



Introduction
The time between seed dispersal and completion of germination can be short or long, thus, seeds have evolved a series of strategies – for example, dormancy – to regulate this interval (Footitt and Finch-Savage, Reference Footitt, Finch-Savage and Clemens2017). These strategies prevent responses to short-lived, out of season environmental changes, making it possible for plants to overcome periods that are unfavourable for seedling establishment (Bentsink and Koornneef, Reference Bentsink and Koornneef2008). Primary dormancy is the innate dormancy possessed by seeds when they are dispersed from the mother plant (Benech-Arnold et al., Reference Benech-Arnold, Sanchez, Forcella, Betina and Ghersa2000). Freshly matured water-permeable seeds can exhibit a strong primary dormancy trait that is determined by the environment during seed development and physiological characteristics of the mother plant (Bewley and Black, Reference Bewley and Black1994). These seeds do not germinate in light or darkness over a range of temperatures (Baskin and Baskin, Reference Baskin and Baskin1989). After the seed has left the mother plant and primary dormancy has been released in response to seasonal environmental changes, non-germinating seeds may enter secondary dormancy (Baskin and Baskin, Reference Baskin and Baskin2014). This entrance into secondary dormancy can be induced in seeds with non-deep physiological dormancy, if the right set of external signals to germinate are absent (Hilhorst, Reference Hilhorst1998). Induction and relief of secondary dormancy can occur during successive seasons, leading to an annual dormancy cycle in the seed bank (Hilhorst, Reference Hilhorst1998). Subtle differences in this behaviour could result in local adaptation and ecotypic differences (Finch-Savage and Footitt, Reference Finch-Savage and Footitt2017).
Wild plant species, especially annual ones which have seeds that persist in a soil seed bank, are better models for physiological studies of seed dormancy than domesticated crops (Hilhorst and Toorop, Reference Hilhorst and Toorop1997). Capsella bursa-pastoris (L.) Medik. [shepherd's purse (Brassicaceae)] is an annual or biennial herb that can form a persistent seed bank in the soil (Hurka and Haase, Reference Hurka and Haase1982). The species displays primary seed dormancy (non-deep physiological) as well as an annual cycle of secondary dormancy/non-dormancy (Neuffer and Hurka, Reference Neuffer and Hurka1986; Baskin and Baskin, Reference Baskin and Baskin1989; Toorop et al., Reference Toorop, Campos Cuerva, Begg, Locardi, Squire and Iannetta2012). It has a worldwide distribution, with the exception of extremely dry tropical environments (Neuffer and Eschner, Reference Neuffer and Eschner1995; Hurka and Neuffer, Reference Hurka and Neuffer1997), and has become one of the five most widely distributed flowering plants on our planet, preferring disturbed, ‘man-made’ habitats, like the margins of agricultural fields (Hintz et al., Reference Hintz, Bartholmes, Nutt, Ziermann, Hameister, Neuffer and Theissen2006). C. bursa-pastoris has a very small phylogenetic distance to the academic model species Arabidopsis thaliana. However, in comparison, the latter appears to be uncompetitive and is actually relatively rare in the wild (Hintz et al., Reference Hintz, Bartholmes, Nutt, Ziermann, Hameister, Neuffer and Theissen2006). Due to its wild nature, cosmopolitan distribution, and complex dormancy traits, we selected C. bursa-pastoris as a model species for our study on the possible (epi)genetic mechanisms involved in secondary seed dormancy induction, including differences in secondary seed dormancy depth between ecotypes.
The first aim of this study was to test the hypothesis that exposing C. bursa-pastoris seeds to the histone deacetylase (HDAC) inhibitors trichostatin A (TSA) and valproic acid during the induction of secondary dormancy prevents the seeds from entering this dormancy state. In addition, we reasoned that exposure to these compounds would lead to a higher germination speed compared with control conditions.
Acetylation of lysines at the N-terminal tails of histones removes their positive charge, altering the histone–histone and DNA–histone interaction and changing the accessibility of DNA to the chromatin-binding proteins (Turner, Reference Turner2000). It is associated with an open chromatin state (euchromatin) and activation of gene transcription, while hypo-acetylation is related to chromatin condensation (heterochromatin) and consequently gene silencing (Wójcikowska et al., Reference Wójcikowska, Botor, Morończyk, Wójcik, Nodzyński, Karcz and Gaj2018). Acetylation of lysine residues is a reversible process and there are two families of enzymes involved in the acetylation state of histones: HISTONE ACETYLTRANSFERASES (HATs) and HISTONE DEACETYLASES (HDACs). The interplay between these enzymes is implicated in the control of many biological processes, such as embryo development, dormancy, germination and morphogenesis (Cadman et al., Reference Cadman, Toorop, Hilhorst and Finch-Savage2006; Wang et al., Reference Wang, Cao, Chen and Liu2014). The best-studied HDACs belong to RPD3 Class 1, which includes HDA6, HDA19, HDA7, HDA9 and the pseudogenes HDA10 and HDA17 (Van Zanten et al., Reference Van Zanten, Zöll, Wang, Philipp, Carles, Li, Kornet, Liu and Soppe2014).
HDAC inhibitors shift a reversible histone acetylation/deacetylation state towards a condition of histone hyper-acetylation (Koeller et al., Reference Koeller, Haggarty, Perkins, Leykin, Wong, Kao and Schreiber2003). HDACs can be pharmacologically inhibited by TSA (Yoshida et al., Reference Yoshida, Horinouchi and Beppu1995) and other compounds, such as valproic acid (Göttlicher et al., Reference Göttlicher, Minucci, Zhu, Krämer, Schimpf, Giavara, Sleeman, Lo Coco, Nervi, Pelicci and Heinzel2001), suberoylanilide hydroxamic acid (SAHA) (Richon et al., Reference Richon, Emiliani, Verdin, Webb, Breslow, Rifkind and Marks1998) and anacardic acid (Cui et al., Reference Cui, Miao, Furuya, Fan, Li, Rathod, Su and Cui2008). They can, for example, be used for determining the role that histone acetylation plays in chromatin structure and remodelling and for finding genes regulated by histone acetylation (Yoshida et al., Reference Yoshida, Horinouchi and Beppu1995; Wójcikowska et al., Reference Wójcikowska, Botor, Morończyk, Wójcik, Nodzyński, Karcz and Gaj2018).
Valproic acid is known to be an inhibitor of histone deacetylases capable of reducing tumour growth and metastasis formation in animals (Göttlicher et al., Reference Göttlicher, Minucci, Zhu, Krämer, Schimpf, Giavara, Sleeman, Lo Coco, Nervi, Pelicci and Heinzel2001). It is used as an anti-epileptic drug and to treat bipolar disorder (Phiel et al., Reference Phiel, Zhang, Huang, Guenther, Lazar and Klein2001). It inhibits the catalytic activity of Class I HDACs in mammals but also induces the proteasomal degradation of HDAC2, in contrast to TSA (Krämer et al., Reference Krämer, Zhu, Ostendorff, Golebiewski, Tiefenbach, Peters, Brill, Groner, Bach, Heinzel and Göttlicher2003). It has been shown to down-regulate the expression of proteins essential for chromatin maintenance in animal cells such as the STRUCTURAL MAINTENANCE OF CHROMATIN 1 TO 6, DNA METHYL TRANSFERASE-1 and HETEROCHROMATIN PROTEIN-1 (Chateauvieux et al., Reference Chateauvieux, Morceau, Dicato and Diederich2010). It is also capable of inducing mono-, di- or tri- methylation of histone H3 at lysine 9 (H3K4) (Chateauvieux et al., Reference Chateauvieux, Morceau, Dicato and Diederich2010). However, no previous studies in plants have reported the effects of this HDAC inhibitor.
The second aim of this study was to test the hypothesis that genes implicated in epigenetic regulation processes would be differentially expressed between C. bursa-pastoris accessions previously shown to contrast in secondary dormancy depth (Gomez-Cabellos et al., Reference Gomez-Cabellos, Toorop, Cañal, Iannetta, Fernández-Pascual, Pritchard and Visscher2021). In particular, histone acetylases were expected to be down-regulated in a relatively deep-dormant accession compared with a non-deep dormant one. In addition, differential gene expression induced by valproic acid exposure was studied in the non-deep accession to identify gene regulation by histone deacetylases.
While (genetic) research on the processes underlying primary dormancy is extensive, the molecular mechanisms controlling secondary seed dormancy are still poorly understood (Cadman et al., Reference Cadman, Toorop, Hilhorst and Finch-Savage2006; Finch-Savage and Leubner-Metzger, Reference Finch-Savage and Leubner-Metzger2006; Holdsworth et al., Reference Holdsworth, Bentsink and Soppe2008b; Footitt et al., Reference Footitt, Huang, Clay, Mead and Finch-Savage2013; Ibarra et al., Reference Ibarra, Tognacca, Dave, Graham, Sánchez and Botto2016; Coughlan et al., Reference Coughlan, Saha and Donohue2017; Buijs, Reference Buijs2020; Laspina et al., Reference Laspina, Batlla and Benech-Arnold2020; Hourston et al., Reference Hourston, Steinbrecher, Chandler, Pérez, Dietrich, Turečková, Tarkowská, Strnad, Weltmeier, Meinhard, Fischer, Fiedler-Wiechers, Ignatz and Leubner-Metzger2022). According to the hormonal balance model (Finkelstein et al., Reference Finkelstein, Reeves, Ariizumi and Steber2008), the ratio of abscisic acid (ABA) (promoting primary dormancy induction and dormancy maintenance) and gibberellic acid (GA) (promoting release of dormancy and germination) is the main determinant of the level of dormancy and seed germination (Finkelstein et al., Reference Finkelstein, Reeves, Ariizumi and Steber2008; Buijs et al., Reference Buijs, Vogelzang, Nijveen and Bentsink2020). Environmental signals regulate the balance between ABA and GA levels, whose changes seem to be caused by alterations in the expression patterns of their metabolic genes and positive and negative regulators of both hormones (Finkelstein et al., Reference Finkelstein, Reeves, Ariizumi and Steber2008; Tuan et al., Reference Tuan, Kumar, Rehal, Toora and Ayele2018). However, subsequent ABA and GA signalling and sensitivity are the more likely regulators of dormancy than the absolute level of these hormones (Ali-Rachedi et al., Reference Ali-Rachedi, Bouinot, Wagner, Bonnet, Sotta, Grappin and Jullien2004; Ibarra et al., Reference Ibarra, Tognacca, Dave, Graham, Sánchez and Botto2016; Laspina et al., Reference Laspina, Batlla and Benech-Arnold2020). In relation to secondary seed dormancy, Cadman et al. (Reference Cadman, Toorop, Hilhorst and Finch-Savage2006) suggested that its control mechanisms differ from primary dormancy. Ibarra et al. (Reference Ibarra, Tognacca, Dave, Graham, Sánchez and Botto2016) demonstrated that the entrance into secondary seed dormancy reduced the content and sensitivity to GA, but not the content and sensitivity to ABA. However, Laspina et al. (Reference Laspina, Batlla and Benech-Arnold2020) and Footitt et al. (Reference Footitt, Walley, Lynn, Hambidge, Penfield and Finch-Savage2020) did find that ABA sensitivity plays an important role in secondary dormancy induction/dormancy cycling.
In addition to ABA and GA, there are other phytohormones with implications in the regulation of primary seed dormancy and germination. Brassinosteroids (BRs) are plant steroid hormones involved in stem elongation and leaf unfurling that promote germination. BR mutants are hypersensitive to inhibition of germination by ABA in comparison with wild-type seeds (Finkelstein et al., Reference Finkelstein, Reeves, Ariizumi and Steber2008). BRs were discovered to be implicated in the promotion of seed germination by modulating ABA signalling with a negative feedback loop (Xi et al., Reference Xi, Liu, Hou and Yu2010). BR molecules may promote seed germination by enhancing embryo growth potential in a gibberellin-dependent manner (Leubner-Metzger, Reference Leubner-Metzger2001). However, the detailed mechanisms underlying the BR and GA crosstalk are still not well understood (Shu et al., Reference Shu, Liu, Xie and He2016).
