division of Engineering and Architecture UJAT-DAIA). The samples were analysed by SEM (Fig. 2(a)). For morphological analysis, the samples were mounted on conductive carbon tape in an aluminium sample holder. The samples were observed on the JEOL JSM-6010LA SEM (Japan), at 20 kV accelerating voltage under high vacuum. For the semi-quantitative and element composition energy dispersive detector (EDS) coupled to the SEM was employed (Fig. 2(b)). For processing the images, the InTouchScope™ software was used.
UV activation
Activation by UV light was performed with a mercury lamp (Pen ray UVP, UVC Light Sources, with fixed parameters, Cambridge, UK) with a primary energy being 254 nm (5.5 W m−2), the irradiation period was 1 up to 24 h approximately, with intervals for manual mixing of the solid mixture. Analysis was performed over the different samples as follows (Table 1): pyrite (P), pyrite UV (PUV), glycine (G), glycine UV (GUV), pyrite–glycine (PG) and glycine–pyrite UV (PGUV).
ATR-FTIR spectroscopy
IR spectra were obtained by using an ATR-FTIR equipment (Spectrum 100 FTIR Spectrometer PerkinElmer, Waltham, Massachusetts) from 4000 to 650 cm−1 scan (over four scans). Analysis of the ATR-FTIR-obtained data, showed the typical bands for glycine and pyrite (Fig. 3(a) and (c)). The penetration depth of the laser beam in ATR-FTIR is about 2 µm making this method a proper technique for the analysis of surfaces (Mourant et al. Reference Mourant, Yamada, Carpenter, Dominique and Freyer2003; Heredia et al. Reference Heredia, van der Strate, Delgadillo, Basiuk and Vrieling2008). To perform a more detailed analysis, we used the method of the second derivative to determine most of the band positions (Fig. 3(b)). The second-derivative graphs have sharper peaks (bands) at the same frequency values, making their potential use of main significance during the analysis of fine details of IR spectra (for comparison see Fig. 3(a)–(c)).
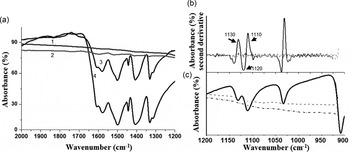
Fig. 3. (a) Characteristic ATR-FTIR spectra of glycine (1), glycine interacting with pyrite (2), UV-irradiated glycine interacting with pyrite (3), and (4) UV-irradiated glycine (see also Table 1 for the sampling strategy). The most clear-cut bands of the α-glycine indicated and were assigned to previously reported data (Table 1). (b) The second derivative method was used to confirm the presence of the α and γ glycine polymorphs, which are not seen directly in the FTIR (c).
Second-derivative method
Some steps are performed to investigate complex FTIR spectra, especially the overlapped bands. In order to gather all potential chemical and structurally useful information on the glycine and pyrite, the second-derivative step is used. This method involves band resolution enhancement. Here, the overlapping of bands are analysed by spectra differentiation. In general, all data points of the second derivative decrease in intensity in each derivative, but the relative resolutions are increased. The analysis of the second derivative and deconvolution of IR spectra were applied first in the early 1980s. Each method presented its own advantages for the exploration of the local changes in the spectra, although the theory and application of derivatives proved to be simpler. For some authors (Barth & Haris Reference Barth and Haris2009), this advantage consists of a second derivative that is straightforward and requires no a priori knowledge of band parameters. An additional application of these procedures is that the solute concentration in the Beer–Lambert law (1) lc or in its second derivative (2) is not affected (Gallignani et al. Reference Gallignani, Rondón, Ovalles and Brunetto2014).
 $$A(\tilde v) = \alpha (\tilde v)lc,$$
$$A(\tilde v) = \alpha (\tilde v)lc,$$
 $$\displaystyle{{{d^2}A(\tilde v)} \over {d{{\tilde v}^2}}} = \displaystyle{{{d^2}\alpha (\tilde v)} \over {d{{\tilde v}^2}}}lc.$$
$$\displaystyle{{{d^2}A(\tilde v)} \over {d{{\tilde v}^2}}} = \displaystyle{{{d^2}\alpha (\tilde v)} \over {d{{\tilde v}^2}}}lc.$$
Here, A is the wavenumber,
 $\bar v$
is the dependent absorbance, α is the wavenumber-dependent absorption coefficient, l is the section thickness, and c is the concentration.
$\bar v$
is the dependent absorbance, α is the wavenumber-dependent absorption coefficient, l is the section thickness, and c is the concentration.
Computational methods
Molecular modelling MM+ was performed starting with molecular mechanics and, afterwards, by semi-empirical quantum mechanics methods as implemented in the HyperChem program Version 8.0 (HyperCube, Canada). Geometry optimizations were used to obtain the coordinates of molecular structures at potential energy minima (Colin-Garcia et al. Reference Colin-Garcia, Heredia, Negron-Mendoza, Ortega, Pi and Ramos-Bernal2014). Full geometry optimization of the organic and inorganic units were performed using the HyperChem settings for the MM+ force field, the Polak–Ribiere conjugate gradient algorithm, and a root-mean-square gradient (RMS) of 0.0001 kcal Å−1 mol−1. Molecular dynamics relaxation of the optimized structures was employed to look for possible local minima (step size of 0.001 ps, constant simulation temperature of 300 K and ca. 12 ps each model). The glycine model was obtained from the database in the software and edited to obtain the zwitterion and the geometrically optimized glycine structure to determine the most stable conformation (MM+ method). Glycine was chosen to solely interact with the pyrite unit with no water to simulate the simplest model of chemical activity.
The PM3 and AM1 semi-empirical methods
The optimized geometries that were obtained by the MM+ molecular mechanics method were further optimized with the PM3 and AM1 semi-empirical methods. In semi-empirical calculations, full geometry optimizations were performed on the restricted Hartree–Fock (RHF) basis, Polak–Ribiere conjugate gradient algorithm, and total RMS gradient of 0.0001 kcal Å−1 mol−1, as is suggested in the HyperChem manual, and mentioned previously as the most suitable methods for long biomolecules (Heredia et al. Reference Heredia, van der Strate, Delgadillo, Basiuk and Vrieling2008).
Results and discussion
Scanning electron micrographs, optical microscope images and EDS results (Figs 2 and 3) show the morphological and chemical features of the pyrite and glycine samples. The EDS elemental relative content expressed as carbon/nitrogen (C/N) ratio shows no organic content after in-pyrite samples (Table 2). On the contrary, the C/N ratio content in the self-assembled glycine on pyrite 6.7%, whereas it is 18.64% in the pyrite with glycine irradiated with UV. This increment is probably due to the fact that the pyrite crystal stabilizes the glycine molecule on its surface (Table 2). The crystal in PGUV shows a remarkable content of glycine, as can be seen from the C/N ratio in Table 2.
Table 2. Elemental composition (in %) of the pyrite samples with and without glycine

n.a. not detected, glycine exposed to UV was not measured.
The ATR-FTIR spectra revealed all the bands from glycine and pyrite (Fig. 3(a)) with a main composition of α-glycine (Fig. 3(b) and 4). After GUV irradiation, a change in intensities in the bands was noticeable (Fig. 4). The changes in the samples after irradiation appeared to originate from the UV activation of pyrite (Figs. 3(a) and 4) (Zhang et al. Reference Zhang, Borda, Schoonen and Strongin2003). Already after UV irradiation, the bands were recognized as chemical changes, since their areas are influenced by UV activation (Fig. 3(b)). According to other theoretical studies, an intricate interaction among water molecules, mineral surfaces and glycine might be responsible for this behaviour (Folliet et al. Reference Folliet, Gervais, Costa, Laurent, Babonneau, Stievano, Lambert and Tielens2013).