Ethylene stimulates germination and breaks primary dormancy establishment in seeds by antagonizing the ABA pathway (Linkies and Leubner-Metzger, Reference Linkies and Leubner-Metzger2012; Corbineau et al., Reference Corbineau, Xia, Bailly and El-Maarouf-Bouteau2014). Seeds of ethylene resistant 1 receptor (etr1) mutants display increased dormancy and their germination is ABA hypersensitive (Beaudoin et al., Reference Beaudoin, Serizet, Gosti and Giraudat2000). Mutations in ENHANCED RESPONSE TO ABA 3 (ERA3)/ETHYLENE INSENSITIVE 2 (EIN2) genes lead to an overaccumulation of ABA and increased seed dormancy, suggesting that ERA3/EIN2 is a negative regulator of its synthesis (Ghassemian et al., Reference Ghassemian, Nambara, Cutler, Kawaide, Kamiya and McCourt2000). The final step in the biosynthesis of ethylene during seed germination is regulated by 1-AMINOCYCLOPROPANE-1-CARBOXYLIC ACID OXIDASE (ACO) and is involved in counteracting the inhibitory effects of ABA (Linkies et al., Reference Linkies, Muller, Morris, Tureckova, Wenk, Cadman, Corbineau, Strnad, Lynn, Finch-Savage and Leubner-Metzger2009). On the other hand, whether ethylene affects GA biosynthesis and signalling in relation to seed dormancy and germination is still poorly understood (Shu et al., Reference Shu, Liu, Xie and He2016).
Other hormones that are known for inhibiting seed germination and promoting primary seed dormancy are auxins (Holdsworth et al., Reference Holdsworth, Finch-Savage, Grappin and Job2008a; Liu et al., Reference Liu, Zhang, Zhao, Feng, Li, Yang, Luan, Li and He2013). Auxins are required for the maintenance of ABSCISIC ACID INSENSTIVE 3 (ABI3) expression by recruiting AUXIN RESPONSE FACTOR 10 (ARF10) and ARF16 (Liu et al., Reference Liu, Zhang, Zhao, Feng, Li, Yang, Luan, Li and He2013). ABI3 is recognized as a major regulator of primary seed dormancy and acts upstream of ABI5 to execute ABA-responsive seed germination inhibition (Lopez-Molina et al., Reference Lopez-Molina, Mongrand, McLachlin, Chait and Chua2002). Another upstream regulator of ABI5 is RGL2, which was found to be important for secondary dormancy induction in Arabidopsis (Ibarra et al., Reference Ibarra, Tognacca, Dave, Graham, Sánchez and Botto2016). In addition, ABI4 plays a role in regulating primary seed dormancy through the regulation of ABA and GA homeostasis (Shu et al., Reference Shu, Zhang, Wang, Chen, Wu, Tang, Liu, Feng, Cao and Xie2013). Together with ABI3, FUSCA 3 (FUS3), LEAFY COTYLEDON 1 (LEC1) and LEC2 (LAFL genes) encode master transcriptional regulators that form a network that is key in regulating several important seed processes, including establishing primary dormancy (Carbonero et al., Reference Carbonero, Iglesias-Fernández and Vicente-Carbajosa2017; Lepiniec et al., Reference Lepiniec, Devic, Roscoe, Bouyer, Zhou, Boulard, Baud and Dubreucq2018). For example, in A. thaliana, lec1, lec2, fus3 and abi3 mutants are severely affected in seed maturation and share some common phenotypes, such as decreased dormancy at maturation (Raz et al., Reference Raz, Bergervoet and Koornneef2001; Bentsink and Koornneef, Reference Bentsink and Koornneef2008). LEC1 and LEC2 seem involved in maintaining or inducing a totipotent cell state during embryogenesis through the control of auxin biosynthetic genes (Stone et al., Reference Stone, Braybrook, Paula, Kwong, Meuser, Pelletier, Hsieh, Fischer, Goldberg and Harada2008; Wójcikowska and Gaj, Reference Wójcikowska and Gaj2015; Lepiniec et al., Reference Lepiniec, Devic, Roscoe, Bouyer, Zhou, Boulard, Baud and Dubreucq2018). FUS3 is known to modulate the ABA/GA balance by increasing ABA levels and repressing the synthesis of GA, while ABI3 integrates ABA signalling (Curaba et al., Reference Curaba, Moritz, Blervaque, Parcy, Raz, Herzog and Vachon2004; Gazzarrini et al., Reference Gazzarrini, Tsuchiya, Lumba, Okamoto and McCourt2004; Braybrook et al., Reference Braybrook, Stone, Park, Bui, Le, Fischer, Goldberg and Harada2006). In addition, LAFL activities are themselves modulated by hormone signalling feedbacks involving ABA, GA, BR or auxins (Braybrook and Harada, Reference Braybrook and Harada2008; Carbonero et al., Reference Carbonero, Iglesias-Fernández and Vicente-Carbajosa2017; Lepiniec et al., Reference Lepiniec, Devic, Roscoe, Bouyer, Zhou, Boulard, Baud and Dubreucq2018).
Another important gene known to regulate primary dormancy in Arabidopsis is DELAY OF GERMINATION 1 (DOG1) (Bentsink et al., Reference Bentsink, Jowett, Hanhart and Koornneef2006). In parallel to Arabidopsis, DOG1 genes have been found in other species of Brassicaceae and some of Latuca, with high similarity between amino acid sequences (Graeber et al., Reference Graeber, Linkies, Müller, Wunchova, Rott and Leubner-Metzger2010, Reference Graeber, Linkies, Steinbrecher, Mummenhoff, Tarkowská, Turečková, Ignatz, Sperber, Voegele, De Jong, Urbanová, Strnad and Leubner-Metzger2014; Huo et al., Reference Huo, Wei and Bradford2016; Carrillo-Barral et al., Reference Carrillo-Barral, Del Carmen Rodríguez-Gacio and Matilla2020). DOG1 is linked to accumulation of thermal time (Footitt et al., Reference Footitt, Müller, Kermode and Finch-Savage2015) and mutations in the gene can completely remove primary seed dormancy (Bentsink et al., Reference Bentsink, Jowett, Hanhart and Koornneef2006). The relationship between DOG1 and phytohormones is beginning to be elucidated. DOG1 physically interacts with two phosphatases, ABA-HYPERSENSITIVE GERMINATION 1 (AHG1) and AHG3, to block their downstream roles in the release of seed dormancy (Née et al., Reference Née, Kramer, Nakabayashi, Yuan, Xiang, Miatton, Finkemeier and Soppe2017). Besides, PROTEIN PHOSPHATASE 2A SUBUNIT A2 (PP2AA/PDF1) also physically interacts with DOG1, although acting upstream to have a negative role in seed dormancy (Née et al., Reference Née, Kramer, Nakabayashi, Yuan, Xiang, Miatton, Finkemeier and Soppe2017). DOG1 transduces environmental effects during seed maturation to alter the depth of primary dormancy (Kendall and Penfield, Reference Kendall and Penfield2012), but its expression in Arabidopsis does not seem to determine the pattern of dormancy cycling in the soil seed bank (Footitt et al., Reference Footitt, Huang, Clay, Mead and Finch-Savage2013). Indeed, Footitt et al. (Reference Footitt, Walley, Lynn, Hambidge, Penfield and Finch-Savage2020) demonstrated that seedling emergence timing is not directly controlled by the amount of DOG1 or concentration of ABA, but through the ratio of DOG1 to negative regulators of ABA sensitivity (e.g. AHG1, PDF1) (Footitt et al., Reference Footitt, Douterelo-Soler, Clay and Finch-Savage2011, Reference Footitt, Walley, Lynn, Hambidge, Penfield and Finch-Savage2020).
Evidence for epigenetic regulation of gene expression in controlling dormancy has only emerged recently. Early studies were carried out by Law and Suttle (Reference Law and Suttle2002, Reference Law and Suttle2004), elucidating the implications of 5-mC and histone H3 and H4 multi-acetylation in potato meristems during dormancy progression. With respect to histone acetylation in seeds, different expression patterns of histone acetyltransferases and deacetylases were found between dormant and non-dormant seeds of Arabidopsis by Cadman et al. (Reference Cadman, Toorop, Hilhorst and Finch-Savage2006). The HD2-LIKE family are plant specific deacetylases with specific roles in seeds and seedling growth (Berr et al., Reference Berr, Shafiq and Shen2011; Colville et al., Reference Colville, Alhattab, Hu, Labbé, Xing and Miki2011; Yano et al., Reference Yano, Takebayashi, Nambara, Kamiya and Seo2013). Mutation analysis of genes encoding for this family of HDACs showed that histone acetylation is involved in seed dormancy. For example, Berr et al. (Reference Berr, Shafiq and Shen2011) demonstrated that HD2A deacetylates HISTONE 3 LYSINE 9 (H3K9), a methylation target for KYP/SUVH4. Moreover, while seed germination is enhanced in hd2a mutants, hd2c mutants are restrained in germination in comparison with wild-type seeds (Colville et al., Reference Colville, Alhattab, Hu, Labbé, Xing and Miki2011). Overexpression of HD2C confers an ABA-insensitive phenotype, as seeds present enhanced germination and expression of LATE EMBRYOGENESIS ABUNDANT PROTEIN (LEA) class genes (Sridha and Wu, Reference Sridha and Wu2006).
SWI-INDEPENDENT 3 (SIN3)-LIKE 1 (SNL1) and SNL2 belong to a protein family that contains a paired amphipathic helix repeat (Bowen et al., Reference Bowen, Gonzalez, Mullins, Bhatt, Martinez and Conlan2010). In Arabidopsis, Wang et al. (Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013) demonstrated their redundant role in the regulation of seed dormancy as components of the HDAC-SNL complex, regulating the transcription of genes implicated in the antagonism between ethylene and ABA pathways (Linkies et al., Reference Linkies, Muller, Morris, Tureckova, Wenk, Cadman, Corbineau, Strnad, Lynn, Finch-Savage and Leubner-Metzger2009) by modifying their histone acetylation levels. The snl1 snl2-1 double mutant exhibited decreased dormancy and showed increased expression of genes involved in ethylene biosynthesis (like ACO1, ACO4 or ERF105) and downstream ethylene-responsive genes (Β-1,3-GLUCANASE and EXPANSINS) (Wang et al., Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013). Moreover, enhanced levels of SNL1 and SNL2 inhibited ABA hydrolysis and promoted its synthesis by histone deacetylation of certain target genes (Wang et al., Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013). In addition, Wang et al. (Reference Wang, Chen, Li, Cao, Ding, Zhang, Zuo, Xu, Xu, Deng, Xiang, Soppe and Liu2016) discovered the regulation of radicle promotion and early growth in a manner dependent on AUX1, with SNLs involved in histone deacetylation of AUX1 H3K9K18ac and repression of AUX1 expression. After-ripened double snl1 snl2-1 mutant seeds presented accelerated radicle protrusion and growth and increased transcript levels of a high number of auxin-related genes. Furthermore, CYCLIN D-TYPE (CYCD) 1;1 and CYCD 4;1, which are involved in cell cycling and seed germination, showed an important role in radicle promotion and growth downstream of AUX1, SNL1 and SNL2. With all these results, a final model was proposed in which the complexes associated with SNL proteins play an essential role in the establishment of primary seed dormancy and the regulation of germination in Arabidopsis seeds.
With respect to histone methylation, its implication in seed dormancy was demonstrated when mutations in KRYPTONITE/SU(VAR)3-9 HOMOLOG4 (KYP/SUVH4), encoding a histone methyltransferase required for H3K9me2, resulted in increased primary seed dormancy (Jackson et al., Reference Jackson, Lindroth, Cao and Jacobsen2002). Sites of H3K9 methylation recruit DNA methyltransferases CMT3 and CMT2, forming a self-reinforcing loop of repressive epigenetic marks (Molitor et al., Reference Molitor, Bu, Yu and Shen2014). The seeds of SUVH4 mutants had increased expression of DOG1 and dormancy-associated genes (Liu et al., Reference Liu, Koornneef and Soppe2007), which indicates that H3K9me2 caused by KYP/SUVH4 induces their silencing through DNA methylation (Katsuya-Gaviria et al., Reference Katsuya-Gaviria, Caro, Carrillo-barral and Iglesias-fernández2020). In addition, PICKLE (PKL) is a chromatin-remodelling factor that affects the levels of H3K27me3 and plays essential roles in regulating various developmental processes and environmental responses, including embryonic development, root meristem activity, photomorphogenesis, and thermomorphogenesis (Zhang et al., Reference Zhang, Jing, Jiang and Lin2014; Zha et al., Reference Zha, Liu, Li, Ma, Yang, Jing and Lin2020). Mutants with a loss or reduced function of PKL showed increased seed dormancy as PKL inhibits DOG1 transcription (Katsuya-Gaviria et al., Reference Katsuya-Gaviria, Caro, Carrillo-barral and Iglesias-fernández2020; Zha et al., Reference Zha, Liu, Li, Ma, Yang, Jing and Lin2020). Zhao et al. (Reference Zhao, Yang, Liu and Wu2015) discovered that LYSINESPECIFIC DEMETHYLASE LIKE 1 and 2 (LDL1 and LDL2) act redundantly in repressing seed primary dormancy. The ldl1 ldl2 double mutant displayed increased seed dormancy, whereas overexpression of LDL1 or LDL2 caused reduced primary dormancy.
The POLYCOMB REPRESSIVE COMPLEX 2 (PRC2) is responsible for the deposition of H3K27me3, which regulates major phase transitions in plant development such as the switch from embryonic to vegetative growth (Bouyer et al., Reference Bouyer, Roudier, Heese, Andersen, Gey, Nowack, Goodrich, Renou, Grini, Colot and Schnittger2011; Müller et al., Reference Müller, Bouyer, Schnittger and Kermode2012; Engelhorn et al., Reference Engelhorn, Blanvillain and Carles2014). In Arabidopsis, there are 12 homologs of the Drosophila PRC2 subunits and, in particular, the histone methyltransferase EZ is encoded by the three homologs CURLY LEAF (CLF), MEDEA (MEA) and SWINGER (SWN). Different combinations of the four subunits result in three different PRC2-like complexes (Ruta et al., Reference Ruta, Longo, Boccaccini, Madia, Saccoliti, Tudino, Di Santo, Lorrai, Dello Ioio, Sabatini, Costi, Costantino and Vittorioso2019). Mutants in FERTILIZATION INDEPENDENT ENDOSPERM (FIE), which is an essential component of the PRC2, display a genome-wide abolition of H3K27me3 and exhibit increased primary seed dormancy and germination defects (Bouyer et al., Reference Bouyer, Roudier, Heese, Andersen, Gey, Nowack, Goodrich, Renou, Grini, Colot and Schnittger2011). The LAFL genes are epigenetically repressed by the E3 H2A monoubiquitin ligase activity of PRC1 and by the H3K27me3 activity of PRC2 (Carbonero et al., Reference Carbonero, Iglesias-Fernández and Vicente-Carbajosa2017). The canonical role of PRC1 is to recognize the H3K27me3 marks and confer chromatin compaction (Lepiniec et al., Reference Lepiniec, Devic, Roscoe, Bouyer, Zhou, Boulard, Baud and Dubreucq2018). PRC1 mutants also exhibit delays in both germination and transcriptional repression, with a delayed switch in chromatin from the H3K4me3-associated active to the H3K27me3-associated repressive transcription state of DOG1 and seed development genes (Molitor et al., Reference Molitor, Bu, Yu and Shen2014). Moreover, ARABIDOPSIS TRITHORAX 4 (ATX4), CLF and SWN are expressed in opposite phases to each other during the dormancy cycle (Footitt et al., Reference Footitt, Müller, Kermode and Finch-Savage2015) and during dormancy breaking and germination (Müller et al., Reference Müller, Bouyer, Schnittger and Kermode2012).
The involvement of DNA methylation in dormancy has also been shown by different investigations. Research in cereals has implicated the RNA-directed DNA methylation pathway (RdDM) in silencing genes linked to seed dormancy (Katsuya-Gaviria et al., Reference Katsuya-Gaviria, Caro, Carrillo-barral and Iglesias-fernández2020). For example, the gene AGO1003, which is an AGO4_9 class of ARGONAUTE, is differentially expressed in the embryos of primary dormant and non-dormant grains (Singh and Singh, Reference Singh and Singh2012). In addition, DNA demethylation driven by the DNA glycosylase REPRESSOR OF SILENCING 1 (ROS1) regulates seed dormancy and the response to ABA by controlling the expression of DOG1-LIKE 4 (DOGL4) (Zhu et al., Reference Zhu, Xie, Xu, Miki, Tang, Huang and Zhu2018). However, the presence of specific DNA methylation markers associated with dormancy or germination transcriptomes remains to be elucidated (Matilla, Reference Matilla2020).
Additional epigenetic regulators of primary dormancy have been identified through mutant screens, such as HISTONE MONO UBIQUITINATION 1 (HUB1) and HUB2, which encode two C3HC4 RING finger proteins with homology to the histone-modifying enzymes BRE1 in yeast and RNF20/RNF40 in humans (Liu et al., Reference Liu, Koornneef and Soppe2007). HUB1 is required for monoubiquitination of histone H2B at Lys-143 (H2BK143), which is a prerequisite for histone H3 methylation at Lys4 (H3K4me3) and Lys79 (H3K79me3), both associated with gene activation (Du, Reference Du2012). Elimination of HUB1 in seeds causes lower primary dormancy through decreased expression of genes related to ABA metabolism and response, such as NCED9 and ABI4, and also reduced ABA levels (Peeters et al., Reference Peeters, Vries, Hanhart, Léon-Kloosterziel, Zeevaart and Koornneef2002).
Finally, REDUCED DORMANCY 2 (RDO2) is related to the POLYMERASE II-ASSOCIATED FACTOR 1 C (PAF1C) and encodes the TRANSCRIPTION ELONGATION FACTOR S-II (TFIIS). Mutations in RDO2 and other PAF1C associated factors, such as VERNALIZATION INDEPENDENT 4 (VIP4), VIP5, EARLY FLOWERING 7 (ELF7), ELF8 and ARABIDOPSIS TRITHORAX-RELATED 7 (ATRX7), cause a reduced seed dormancy phenotype and several dormancy-related genes, such as DOG1, are down-regulated in the rdo mutant (Liu et al., Reference Liu, Geyer, van Zanten, Carles, Li, Hörold, van Nocker and Soppe2011). TFIIS (RDO2) and HUB1 are induced during the same stages of seed maturation, and a significant overlap of differentially expressed genes was observed in tfIIs and hub1 mutants, which indicates that they share common targets, such as DOG1 (Liu et al., Reference Liu, Geyer, van Zanten, Carles, Li, Hörold, van Nocker and Soppe2011).
The transcriptome analysis performed as part of our study aimed to elucidate whether (epigenetic regulatory) genes identified in primary and secondary dormancy research to date also play a role in secondary dormancy induction and depth of C. bursa-pastoris.
Materials and methods
Germination and secondary dormancy induction in the presence or absence of histone deacetylase inhibitors
For the first aim of our study, C. bursa-pastoris seeds from nine James Hutton Institute (then, ‘Scottish Crop Research Institute’) accessions (SCRI -156, -177, -367, -416, -469, -707, -773, -799 and -937: Champion et al., Reference Champion, May, Bennett, Brooks, Clark, Daniels, Firbank, Haughton, Hawes, Heard, Perry, Randle, Rossall, Rothery, Skellern, Scott, Squire and Thomas2003; Iannetta et al., Reference Iannetta, Begg, Hawes, Young, Russell and Squire2007; Gomez-Cabellos et al., Reference Gomez-Cabellos, Toorop, Cañal, Iannetta, Fernández-Pascual, Pritchard and Visscher2021) were used to assess the effects of different HDAC inhibitors on the induction and maintenance of secondary dormancy. Seeds of all accessions were imbibed in either 1.5 ml of 10 mM valproic acid (control: water) or 33 μM TSA (control: 1:100 DMSO). The osmotic potential (MPa) of all solutions was tested with a Micro-Osmometer (Roebling Autocal Type 13, Camlab, UK). Incubating conditions for secondary dormancy induction, subsequent germination testing and statistical analysis of the resulting data were previously described (Gomez-Cabellos et al., Reference Gomez-Cabellos, Toorop, Cañal, Iannetta, Fernández-Pascual, Pritchard and Visscher2021). Briefly, seeds were incubated in darkness for 0 d (i.e. went directly into light conditions), 1, 2, 3 or 7 d at 30°C. Each treatment had three replicates (50 seeds each) in separate 50 mm Petri dishes on two layers of Whatman no. 1 filter paper soaked with 1.5 ml of the solution tested. Germination (radicle >1 mm) was scored at the end of each dark-incubation period and following transfer to a 12 h photoperiod (30°C). Non-germinated seeds were checked for viability by after-ripening for 7 d, soaking in 1.5 ml of a 10 mM KNO3 solution and germination scoring at 25/10°C (12/12 h light/dark). Seed germination was quantified as final germination (%), while times to 50% (of total number of seeds) testa rupture and germination (t50s) were estimated by linear interpolation of the empirical cumulative germination curves. Results were analysed by fitting Generalized Linear Mixed Models (for germination %, with logit link and binomial error) or Linear Mixed Models (for t50s); with compound, darkness (at 30°C), and their interaction as fixed factors; accession and individual nested within accession as random factors; and 0 d of incubation in water as the contrast level. We only used data points for which it was possible to calculate the t50 of both testa rupture and germination, that is, Petri dishes in which final germination had been at least 50% (=487 Petri dishes). The total number of Petri dishes was 540 (9 accessions × 3 dishes/individuals × 4 compounds tested × 5 dark incubation times = 540).
Whole seed mRNA extraction and sequencing
For the second aim of our study, RNA was extracted from seeds of two accessions contrasting in secondary dormancy depth (Gomez-Cabellos et al., Reference Gomez-Cabellos, Toorop, Cañal, Iannetta, Fernández-Pascual, Pritchard and Visscher2021) that were imbibed in either water or valproic acid during secondary dormancy induction (Fig. 1). Mother plants 367.1, 367.2, 367.3, 799.1, 799.2 and 799.4 were chosen for RNA extraction based on the individual germination tests (Supplementary Data S1). After 3 d of imbibition in darkness at 30°C in water or in valproic acid, seeds wrapped in aluminium foil were opened under a green safe light in a dark room. At this point, any germinated seeds were removed. For each extraction, 50–100 mg of seeds were weighed and immediately frozen and ground to a fine powder in liquid nitrogen with a mortar and pestle. The homogenized material was incubated with 1 ml of lysis extraction buffer AP1 (QIAGEN) at 56°C for 10 min. The sample was cooled down and mixed with 425 μl of potassium acetate (3 M C2H3KO2) by vortex. After incubation on ice for 20 min, the solution was centrifuged at 22,000g for 5 min at 4°C. The supernatant was transferred to a new collection tube and the previous step repeated. To the solution, 450 μl of lysis extraction buffer RLT (QIAGEN) and 5% β-mercaptoethanol were added and the RNase Plant Mini Kit (QIAGEN) protocol was followed according to the manufacturer's instructions. Two samples of RNA from each mother plant were extracted, with each mother plant considered a biological replicate and the two samples per mother plant technical replicates. The technical replicate from each mother plant that showed the best quality based on agarose gel assessment was used for sequencing (Fig. 1). The amount of RNA in each sample was quantified (≥1 μg total RNA per sample) and its quality further analysed via RNA integrity number (RIN: ≥7 for each sample).
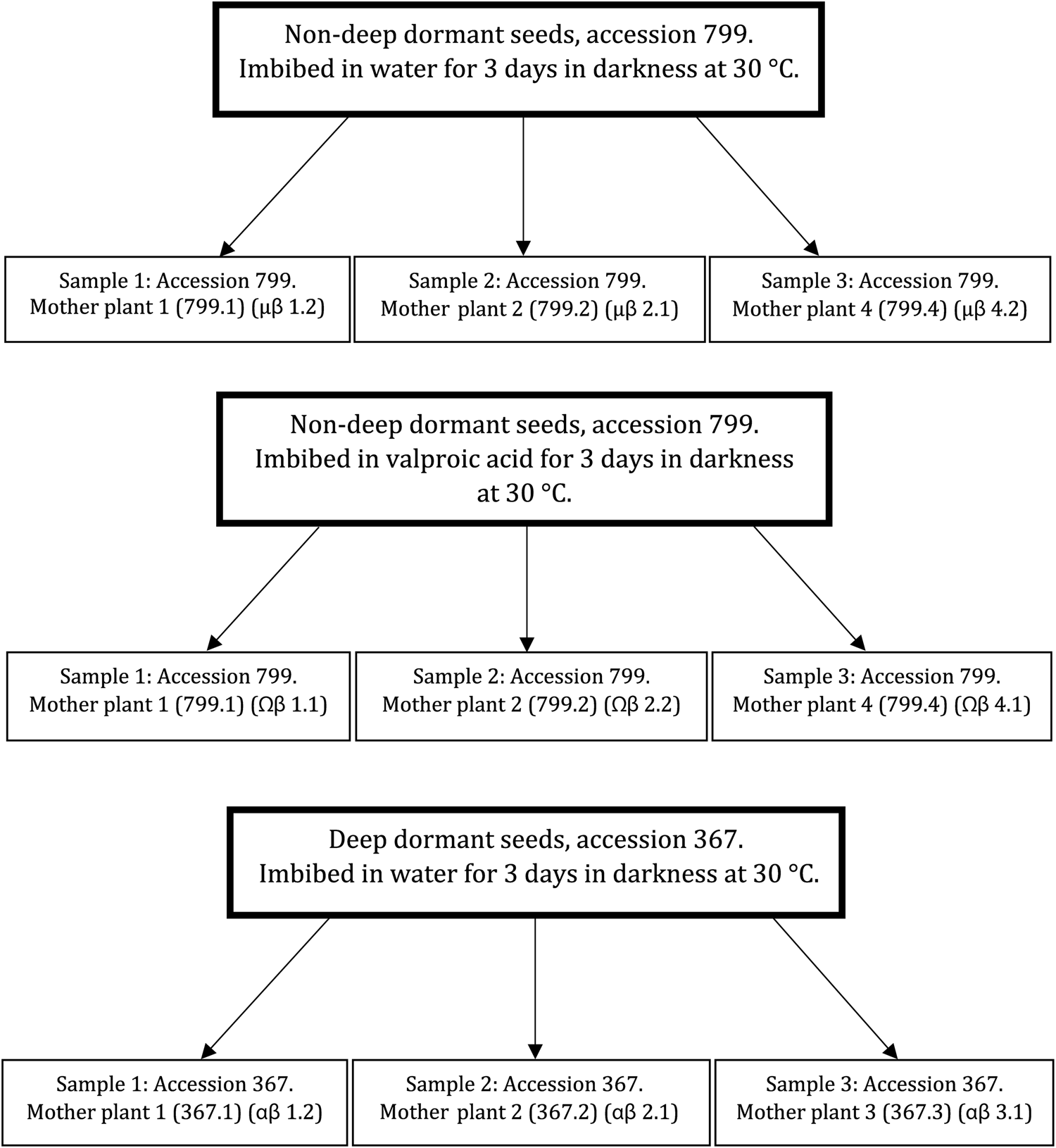
Fig. 1. Schematic representation of the nine samples used for transcriptome sequencing.