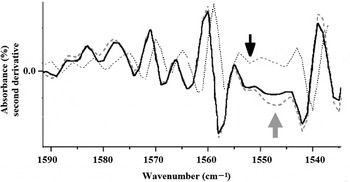
Fig. 4. Second-derivative ATR-FTIR of glycine (continuous line), UV treated glycine (dotted line) and GUV treated (discontinuous line) assembled in pyrite suggests a trend to transform glycine from α to γ polymorph (grey arrow). No amide bands (1560 and 1550 cm−1) are present (black arrow).
The ATR-FTIR spectra were collected from ground pyrite samples and further analysed to study the assembly and subsequent polymorph formation of glycine.
The spectra obtained displayed the most important fingerprints of glycine. The bands at ~1131 cm−1 were assigned as rocking vibrations of NH+ 3 groups, and were clearly identified over all of the samples. For glycine irradiated in the UV, however, no clear changes were noticed, which is consistent with the chemical stability of glycine against the exposure to ionizing radiation; this is in agreement with earlier observations (Pernet et al. Reference Pernet, Pilmé, Pauzat, Ellinger, Sirotti, Silly, Parent and Laffon2013). Other fingerprint bands that were absent in all spectra were those of amide I at ~1650 cm−1 and amide II at ~1550 cm−1 (black arrow in Fig. 4); these bands refer specifically to the peptide bonds. This shows that no dimerization of glycine units occurred.
The second derivative analysis of the ATR-FTIR spectra shows that the effects of UV irradiation are in bands near 1060 cm −1 (Fig. 4). In this region, there is a change in the band of glycine irradiated with UV, compared with the control sample. This result reveals a possible increase in glycine γ polymorph due to the importance of the activation generated by UV irradiation (dotted line, grey arrow in Fig. 4).
Computation simulations
To obtain insight into the viable molecular interactions between chemical groups from glycine to pyrite or glycine self-assembly, we have performed MM+ simulations. The combination of geometry optimization and molecular dynamics for establishing relaxation of the optimized structures, is useful to determine the local energy minima for every tested model (see Table 3) and represented graphically (see profile of heats of formation in Fig. 5). The most stable structure that was obtained was the model of −1832.52 kcal mol−1 corresponding to a semi-empirical method AM1 from 2GP unit (Table 3 cell in grey).
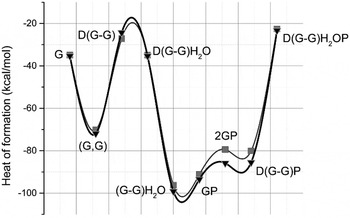
Fig. 5. Heats of formation from PM3 (![]() ) and AM1 (▼) semi-empirical methods. Both semi-empirical methods, AM1 and PM3 result in favoured heats of formation for (G–G)H2O, GP, 2GP and D(G–G)P molecules. So, it is important to note that using the method AM1, more heat of formation is shown, specifically for the 2GP molecule relative to the PM3 method.
) and AM1 (▼) semi-empirical methods. Both semi-empirical methods, AM1 and PM3 result in favoured heats of formation for (G–G)H2O, GP, 2GP and D(G–G)P molecules. So, it is important to note that using the method AM1, more heat of formation is shown, specifically for the 2GP molecule relative to the PM3 method.
Table 3. Energies obtained from MM+ and semi-empirical procedures and the corresponding heats of formation from semi-empirical methods
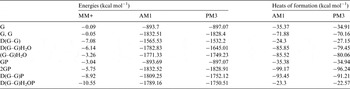
G, one unit of glycine; G,G, two units of zwitterionic glycine; D(G–G), glycine dipeptide; D(G–G)H2O, glycine dipeptide with one molecule of water; (G–G)H2O, two unites of zwitterionic glycine with one molecule of water; GP, one unit of glycine and one unit of pyrite; 2GP, two units of zwitterionic glycine and one unit of pyrite; D(G–G)P, glycine dipeptide in one unit of pyrite and D(G–G)H2OP, glycine dipeptide and one unit of pyrite with one molecule of water.
In this conformation, it was possible for glycine units to approach the Fe-rich regions (Fig. 6).