The isolation of mRNA and library construction of cDNA molecules was carried out by STAB VIDA (Caparica, Portugal) using a Kapa Stranded mRNA Library Preparation Kit (poly-A selected) and the generated DNA fragments were sequenced on an Illumina Hiseq 4000 platform, using 150 bp paired-end sequencing reads. The RNA-Seq raw reads were processed using CLC Genomics Workbench 11.0.1 and the bioinformatics analysis started with trimming of raw sequences to ensure the generation of high-quality data. 77.11–88.92% of the resulting paired high-quality reads were mapped against the reference genome of C. bursa-pastoris [GenBank assembly accession GCA_001974645.1 (Kasianov et al., Reference Kasianov, Klepikova, Kulakovskiy, Gerasimov, Fedotova, Besedina, Kondrashov, Logacheva and Penin2017)] using the following parameters: length fraction = 8; similarity fraction = 8. The result of the mapping served to determine the gene expression levels based on the Transcripts per Million (TPM) method, which is a variation of the commonly used Reads per Kilobase of exon model per Million (RPKM).
mRNA sequencing data analysis: differential expression, annotation and GO-term abundance
A multi-factorial statistical tool based on a negative binomial model was used for the analysis of the differential expression of RNA-seq, using a Generalized Linear Model (GLM) approach influenced by the EdgeR method. Differential expression analysis was performed comparing (1) deep dormant accession -367 imbibed in darkness for 3 d at 30°C in water versus non-deep dormant accession -799 imbibed in darkness for 3 d at 30°C in water (D water vs ND water) and (2) non-deep dormant accession -799 imbibed in darkness for 3 d at 30°C in valproic acid versus non-deep dormant accession -799 imbibed in darkness for 3 d at 30°C in water (ND valproic acid vs ND water). Replicate sample 3 from the non-deep dormant accession -799 imbibed in water for 3 d was not included in this analysis as it clustered with the non-deep dormant samples imbibed in valproic acid and was considered an outlying sample by STAB VIDA (PCA: Supplementary Fig. S1). Those genes with a fold change ≥ |1| and a false discovery rate (FDR)-adjusted P-value < 0.05 were considered as statistically differentially expressed (differentially expressed genes, DEGs).
For functional annotation, the full set of 52,597 C. bursa-pastoris gene sequences was analysed with OmicsBox version 1.2.4 (BioBam Spain, https://www.biobam.com/omicsbox/) (Götz et al., Reference Götz, García-Gómez, Terol, Williams, Nagaraj, Nueda, Robles, Talón, Dopazo and Conesa2008). C. bursa-pastoris sequences were used as queries in a BlastX search launched via CloudBlast. The Blast search was run against the non-redundant (nr) reference protein sequences database with normal speed BlastX, an expectation value (e-value) threshold of 1.0 × 10−3, keeping the top 20 alignments for each sequence and a minimal alignment length (HSP length) cut-off of 33. In some cases, several C. bursa-pastoris sequences shared the same Blast result, possibly due to multiple gene copies (with sequence variation between the duplicated genes).
Following the first step, Gene Ontology (GO) mapping allowed the retrieval of the functional information for all of the Blast Hits, obtaining a set of GO candidate annotation terms for each shepherd's purse query sequence. Default weights of the evidence codes were used. The annotation algorithm in the Blast2GO module selected GO-terms from the pool of candidate GOs obtained by the previous mapping step and assigned them to the query sequences (Conesa and Götz, Reference Conesa and Götz2008; Götz et al., Reference Götz, García-Gómez, Terol, Williams, Nagaraj, Nueda, Robles, Talón, Dopazo and Conesa2008). The Blast2GO annotation module applies an annotation rule on the found ontology terms in order to find the most specific annotations with a certain level of reliability (Conesa and Götz, Reference Conesa and Götz2008; Götz et al., Reference Götz, García-Gómez, Terol, Williams, Nagaraj, Nueda, Robles, Talón, Dopazo and Conesa2008). The default values of Blast2GO annotation parameters were chosen (e-value Hit filter of 1.0 × 10−6; annotation score of 55 as cut-off value; GO-weight of 5 to mapped children terms).
Afterwards, an InterPro domain and motif search were performed via CloudInterProScan (implemented in OmicsBox software version 1.2.4) with the default parameters. The identified domains and motifs were directly translated into GO-terms and this information was combined with the previous Blast searches. Finally, eggNOG mapper was run against the sequences followed by GO-enzyme code (both implemented in OmicsBox software version 1.2.4) and the results were merged with the previous data providing only one integrated functional annotation result. The possible parent–child relationships that had originated from the merging process were removed.
OmicsBox software version 1.2.4 was used for analysis of GO-term abundance. Only functionally annotated sequences from the DEGs and the whole genome were used to carry out the analysis. The GO-terms of the different up- and down-regulated subsets of annotated DEGs (water vs ND water and ND valproic acid vs ND water) were compared against those of the whole annotated genome using a Fisher's Exact Test with Multiple Test Correction of FDR at the significance threshold of <0.05. The setting of FDR < 0.05 is in general 1000 times more stringent than the P-value ≤ 0.05 (Chang and Scharfenstein, Reference Chang and Scharfenstein2014). FDR is used to control the expected proportion of incorrectly rejected null hypothesis, with a Benjamin-Hochberg correction. The results of the analysis show over- and under-represented GO biological process, cellular compartment, and molecular function categories for the different subsets of DEGs (Supplementary Data S5).
Results and discussion
The effect of histone deacetylase inhibitors on secondary seed dormancy induction and germination
A broad range in secondary seed dormancy potential was previously observed among the nine C. bursa-pastoris ecotypes examined, with accessions -367 and -799 showing the deepest and shallowest secondary dormancy, respectively (Gomez-Cabellos et al., Reference Gomez-Cabellos, Toorop, Cañal, Iannetta, Fernández-Pascual, Pritchard and Visscher2021). For the current study, we tested the effect of two HDAC inhibitors (TSA and valproic acid) on secondary seed dormancy induction and germination of the nine genotypes. The final mean germination percentages in response to the different compounds and times of incubation in darkness at 30°C are shown in Fig. 2. Accessions -367 and -799 showed the lowest and highest final germination across treatments (Fig. 3A), while accessions -156 and -469 germinated the fastest (Fig. 3B).

Fig. 2. Final mean germination percentages of seeds from all the accessions (SCRI -156, -177, -367, -416, -469, -707, -773, -799 and -937) of C. bursa-pastoris studied in the different compounds (water, DMSO, TSA and valproic acid) and times of incubation in darkness at 30°C tested (0, 1, 2, 3 and 7 d), after being exposed to germination-promoting conditions. Bars represent means and brackets the 95% binomial confidence interval. The most frequent number of viable seeds per condition tested was 200 (4 × 50 = 200).
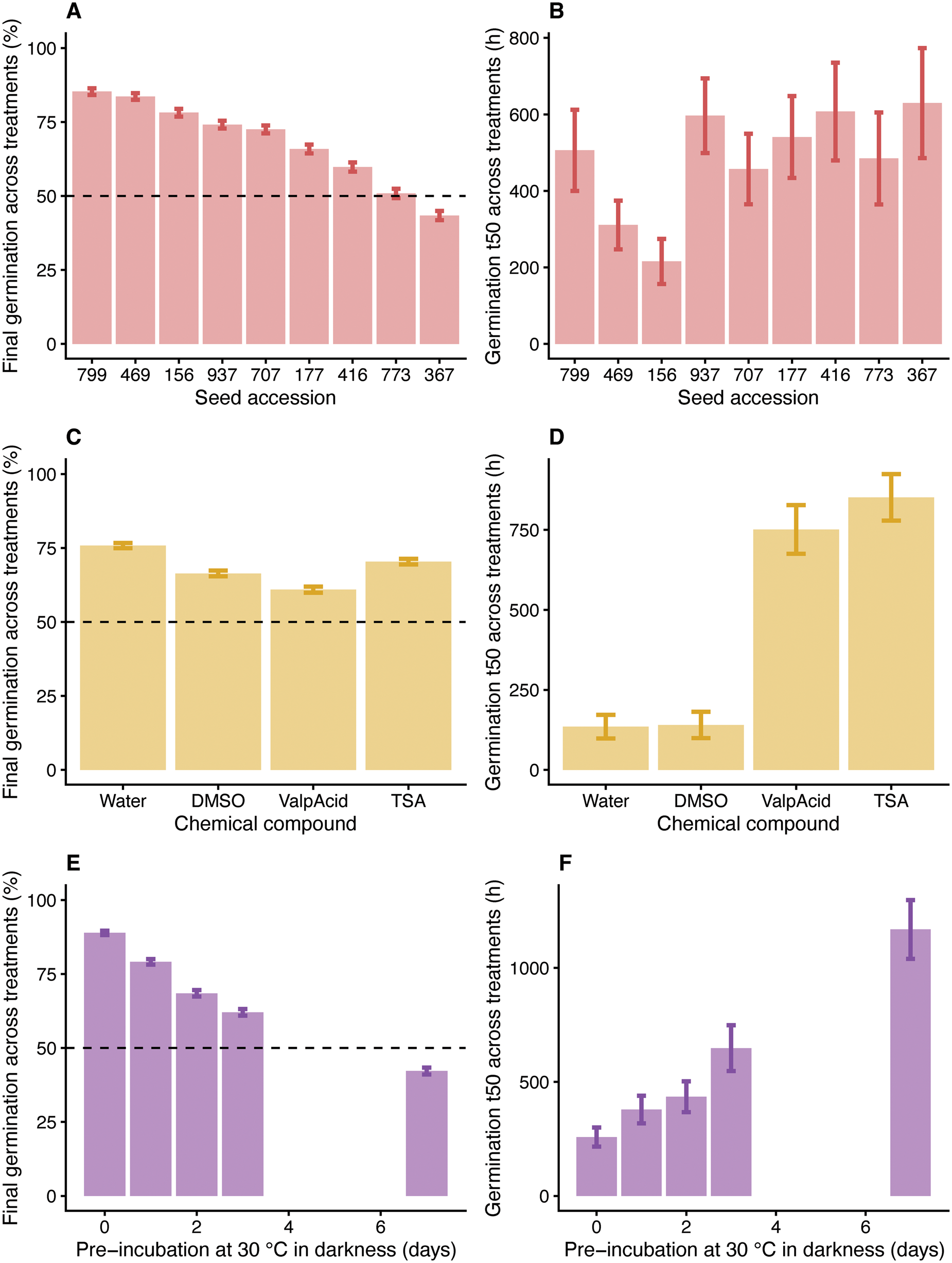
Fig. 3. Generalized Linear Model (GLM) fitted to the data, taking all the accessions analysed (SCRI -156, -177, -367, -416, -469, -707, -773, -799 and -937). (A) Final mean germination percentages (%) across all treatments studied in relation to the accession analysed. (B) t50s in hours to germination across all treatments studied in relation to the accession analysed. (C) Final mean germination percentages (%) across all treatments studied in relation to the compound used. (D) t50 for germination in hours for all the accessions and treatments analysed in relation to the compound used. (E) Final mean germination percentages (%) for all the accessions and compounds used in relation to the period of incubation in darkness at 30°C. (F) t50 for germination in hours for all the accessions and compounds used in relation to the period of incubation in darkness at 30°C. For (A, C and E), brackets represent the 95% binomial confidence interval and for (B, D and F), brackets represent standard errors.
In general terms, darkness and 30°C had a strongly significant and almost linear effect on final germination. The longer the period in darkness at 30°C, the lower the subsequent final germination percentages across all treatments of all the accessions studied (Fig. 3E). The period of time needed for 50% of the total number of seeds to germinate is denominated t50. If we compare the germination t50s, the longer the period of incubation in darkness at 30°C, the longer the time for 50% of seeds to germinate, across all treatments and accessions (Fig. 3F). These results indicate that with longer incubation periods in darkness at 30°C, both final germination and germination speed are significantly reduced.
All the compounds, including the control for TSA (DMSO), reduced the final germination percentages in comparison with water significantly (Figs 3C and 4A). TSA dissolved in DMSO showed higher final germination than DMSO by itself (Figs 3C and 4A), while valproic acid seemed to cause a deeper secondary dormancy [i.e. lower final germination in comparison with its control (water); Figs 3C and 4A], which was contrary to our hypothesis that blocking deacetylation would reduce secondary dormancy induction. Valproic acid and TSA had no significant interaction with darkness at 30°C (Fig. 4A), meaning that the negative response to darkness at 30°C seen in water was not affected by the presence of these compounds. However, DMSO (used as a control for TSA) was more negatively affected by darkness at 30°C than water (P < 0.001; Fig. 4A).
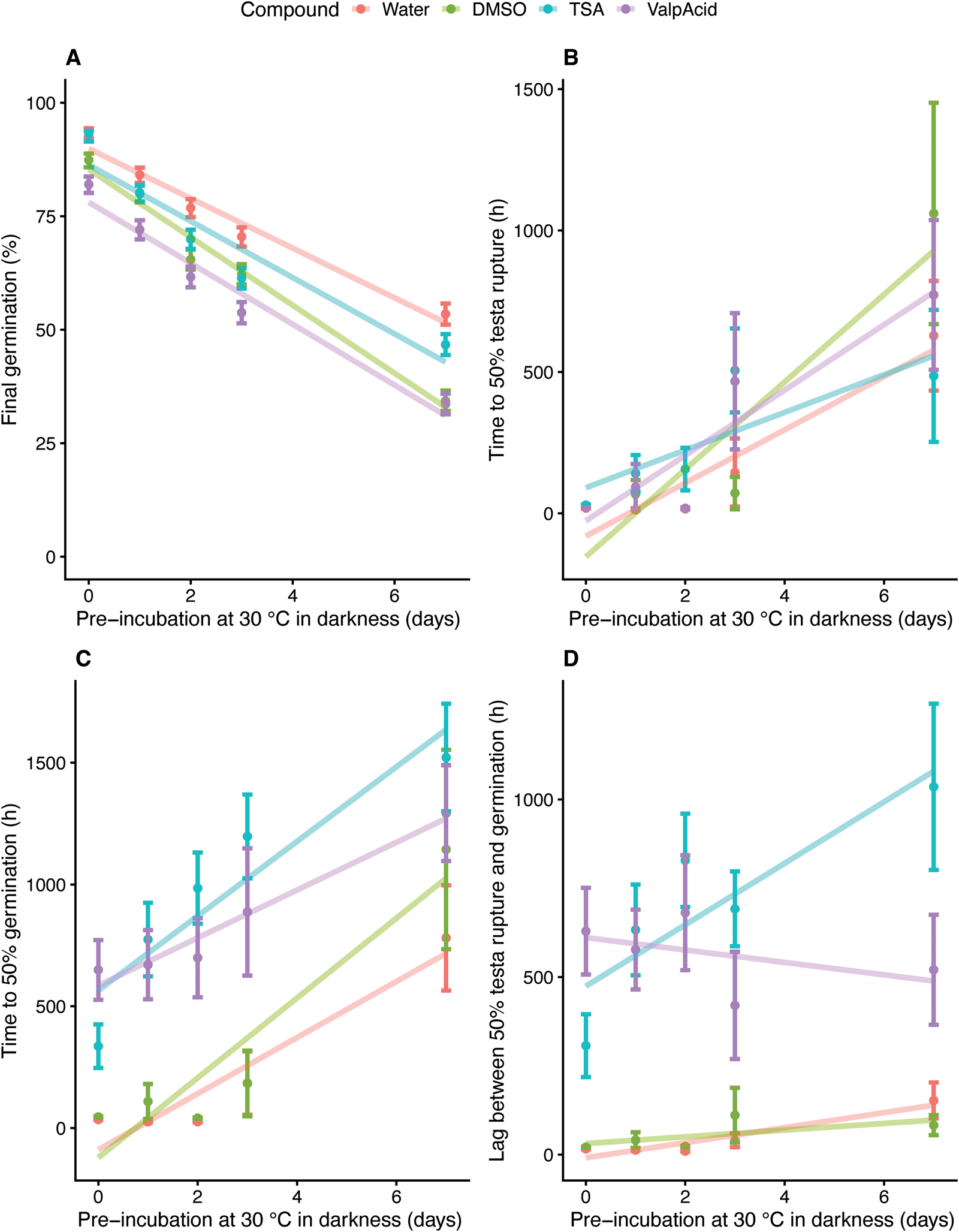
Fig. 4. Generalized Linear Model (GLM) fitted to the data, taking all the accessions analysed (SCRI -156, -177, -367, -416, -469, -707, -773, -799 and -937). (A) Final mean germination percentages (%) in relation to the period of incubation in darkness at 30°C and separated by the compound used. (B) t50s in hours to testa rupture in relation to the period of incubation in darkness at 30°C and separated by the compound used. (C) t50s in hours to germination in relation to the period of incubation in darkness at 30°C and separated by the compound used. (D) Differences between t50s of testa rupture and germination, with the mean of all the accessions together. For (A), brackets represent the 95% binomial confidence interval and for (B, C and D), brackets represent standard errors.
Analysing the germination t50s according to compound (vs water), significant effects were observed for valproic acid and TSA (P < 0.001; P < 0.001), but not for DMSO (Figs 3D and 4C), revealing that both valproic acid and TSA reduced the speed of germination significantly compared with their controls. Testa rupture was also measured, with none of the compounds significantly affecting t50s for testa rupture in comparison with water (Fig. 4B). Therefore, valproic acid and TSA had a strongly significant retarding effect on the germination speed (germination t50s) but did not affect the speed of testa rupture (testa rupture t50s) (Figs 3D and 4B, C). The differences between germination t50s and testa rupture t50s in relation to the compounds are represented in Fig. 4D. The osmotic potential of all solutions was 0 MPa, discarding an osmotic potential effect rather than the results being due to the compounds’ inhibitory characteristics. The viability of the non-germinated seeds, after drying and treatment with KNO3, is represented in Supplementary Fig. S2, with 97% being the lowest viability. Application of exogenous nitrate alleviates seed dormancy and stimulates germination through transcriptional changes of several genes involved in ABA (e.g. CYP707A2) and GA metabolism and sensitivity (Matilla et al., Reference Matilla, Carrillo-Barral and Rodríguez-Gacio2015; Sano and Marion-Poll, Reference Sano and Marion-Poll2021).
In this study, TSA and valproic acid caused a higher t50 for germination but the speed of testa rupture (t50 for testa rupture) was not affected (Figs 3D and 4B, C). Germination has two visible physical stages: testa and endosperm rupture, the latter being completed with the micropylar endosperm rupture by the radicle (Bentsink and Koornneef, Reference Bentsink and Koornneef2008). Our results indicate that, contrary to our hypothesis, exposure to HDAC inhibitors slowed germination speed (higher t50 for radicle emergence) of the non-dormant seeds (Figs 3D and 4C), while valproic acid also increased secondary dormancy depth (lower final germination) (Figs 3C and 4A).
Several reports on germination of non-dormant seeds have shown inhibiting effects of TSA. For example, Tanaka et al. (Reference Tanaka, Kikuchi and Kamada2008) exposed non-dormant Arabidopsis seeds to 5–50 μM TSA and found a delay in germination in comparison with the control after 3 d of sowing. Zhang et al. (Reference Zhang, Qiu, Hu, Yang, Yan, Zhao, Li, He, Huang, Li and Li2011) applied 1–50 μM TSA to non-dormant Zea mays L. (maize) seeds, observing lower germination rates in seeds imbibed in TSA compared with seeds imbibed in the control (a DMSO control was not indicated). However, Nelson et al. (Reference Nelson, Ariizumi and Steber2017) rescued germination of dormant and gibberellic acid (GA) insensitive sly1-2 mutant seeds with 2 μM TSA, while 4 and 6 μM led to decreasing germination capacity.
The probable involvement of a deacetylation event in seed germination has been described in previous studies (Perrella et al., Reference Perrella, Consiglio, Aiese-Cigliano, Cremona, Sanchez-Moran, Barra, Errico, Bressan, Franklin and Conicella2010; Cigliano et al., Reference Cigliano, Cremona, Paparo, Termolino, Perrella, Gutzat, Consiglio and Conicella2013; Wang et al., Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013). HDACs contribute to the repression of embryogenesis related gene expression during germination (Tai et al., Reference Tai, Tai and Beardmore2005; Tanaka et al., Reference Tanaka, Kikuchi and Kamada2008). This indicates that the action of TSA and valproic acid in the increased t50 for germination could possibly be related to the positive regulation of embryo-specific transcription factors implicated in the maintenance of embryonic properties, such as LEC1, LEC2, FUS3 or ABI3, hence promoting a delay of germination (Carbonero et al., Reference Carbonero, Iglesias-Fernández and Vicente-Carbajosa2017; Lepiniec et al., Reference Lepiniec, Devic, Roscoe, Bouyer, Zhou, Boulard, Baud and Dubreucq2018). In addition, we hypothesized that enhanced secondary dormancy and delay in germination observed in seeds treated with HDAC inhibitors could involve mis-regulation of genes related to hormone (ABA, GAs) biosynthesis or signalling pathways (Lepiniec et al., Reference Lepiniec, Devic, Roscoe, Bouyer, Zhou, Boulard, Baud and Dubreucq2018).
At the same time, these results could indicate that acetylation/deacetylation of histones plays a role in the elongation of the transition zone and lower hypocotyl cells necessary for radicle protrusion (Sliwinska et al., Reference Sliwinska, Bassel and Bewley2009). The main hormones implicated in these processes are auxins (Fu and Harberd, Reference Fu and Harberd2003). Their relation with histone deacetylation was unveiled by Wang et al. (Reference Wang, Chen, Li, Cao, Ding, Zhang, Zuo, Xu, Xu, Deng, Xiang, Soppe and Liu2016), who demonstrated the negative regulation of radicle promotion and early growth of Arabidopsis seeds by PAIRED AMPHIPATHIC HELIX SWI-INDEPENDENT3 (SIN3)-LIKE (SNL) 1 and SNL2 in a manner dependent on AUXIN RESISTANT 1 (AUX1). SNL1 and SNL2 can act as components of a HDAC-SNL complex capable of modulating the transcription of genes through histone deacetylation (Wang et al., Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013). Thus, we could hypothesize that HDAC inhibitors, such as TSA and valproic acid, act on histone deacetylases that are implicated in the regulation of auxin pathways and signals, which at the same time regulate specific steps in the seed germination process.
(Epigenetic) gene expression associated with exposure to histone deacetylase inhibitors during secondary seed dormancy induction
In order to test the hypotheses mentioned in the section ‘The effect of histone deacetylase inhibitors on secondary seed dormancy induction and germination’, we compared gene expression of a non-deep dormant accession (-799) exposed to either an HDAC inhibitor or a control. Valproic acid instead of TSA was used to treat the seeds as it caused a deeper secondary dormancy and stronger delay of germination in all the accessions and does not need to be dissolved in DMSO. In the case of the ND valproic acid versus ND water comparison, the number of genes differentially expressed was 393, of which 122 were up-regulated and 271 down-regulated (Table 1). The DEGs highlighted below are listed in Supplementary Data S2 and S3. GO-term abundance analysis was also performed, with results presented in Supplementary Data S4 and S5.
Table 1. Number of DEGs between conditions in C. bursa-pastoris seeds

Total number of DEGs between comparison pairs, significantly up-regulated and down-regulated and final number of annotated genes.