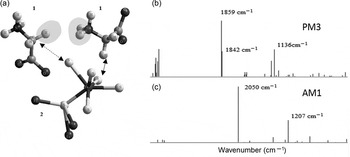
Fig. 6. Glycine molecules (1) interacting with a pyrite unit (2). (a) Two molecules of glycine interacting with the pyrite unit at the region close to the Fe–S bond. The distance between the chemical groups –CH2 from glycine to S is 3.24 Å (arrow with discontinuous line) and 3.04 Å for the –CH2 to S in another glycine molecule (arrow with solid line). The shortest distance between the two glycine molecules is 4.5 Å (shadowed regions). (b) PM3 and (c) AM1 IR simulations show clear-cut differences with the experimental results.
At the molecular scale, pyrite might interact with organics such as glycine through its thiol or iron surface sites rising to chemical reactivity (Bebié & Schoonen Reference Bebié and Schoonen2000).
In the MM+ method, the glycine zwitterion was optimized. This conformation was, in addition, used for the semi-empirical methods. For this, two glycine molecules were put in the workspace to approach the regions of the unit of pyrite (Fig. 6).
The preliminary results have significance in prebiotic chemistry, specifically about the progressive increasing transformation from α to the γ glycine polymorph (a change in molecular packing) due to a non-biological mechanism. At the molecular scale, nonetheless, the possible glycine dimerization must be discussed in depth, since published works report the overwhelming stability of glycine under UV radiation (Okihana & Ponnamperuma Reference Okihana and Ponnamperuma1982) and no polymerization under Co-60 radiation (Draganić et al. Reference Draganić, Niketić and Vujošević1985). The role of water molecules in the heat of formation and the subsequent molecular transformation of glycine is of main importance. Specifically, the heat of formation in the AM1 and PM3 computer molecular simulations without water molecules favours the model in which two glycine molecules interact with pyrite (from Table 3, the difference in heats of formation between 2GP and D(G–G)P in AM1 corresponds to 5.76% and PM3 to 5.22%) over the glycine–glycine dimers. These results confirm that the lowest minima of heats of formation in the simulations favour the interaction of separated glycine units with pyrite in the absence of water molecules. Our ATR-FTIR further substantiates such observation, showing no peptide bonds (Figs. 3 and 4) under experimental conditions having water solutions (Fig. 3).
Simulated IR bands (Fig. 6(b) and (c)) show a divergence with the experimental ATR-FTIR that might originate in the lack of water molecules in the simulations. Besides, the computational model consists of single molecules, causing the local minima to produce just clear specific bands with no more complex molecular interactions. Although FTIR bands obtained here are not accurate, PM3 requires only a minor horizontal correction compared with the experimental results, and as such, might be used to explore the theoretical formation of the IR bands in future computational modelling efforts.
Conclusions
Here, the UV radiation was used as a driving force together with a mineral surface to impact the self-assembly of a specific amino acid polymorph. The mechanism of molecular interaction of glycine and pyrite is a matter of further investigation, although our computational simulations suggest a potential role of the complexation of sulphur-rich regions in the pyrite model. These preliminary results should be examined, thoroughly and additionally, under other radiation sources such as Co-60, and other physicochemical conditions (different pH, temperatures and pressures) that will lead to understand the prebiotic conditions of astrobiological importance capable of yielding amino acids polymorphs and oligomers. These new conditions will be studied at the molecular level by means of molecular simulations to understand the chemical complexity of prebiotic routes of complexation of molecules, important for living systems such as glycine. Molecular simulations of free radicals in heterogeneous systems (pyrite–glycine–water) and the use of γ radiation are of main relevance for these studies and are part of our current research.
Acknowledgements
ACL thanks Universidad Juárez Autónoma de Tabasco for the grant, and Dr Angel Meraz for supporting the Verano Científico program at ICN-UNAM under the advice of Dr Heredia. ACL also thanks co-workers of the Lab. of Chemical Evolution at Instituto de Ciencias Nucleares, UNAM for all the support. We thank Luciano Díaz González and Martín Cruz Villafañe for their technical support. We thank a lot the reviewers for the important improvements in our manuscript.