Our first hypothesis regarding the delay in germination caused by valproic acid involved a positive regulation of embryo-specific transcription factors implicated in the maintenance of embryonic properties. However, the results showed no significant differential expression of genes such as LEC1, LEC2, FUS3 or ABI3, despite the up-regulation of CHROMATIN REMODELING 5 (CHR5) (1.62) (Shen et al., Reference Shen, Devic, Lepiniec and Zhou2015). We, therefore, assessed the possibility that mis-regulation of phytohormone genes could underlie increased secondary dormancy depth and germination delay. The phytohormone ABA has been found to play a key role in the regulation of seed dormancy and germination. It can inhibit germination and its accumulation correlates with the onset of primary dormancy (Tuan et al., Reference Tuan, Kumar, Rehal, Toora and Ayele2018). In the up-regulated DEGs, only phytohormone GOs related to ABA transmembrane transport were enriched. Overall, several positive regulators of ABA were down-regulated, including PYL4 (-2.02), which is a receptor of ABA required for ABA-mediated response that is crucial for seed germination (Wang et al., Reference Wang, Ren, Cheng, Wang, Ji, Zhao, Deng, Zhi, Lu, Wu, Xu, Cao, Zhao, Liu, Zhu and Li2020). Another example is ABI5 (-4.05), which is essential to execute an ABA-dependent growth arrest that sets in after breakage of seed dormancy but prior to autotrophic growth (Lopez-Molina et al., Reference Lopez-Molina, Mongrand and Chua2001, Reference Lopez-Molina, Mongrand, McLachlin, Chait and Chua2002). In accordance with this, a sequence implicated in the degradation of ABI5 was up-regulated: E3 ubiquitin-protein ligase KEEP ON GOING (KEG) (1.33) (Liu and Stone, Reference Liu and Stone2010). UDP-GLUCOSYLTRANSFERASE 75B1 (UGT75B1) (1.88), which is the main enzyme responsible for pABA-Glc formation in Arabidopsis, was also up-regulated. ABA can be transformed into inactive forms by glycosylation, which is a flexible way of maintaining ABA homeostasis, and this is catalyzed by the plant family of UDP-GLYCOSYLTRANSFERASES (UGTs). Overexpression of UGT75B1 leads to overproduction of ABA-Glc in Arabidopsis and lowers the active ABA levels, allowing seed germination and seedling greening (Chen et al., Reference Chen, Liu, Xiao, Jiang, Li, Zhao, Hou and Li2020). However, we also observed up-regulation of genes implicated in positive signalling of ABA [SNL1 (1.39) (Wang et al., Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013, Reference Wang, Chen, Li, Cao, Ding, Zhang, Zuo, Xu, Xu, Deng, Xiang, Soppe and Liu2016)] and down-regulation of genes implicated in negative signalling, such as WRKY29 (Zhou et al., Reference Zhou, Lin, Lan, Zhang, Liu, Miao, Mou, Nguyen, Wang, Zhang, Zhou, Zhu, Wang, Zhang, Guo, Liu, Jiang and Wan2020) (-7.94), WRKY18-LIKE (-1.89) and WRKY60 (-1.69).
The enriched GOs related to phytohormones in the down-regulated DEGs were linked to cytokinin and indoleacetic acid, such as the GO-term ‘indoleacetic acid biosynthesis’. Analysing auxin-related sequences within the up-regulated DEGs, there was one gene implicated in auxins efflux: the protein BIG (1.63), which is involved in cell elongation, lateral root promotion and general growth and development (Kanyuka et al., Reference Kanyuka, Praekelt, Franklin, Billingham, Hooley, Whitelam and Halliday2003). Nonetheless, TRYPTOPHAN N-MONOOXYGENASE 2 (CYP79B3), which catalyzes the first step of IAA biosynthesis (Wang et al., Reference Wang, Chen, Li, Cao, Ding, Zhang, Zuo, Xu, Xu, Deng, Xiang, Soppe and Liu2016), presented a down-regulation of -2.29-fold. In addition, SNL1 (1.39), which is implicated in the repression of sequences related to the biosynthesis of auxins (Wang et al., Reference Wang, Chen, Li, Cao, Ding, Zhang, Zuo, Xu, Xu, Deng, Xiang, Soppe and Liu2016), was up-regulated. We could hypothesize that, due to the up-regulation of SNL1, there may be no up-regulation of auxins biosynthesis genes. The reason behind the up-regulation of this histone deacetylase when seeds were treated with valproic acid could potentially be a compensation for the inhibition of deacetylation provoked by the exogenously applied compound.
Although no enrichment of GOs related to GAs were found, genes such as GA20ox1 (-8.24 and -4.17) and GIBBERELLIN-REGULATED-LIKE PROTEIN (-331.29, -91.56, -88.50 and -17.47) were found in the down-regulated list of DEGs. GAs are known to stimulate seed germination in a wide range of plant species (Tuan et al., Reference Tuan, Kumar, Rehal, Toora and Ayele2018). The same situation was found for BRs, with genes implicated in their biosynthesis or response within the down-regulated DEGs, such as CYTOCHROME P450 CYP708A2 (-32.09 and -7.62), EXORDIUM-LIKE 1 (EXL1) (-7.19), EXORDIUM-LIKE 5 (EXL5) (-2.44) or EXORDIUM-LIKE (EXO) (-2.68). BRs are known for promoting seed germination and both BRs and GAs induce the expression of cell elongation-associated genes such as distinct expansin family members (Finkelstein et al., Reference Finkelstein, Reeves, Ariizumi and Steber2008).
Altogether, it could be said that the hyper-acetylation caused by the use of valproic acid provoked alterations in the expression of genes implicated in the biosynthesis and signalling pathways of different phytohormones. As mentioned above, ABA signalling gene expression was both repressed and stimulated in seeds treated with valproic acid. However, genes related to synthesis of GAs, BRs and auxins were down-regulated (Table 2), which might explain the slowing of germination (higher t50) in response to valproic acid.
Table 2. DEGs potentially explaining the observed delay in t50 for germination in C. bursa-pastoris seeds treated with HDAC inhibitors

The selected DEGs are from a comparison of a non-deep dormant accession (-799) exposed to an HDAC inhibitor (valproic acid) versus a control (water): the seeds were imbibed in darkness for 3 d at 30°C in water to induce secondary seed dormancy.
With respect to epigenetic regulation and chromatin remodelling, GOs related to histone acetylation and histone acetyltransferase complexes were within the enriched terms for the up-regulated DEGs, including ‘chromatin remodelling’, ‘lysine-acetylated histone binding’ and ‘acetylation-dependent protein binding’. However, the histone deacetylases SNL1 (1.39) and SNL4 (1.34) were also up-regulated. These results seem to be contradictory but could be indicating that seeds are trying to stabilize histone acetylation levels. In addition, the up-regulation of SNL1 might help explain the increase in secondary dormancy caused by valproic acid exposure.
Regarding methylation, we observed both up-regulation of genes involved in demethylation [LYSINE-SPECIFIC DEMETHYLASE JMJ18 (5.87)] as well as methylation [HISTONE-LYSINE N-METHYLTRANSFERASE ATXR3-LIKE (1.36)]. Finally, the DNA helicase INO80-LIKE was also up-regulated (1.34). INO80 is a chromatin remodelling complex that modulates, together with SWR1, the incorporation of H2A.Z in nucleosomes. The deposition of this histone variant at gene bodies is associated with lower transcription levels (Wang et al., Reference Wang, Gao, Peng, Wu and Yang2019). Overall, the genes involved in epigenetic regulation of transcription show a dynamic pattern of expression in response to valproic acid.
Expression of (epigenetic regulatory) genes associated with secondary seed dormancy depth
Taking two ecotypes that presented extreme responses to the induction of secondary dormancy in water (-367 and -799), a transcriptome analysis was performed. The number of genes with statistically significant differences (fold change ≥|1| and FDR P-value < 0.05) in expression between D water versus ND water was 6,337, of which 2,727 were up-regulated and 3,610 down-regulated (Table 1; Supplementary Data S3). The focus of the results and discussion is on DEGs with GO, terms related to dormancy, phytohormones, as well epigenetic processes that could potentially explain differences in secondary dormancy depth between the accessions analysed. Given that the two ecotypes contrast in their response to secondary seed dormancy induction, the results may reflect differences in germination potential/status as well as secondary dormancy depth. The full results of the GO analysis are available in Supplementary Data S4 and S5. In addition, manual categorization of DEGs according to function was also performed (Supplementary Data S6).
Dormancy and phytohormones
DOG1 has been described as an essential and necessary protein in the establishment of primary seed dormancy over the last decade (Bentsink et al., Reference Bentsink, Jowett, Hanhart and Koornneef2006; Footitt et al., Reference Footitt, Müller, Kermode and Finch-Savage2015; Carrillo-Barral et al., Reference Carrillo-Barral, Del Carmen Rodríguez-Gacio and Matilla2020). Although DOG1 was not found to have direct control of secondary dormancy depth in Arabidopsis, Footitt et al. (Reference Footitt, Walley, Lynn, Hambidge, Penfield and Finch-Savage2020) proposed a model for the regulation of dormancy cycling where a lower AHG1/ANAC060/PDF1:DOG1 ratio is linked to deeper dormancy/lower germination potential. The results in this study on C. bursa-pastoris did not reveal differential expression of DOG1 (there were no sequences annotated as DOG1), while AHG1 (PROBABLE PROTEIN PHOSPHATASE 2C 75) and ANAC060 (NAC DOMAIN-CONTAINING PROTEIN 60 ISOFORM X1) were more highly expressed (1.36 and 1.41) in the deeper dormant ecotype. Several DOG1-LIKE genes were also up-regulated in the deep versus non-deep dormant genotype: DOG1-LIKE 3 (2.02, 1.74), DOG1-LIKE 4 (1.54) and DOG1-LIKE 1 (1.51). Ectopic expression of wheat and barley DOG1-LIKE genes promoted seed dormancy in Arabidopsis (Ashikawa et al., Reference Ashikawa, Abe and Nakamura2010). DOG1-LIKE 3 (DOGL3) is capable, like DOG1, of binding to AHG1 PP2C, thereby playing a similar role to DOG1 in ABA sensitivity and dormancy enhancement (Nonogaki et al., Reference Nonogaki, Nishiyama, Ohshima and Nonogaki2020). For example, the OsDOG1-LIKE 3 gene was found to up-regulate ABA biosynthesis and signalling-related genes, suggesting that its promotion of primary seed dormancy likely occurs by enhancing the ABA pathway (Wang et al., Reference Wang, Lin, Wu, Duan, Huang, Yang, Mou, Lan, Zhou, Xie, Liu, Zhang, Guo, Wang, Jiang and Wan2020). However, DOGL4 is a negative regulator of primary seed dormancy and the ABA response, with mutations in DOGL4 enhancing dormancy (Zhu et al., Reference Zhu, Xie, Xu, Miki, Tang, Huang and Zhu2018; Katsuya-Gaviria et al., Reference Katsuya-Gaviria, Caro, Carrillo-barral and Iglesias-fernández2020).
A high number of over-represented GO-terms in the down-regulated DEGs were related to water and water transport. Footitt et al. (Reference Footitt, Clewes, Feeney, Finch-Savage and Frigerio2019) revealed a role for aquaporins in the induction and relief of secondary seed dormancy. TIP3-2 was identified as a negative regulator of ABA in Arabidopsis, but a PROBABLE AQUAPORIN TIP3-2 was expressed more highly in the deeper dormant accession of C. bursa-pastoris in this study (1.61 and 1.93). In general, the majority of differentially regulated aquaporins showed higher expression in the less deep dormant accession: aquaporin TIP2-1 (-439.48, -286.70 and -223.97), aquaporin TIP1-1 (-221.79 and -199.42), aquaporin TIP1-2 (-90.19, -49.16 and -35.77), aquaporin PIP1-3 (-10.90 and -3.81), probable aquaporin PIP2-5 (-10.38) and probable aquaporin NIP5-1 (-9.86 and -6.51).
With respect to phytohormones, we found that the number of over-represented GO-terms in the up-regulated DEGs was much lower than in the down-regulated list, with the former presenting only the two GO-terms: ‘positive regulation of cytokine production’ and ‘response to ABA’. The GO-term ‘response to ABA’ was also found within the enriched GO-terms of the down-regulated genes. Therefore, there are genes implicated in the positive and negative regulation of this hormone within the up- and the down-regulated sequences.
Looking at specific genes within the up-regulated DEGs, some sequences that have been described as important in the positive regulation of ABA pathways and signalling were found: SERINE/THREONINE-PROTEIN KINASE SRK2A-LIKE (142.97), ABC TRANSPORTER G FAMILY MEMBER 40 (ABCG40) (15.16 and 10.65), 9-CIS-EPOXYCAROTENOID DIOXYGENASE NCED6 (8.51), NCED2 (1.68 and 1.51), B3 DOMAIN-CONTAINING TRANSCRIPTION FACTOR ABSCISIC ACID-INSENSITIVE 3 (ABI3) (2.49) and ABSCISIC ACID-INSENSITIVE 5-LIKE PROTEIN 7 (ABF4) (2.28).
NCED6, together with NCED9, were shown to be up-regulated in secondary dormant versus non-dormant seeds (Cadman et al., Reference Cadman, Toorop, Hilhorst and Finch-Savage2006). The same pattern was found for ABI3, which presented higher expression in dormant (primary and secondary) compared with non-dormant states (Cadman et al., Reference Cadman, Toorop, Hilhorst and Finch-Savage2006). SWI-INDEPENDENT 3 (SIN3)-LIKE (SNL) 1 and SNL2 are histone deacetylases that have an important function in the regulation of primary seed dormancy (Wang et al., Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013). In research by Wang et al. (Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013), enhanced levels of SNL1 and SNL2 inhibited ABA hydrolysis and promoted its synthesis by histone deacetylation of certain target genes. In this study, a potential SNL1 (Cbp42606: Data S7) was highly up-regulated in the deep dormant accession in comparison with the non-deep dormant one (1633.68). In addition, the protein CRUCIFERIN CRU1-LIKE was differentially expressed with a fold change of 2.98. CRUCIFERIN A1 is a storage protein and a downstream target of ABA found to be related to dormant seeds by Gao et al. (Reference Gao, Jordan and Ayele2012).
On the other hand, within the down-regulated DEGs related to ABA, there were mostly negative regulators of its pathways, such as NINJA-FAMILY PROTEIN AFP4 (-211.51), RAC-LIKE GTP-BINDING PROTEIN ARAC7 and ARAC10 (-26.70 and -3.79, respectively), ZINC FINGER PROTEIN 8 and 3 (ZFP8 and ZFP3) (-17.17 and -3.48, respectively), PROTEIN TERMINAL EAR1 HOMOLOG (ENHANCER OF ABA CO-RECEPTOR 1) (-13.42), ABSCISIC ACID 8′-HYDROXYLASE 3 (CYP707A3: -4.96) and 1 (CYP707A1: -2.21), ENHANCED DISEASE RESISTANCE 2-LIKE (EDR2-LIKE) (-6.60 and -2.46) and the key negative regulator PROTEIN PHOSPHATASE 2C 56 (ABI1) (-3.41 and -3.30). Nevertheless, there were also down-regulated sequences related to positive responses to ABA such as the previously mentioned SERINE/THREONINE-PROTEIN KINASE SRK2A-LIKE (-11.94), HVA22E (-10.37 and -4.23), PHOSPHOINOSITIDE PHOSPHOLIPASE C1 (PLC1) (-6.56), ABSCISIC ACID-INSENSITIVE 5-LIKE PROTEIN 1 (-5.98 and -4.75), ABSCISIC ACID-INSENSITIVE 5-LIKE PROTEIN 6 (-4.69), ABI4-LIKE (-4.01 and -2.19), PYL4 (-2.00) and F-BOX/KLECH-REPEAT PROTEIN AT3G16740-LIKE (FOA2) (-505.45 and -471.55). However, the down-regulation of FOA2 expression might be caused by ABA through a feedback regulation mechanism (He et al., Reference He, Yu, Li, Duan, Zhang, Tang, Zhao and Liu2016). Although a fivefold higher expression of PYL4 was found in seeds induced into secondary dormancy compared with an array of primary dormant states (Laspina et al., Reference Laspina, Batlla and Benech-Arnold2020), our results show no evidence for enhanced expression of PYL4 being associated with deeper secondary dormancy.
A large part of ABA accumulation in seeds relies on the regulation of the NCED gene family as the enzymes they encode carry out the first step in the synthesis of ABA (Nambara et al., Reference Nambara, Okamoto, Tatematsu, Yano, Seo and Kamiya2010). The induction of NCED6 during imbibition is sufficient to prevent seed germination (Martínez-Andújar et al., Reference Martínez-Andújar, Ordiz, Huang, Nonogaki, Beachy and Nonogaki2011). On the other hand, the major catabolic route is via the ABA 8′-HYDROXYLASE (Matilla et al., Reference Matilla, Carrillo-Barral and Rodríguez-Gacio2015). The up-regulation of NCED6 and NCED2 and the down-regulation of ABSCISIC ACID 8′-HYDROXYLASE 3 might be indicating higher ABA levels in the deep-dormant accession than in the non-deep one. Taking into account all the ABA-related genes of the DEGs, in the up-regulated list, most of them are promoters of its synthesis or signalling, while in the down-regulated set, the most abundant are repressors or sequences related to its catabolism. However, the number of ABA-related genes with differential expression is more limited than may have been expected, which was also observed in several earlier studies (Cadman et al., Reference Cadman, Toorop, Hilhorst and Finch-Savage2006; Matilla et al., Reference Matilla, Carrillo-Barral and Rodríguez-Gacio2015; Ibarra et al., Reference Ibarra, Tognacca, Dave, Graham, Sánchez and Botto2016; Laspina et al., Reference Laspina, Batlla and Benech-Arnold2020). Footitt et al. (Reference Footitt, Walley, Lynn, Hambidge, Penfield and Finch-Savage2020) proposed a model in which dormancy cycling is regulated via a negative response to ABA.
With respect to the over-represented GO-terms of the down-regulated DEGs that involve hormones, we observed a high number of GOs related to biosynthesis, signalling pathways or metabolism. For example, changes in the balance of catabolism and synthesis of GAs are necessary for the promotion of germination. Genes such as GIBBERELLIN 20-OXIDASE 1 (GA20ox1) (-12.24 and -5.22) and GIBBERELLIN 20-OXIDASE 2 (GA20ox2) (-6.22) were within the down-regulated DEGs. These are key players in the biosynthesis of gibberellins, act partially redundantly and are the most highly expressed of the genes implicated in the synthesis of GAs during vegetative and early reproductive development (Rieu et al., Reference Rieu, Ruiz-Rivero, Fernandez-Garcia, Griffiths, Powers, Gong, Linhartova, Eriksson, Nilsson, Thomas, Phillips and Hedden2008; Tuan et al., Reference Tuan, Kumar, Rehal, Toora and Ayele2018). The transcription factor bHLH93, which is implicated in regulation of flowering time in short days (SD), was also within the down-regulated genes with fold changes of -5.87 and -4.13. Mutants of this gene presented down-regulation of GA biosynthetic genes (GA3ox1, GA3ox2, GA20ox1) and up-regulation of the GA catabolic (GA2ox2 and GA2ox7) and receptor genes in comparison with wild-type plants (Sharma et al., Reference Sharma, Xin, Kim, Sung, Lange and Huq2016). DEGs related to GA-mediated signalling presented contrasting results. For example, DELLA proteins (repressors of GA responses) showed both up- and down-regulation, involving relatively small fold changes [e.g. DELLA proteins GAI (1.48 and 1.39), RGL2 (-1.43 and -1.91) and RGL3 (-1.53 and -1.67)]. Although RGL2 plays an important role in secondary dormancy induction in Arabidopsis as shown by mutant analysis (Ibarra et al., Reference Ibarra, Tognacca, Dave, Graham, Sánchez and Botto2016), our results indicate that its expression is lower in the deeper dormant genotype during secondary dormancy induction. In addition, F-BOX PROTEIN GID2, a positive regulator of gibberellin signalling through degradation of DELLA proteins [including RGL2 (Tyler et al., Reference Tyler, Thomas, Hu, Dill, Alonso, Ecker and Sun2004; Yang et al., Reference Yang, Jiang, Liu and Lin2020)], was highly up-regulated (68.91). Loss of GID2 (SLY1) results in increased primary seed dormancy (Ariizumi et al., Reference Ariizumi, Lawrence and Steber2011), and therefore, its up-regulation in the deeper secondary dormant accession indicates that this gene may also not be a candidate for explaining differences in secondary dormancy depth.
Overall, the down-regulation of genes implicated in the biosynthesis of GAs in the deep dormant accession in comparison with the non-deep dormant one and the up-regulation of genes implicated in the synthesis or signalling of ABA suggest an active involvement of the ABA/GAs balance in the differences in capacity of induction of secondary seed dormancy found between the accessions. However, a larger number of differentially expressed genes between the accessions, especially those related to ABA biosynthesis and signalling pathways, was expected based on the literature related to primary seed dormancy of the last decades (Finkelstein et al., Reference Finkelstein, Reeves, Ariizumi and Steber2008; Dekkers and Bentsink, Reference Dekkers and Bentsink2015; Matilla et al., Reference Matilla, Carrillo-Barral and Rodríguez-Gacio2015; Tuan et al., Reference Tuan, Kumar, Rehal, Toora and Ayele2018; Sano and Marion-Poll, Reference Sano and Marion-Poll2021).
There is abundant evidence that ABA and ethylene play important roles in the regulation of seed dormancy (Finkelstein et al., Reference Finkelstein, Reeves, Ariizumi and Steber2008). Ethylene can promote seed germination and repress seed dormancy establishment as it antagonizes the ABA pathway (Finkelstein et al., Reference Finkelstein, Reeves, Ariizumi and Steber2008; Linkies and Leubner-Metzger, Reference Linkies and Leubner-Metzger2012). However, the ethylene-ABA antagonism during seed dormancy and germination is still poorly understood (Wang et al., Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013). Ethylene's biosynthesis is controlled by the 1-AMINOCYCLOPROPANE-1-CARBOXYLIC ACID OXIDASE (ACO), which is involved in counteracting the inhibiting effects of ABA on endosperm cap weakening and endosperm rupture (Linkies et al., Reference Linkies, Muller, Morris, Tureckova, Wenk, Cadman, Corbineau, Strnad, Lynn, Finch-Savage and Leubner-Metzger2009). The main role of ethylene could be related to the promotion of radial cell expansion in the embryonic hypocotyl, decreasing the seed water potential and increasing the activity of cell wall hydrolases in the endosperm cap (Kucera et al., Reference Kucera, Cohn and Leubner-Metzger2005). In research carried out by Wang et al. (Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013), seeds of the snl1 snl2-1 double mutant (with higher germination than non-mutant Arabidopsis Columbia seeds) presented affected transcription levels of genes regulating the ethylene pathways, including the up-regulation of the genes ACO1, ACO4, ETHYLENE-RESPONSIVE TRANSCRIPTION FACTOR (ERF) 105, ERF9 and ERF112. This pointed to the involvement of SNL1 and SNL2 in seed dormancy by repressing ethylene synthesis and response. As mentioned above, great differences in expression of a potential SNL1 [Cbp42606 (1633.68)] were found within our study, a result that will be discussed later on.
Within the over-represented GO-terms of the down-regulated DEGs, some were related to responses and signalling pathways of ethylene. Specific genes related to its synthesis or responses could be found, such as ETHYLENE RESPONSE FACTORS (ERFs). Some of the down-regulated sequences were 1-AMINOCYCLOPROPANE-1-CARBOXYLATE OXIDASE 1 (ACO1) (-526.98 and -244.23), ERF4-LIKE (-1123.97 and -10.10), ERF109-LIKE (-14.21), ERF105 (-12.27 and -4.61), APETALA 2 (AP2)-LIKE ETHYLENE-RESPONSIVE TRANSCRIPTION FACTOR AIL1 (-4.38 and -1.58), ETHYLENE-RESPONSIVE TRANSCRIPTION FACTOR CRF1 (-3.51), among others. These results are in accordance with the hypothesis of Paul et al. (Reference Paul, Jha, Bhardwaj, Singh, Shankar and Kumar2014) and Wang et al. (Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013), in which ethylene signalling pathways are important in the regulation of dormancy, and also with previous works that demonstrate that ethylene expression is partially regulated by histone acetylation and deacetylation (Wang et al., Reference Wang, Zhang and Qiao2020).
Aside from GAs and ethylene, the roles of BRs in the promotion of germination by improving growth potential in a GA-independent manner have begun to be elucidated (Leubner-Metzger, Reference Leubner-Metzger2001; Hao et al., Reference Hao, Yang, Yue, Wang, Horvath and Wang2017; Xu et al., Reference Xu, Tang, Cui, Chen, Ma, Zhu, Li, Shan, Liu and Wan2020). They constitute another antagonist of ABA (Kucera et al., Reference Kucera, Cohn and Leubner-Metzger2005). Although no GO-terms related to BRs were within the over-represented GOs, multiple key genes implicated in BRs synthesis, signalling pathways and homeostasis were markedly down-regulated. Some of them could be highlighted, for example those encoding BAHD ACYLTRANSFERASE BIA1 (-226.23 and -11.17), CYTOCHROME P450 708A2-LIKE (-161.72 and -89.40), PROBABLE WRKY TRANSCRIPTION FACTOR 46 (-25.35 and -6.99), BRASSINOSTEROID INSENSITIVE 1 (BRI1)-ASSOCIATED RECEPTOR KINASE 1 (BAK1) (-4.47), PROTEIN BRI1-5 ENHANCED 1 (BEN1) (-3.97 and -3.78), BRASSINOSTEROID SIGNALLING POSITIVE REGULATOR (BZR1) FAMILY PROTEIN (BES1) (-3.80 and -3.16) and BRI1 (-2.00 and -1.95), although a negative regulator was also down-regulated: BRI1 KINASE INHIBITOR 1-LIKE (BKI1) (-3.22). Most of these genes were previously found to be up-regulated during the germination of peanut seeds (Xu et al., Reference Xu, Tang, Cui, Chen, Ma, Zhu, Li, Shan, Liu and Wan2020). BES1 forms a transcriptional repressor complex with TPL-HDA19, which directly facilitates the histone deacetylation of ABI3 chromatin, leading to the transcriptional repression of ABI3 and consequently ABI5 (Ryu et al., Reference Ryu, Cho, Bae and Hwang2014). BRI1 has been proposed to play a role in the cold stratification pathway for releasing primary seed dormancy and triggering germination (Kim et al., Reference Kim, Warpeha and Huber2019). These differences of expression in several genes of the BR signalling pathways are consistent with a lower dormancy/higher germination capability of the non-deep accession in comparison with the deep dormant one.
Overall, auxins were the phytohormones with the greatest number of associated GO-terms. They are hormones that can regulate plant development by induction of cell elongation and division (Campanoni and Nick, Reference Campanoni and Nick2005; Chapman and Estelle, Reference Chapman and Estelle2009). Their biosynthesis has been found to be of great importance in seed germination as it is essential for hypocotyl elongation (Kucera et al., Reference Kucera, Cohn and Leubner-Metzger2005; Bai et al., Reference Bai, Novák, Ljung, Hanson and Bentsink2018). Auxins can also affect ABA and GAs signalling pathways through their transport in the root tip, where AUX1 plays an essential role (Wang et al., Reference Wang, Chen, Li, Cao, Ding, Zhang, Zuo, Xu, Xu, Deng, Xiang, Soppe and Liu2016). Low auxins levels during the deep dormant stage of tea buds and other species have been observed (Li et al., Reference Li, Junttila, Ernstsen, Heino and Palva2003; Nagar and Sood, Reference Nagar and Sood2006), suggesting the need of changes in auxins content and auxin-associated genes for dormancy transitions in these systems (Hao et al., Reference Hao, Yang, Yue, Wang, Horvath and Wang2017). Moreover, Carrera et al. (Reference Carrera, Holman, Medhurst, Dietrich, Footitt, Theodoulou and Holdsworth2008) found auxins efflux and influx transporters up-regulated in after-ripened seeds compared with dormant seeds.
A high number of auxin-related genes was found in the list of down-regulated DEGs. Some examples of genes known to play an important role in auxin synthesis or their signalling pathways should be highlighted, such as those encoding INDOLE-3-ACETIC ACID-AMINO SYNTHETASE GH3.6 (-133.53 and -2.38) and GH3.3 (-24.06 and -8.72), ABC TRANSPORTER B FAMILY MEMBER 19 (ABCB19) (-3266.98), MYROSINASE 4 (TGG4) (-1406.78 and -190.45), AUXIN TRANSPORTER PROTEIN 1/AUXIN RESISTANT 1 (AUX1) (-487.14), AUXIN EFFLUX CARRIER COMPONENT 6 (PIN6) (-421.66), PROTEIN WALLS ARE THIN (WAT1) (-172.76) and ERF109-LIKE (-14.21). However, two repressors of auxin response genes were also down-regulated: AUXIN-RESPONSIVE PROTEIN IAA17 and IAA27 (-23.74 and -23.53, respectively). The up-regulation of a potential SNL1 [Cbp42606 (1633.68)] and down-regulation of AUX1 in this study (i.e. lower expression of a potential SNL1 and higher expression of AUX1 in the non-deep accession) would be in accordance with Wang et al. (Reference Wang, Chen, Li, Cao, Ding, Zhang, Zuo, Xu, Xu, Deng, Xiang, Soppe and Liu2016), where AUX1 was proven to be positively implicated in seed germination and negatively regulated by the histone deacetylases SNL1 and SNL2.
Overall, phytohormone synthesis or signalling was generally up-regulated for ABA (e.g. NCED6, NCED2, ABCG40, ABI3) and down-regulated for GAs (GA20ox1, GA20ox2, bHLH93), ethylene (ACO1, ERF4-LIKE, ERF105, ERF109-LIKE), BRs (BIA1, CYP708A2-LIKE, probable WRKY46, BAK1, BEN1, BES1, BRI1) and auxin (GH3.3, GH3.6, ABCB19, TGG4, AUX1, PIN6, WAT1), while several ABA repressors or sequences related to its catabolism were down-regulated (e.g. AFP4, ARAC7, ARAC10, ZFP8, ZFP3, ABI1, CYP707A3 and CYP707A1) (Table 3). Together, these results suggest that phytohormones play an important role in controlling differences in secondary dormancy depth between accessions.
Table 3. DEGs potentially explaining the difference in secondary seed dormancy depth between two accessions of C. bursa-pastoris

The selected DEGs are from a comparison of a deep dormant (-367) versus a non-deep dormant accession (-799): both were imbibed in darkness for 3 d at 30°C in water to induce secondary seed dormancy.
Epigenetic regulation and chromatin remodelling
While the up-regulated DEGs included terms related to epigenetic regulation in the over-represented GOs, the down-regulated DEGs included these terms in the under-represented GOs. This indicates an active expression and implication of these processes in differences in secondary seed dormancy depth between the accessions. The up-regulated DEGs were enriched for ‘chromatin remodelling’, ‘lysine-acetylated histone binding’ and ‘acetylation-dependent protein binding’. Chromatin remodelling factors PROBABLE ATP-DEPENDENT DNA HELICASE CHR23 and CHR12 were both up-regulated (2.82 and 2.29). Overexpression of CHR12 or CHR23 reduced the frequency of seed germination in Arabidopsis up to 30% relative to wild-type (Leeggangers et al., Reference Leeggangers, Folta, Muras, Nap and Mlynarova2015).
Several sequences encoding histone deacetylases were within the up-regulated genes. As HDACs lack intrinsic DNA-binding activity, they are recruited to target genes through association with transcription factors or by incorporation into large multiprotein transcriptional complexes (Luo et al., Reference Luo, Cheng, Xu, Yang and Wu2017). Up-regulated HDACs included HISTONE DEACETYLASE 14 (HDA14) (35.98), HISTONE DEACETYLASE 6-LIKE (HDA6-LIKE) (29.62) and HISTONE DEACETYLASE-LIKE PROTEIN (HDA-LIKE) (3.50 and 2.58). HISTONE DEACETYLASE 6 (HDA6) and HDA19 have partially redundant functions in regulating seed germination, embryo development and salt resistance (Tanaka et al., Reference Tanaka, Kikuchi and Kamada2008) and both can interact with HISTONE DEACETYLASE COMPLEX 1 (HDC1), SNLs and MULTICOPY SUPRESSOR OF IRA1 (MSI) in order to repress gene expression and regulate plant development (Chen and Wu, Reference Chen and Wu2010). The AT-HOOK MOTIF NUCLEAR-LOCALIZED PROTEIN 22 (AHL22) was also highly up-regulated (241.89 and 28.15). This protein acts as a chromatin remodelling factor, interacting with HDA6, and modulating both H3 acetylation (through deacetylation of acetylated histones) and methylation of FLOWERING LOCUS T (FT) (Yu et al., Reference Yu, Liu, Luo, Chen, Lin, Tian, Lu, Cui and Wu2011). Another up-regulated gene, the PHD FINGER PROTEIN ING2 (88.43), codes for a protein that is a native subunit of the repressive complex mSin3a-HDAC1 in mammalian cells (Shi et al., Reference Shi, Hong, Walter, Ewalt, Michishita, Hung, Carney, Peña, Lan, Kaadige, Lacoste, Cayrou, Davrazou, Saha, Cairns, Ayer, Kutateladze, Shi, Côté, Chua and Gozani2006).
The proteins PAIRED AMPHIPATHIC HELIX SWI-INDEPENDENT3 (SIN3)-LIKE (SNL) 1 and SNL2 can act as components of a HDAC-SNL complex capable of modulating the transcription of genes through histone deacetylation (Wang et al., Reference Wang, Cao, Sun, Li, Chen, Carles, Li, Ding, Zhang, Deng, Soppe and Liu2013). Out of 45 annotated PAIRED AMPHIPATHIC HELIX proteins in the C. bursa-pastoris genome, two were up-regulated [Cbp42606 (1633.68) and Cbp47861 (1.41)]. The annotation results identified them as most closely related to SNL4 and SNL6 (Supplementary Data S3), although the most similar sequences in Arabidopsis were SNL1 and SNL5 (Supplementary Data S7). Further research is needed to establish their exact identity in C. bursa-pastoris and their potential role in secondary seed dormancy depth variation. The high up-regulation (1633.68) of a potential SNL1 indicates possible similarities between epigenetic regulation of primary and secondary seed dormancy.
However, in the up-regulated genes, there were also sequences implicated in histone acetyltransferase activity, such as the CHROMATIN MODIFICATION-RELATED PROTEIN EAF6 (3.48). EAF6 is a small 13-kDa protein that in yeast forms part of the PICCOLO NUCLEOSOME ACETYLTRANSFERASE OF HISTONE H4 (NuA4) complex but its contribution to the transcriptional regulation mediated by NuA4 has not been fully addressed (Espinosa-Cores et al., Reference Espinosa-Cores, Bouza-Morcillo, Barrero-Gil, Jiménez-Suárez, Lázaro, Piqueras, Jarillo and Piñeiro2020). Another example is the gene transcription regulator of RNA polymerase II, SPT-ADA-GCN5-ACETYLTRANSFERASE (SAGA) SUBUNIT, with a fold change of 2.55.
As mentioned above, the down-regulated DEGs presented under-representation of GO-terms related to epigenetic regulation, with terms such as: ‘histone acetylation’, ‘peptidyl-lysine acetylation’, ‘internal peptidyl-lysine acetylation’, ‘histone modification’, ‘histone acetyltransferase activity’ or ‘peptide-lysine-N-acetyltransferase activity’, as well as ‘chromosome organization’ and ‘chromatin organization’. Despite the fact that the acetylation categories were under-represented, a few examples were still found, such as BZIP TRANSCRIPTION FACTOR 11 (-8.66 and -8.19), which physically interacts with transcription factor ADAPTOR PROTEIN ADA2b to promote recruitment of SAGA-like histone acetyltransferase complexes to specific auxin-responsive genes (Weiste and Dröge-Laser, Reference Weiste and Dröge-Laser2014). In this way, the bZIP11 transcription factor is able to recruit the histone acetylation machinery to open up packed chromatin. Another example of a down-regulated sequence implicated in histone acetylation is AT-RICH INTERACTIVE DOMAIN-CONTAINING PROTEIN 1A-LIKE (ARID1A-LIKE) (-7.41), which is responsible for maintaining the levels of histone acetylation between the vegetative nucleus and the sperm nuclei in pollen by associating with a histone deacetylation machinery (Zheng et al., Reference Zheng, He, Zheng, Wu and McCormick2014).
With respect to histone methylation, several sequences were within the up-regulated genes, such as HISTONE-LYSINE N-METHYLTRANSFERASE (1.55), SUVH7 (12.22) and SUVH9 (5.93), although others were down-regulated, including CLF ISOFORM X1 (-2.60), ASHH1 (-1.92) and SUVH3-LIKE (-1.56). However, CLF was suggested to be a repressor of a positive regulator of primary dormancy (DOG1), as it was found to be negatively correlated with seed dormancy in the soil seed bank by Footitt et al. (Reference Footitt, Müller, Kermode and Finch-Savage2015) in the Cape Verdi Islands (Cvi) Arabidopsis deep dormant genotype. Demethylases were also both up- and down-regulated, as shown by LYSINE-SPECIFIC HISTONE DEMETHYLASE 1 HOMOLOG 1 (LDL1) (4.06 and -4.43), LYSINE-SPECIFIC DEMETHYLASE JMJ30 (-4.05, -2.33), JMJ25 ISOFORM X2 (-2.80) and JMJ14 ISOFORM X1 (1.53). JMJ30 is a histone demethylase that demethylates Lys-36 of histone H3 with a specific activity for H3K36me3 and H3K36me2. H3K36me3 acts as a mark for HDACs to bind to and deacetylate the histone, which would prevent run-away transcription. Thus, the down-regulation of this demethylase is preventing the removal of methylation from H3 and hence allowing HDACs to bind to and deacetylate the histone (Yan et al., Reference Yan, Shen, Chen, Bao, Thong and Yu2014).
Regarding DNA methylation, we observed differential regulation of DNA (CYTOSINE-5)-METHYLTRANSFERASE CMT3 (-3.42), but not others (e.g. DRM2, MET1). Finally, HEN1 SUPPRESSOR 1 (HESO1) uridylates miRNAs and siRNAs, thereby leading to their degradation. Its low fold change in our results (-158.33) could be indicating an over-accumulation of small RNAs being stabilized by methylation catalyzed by HEN1 (small RNA methyl transferase) (Ren et al., Reference Ren, Xie, Zhang, Vinovskis, Chen and Yu2014).
Altogether, these results indicate the need of a general up-regulation of genes related to histone deacetylation and a down-regulation of specific sequences implicated in histone acetylation processes in the deep dormant accession in comparison with the non-deep dormant one in order to establish a deeper secondary seed dormancy state (Table 3). Nevertheless, as indicated before, some contradictions could be found, such as the up-regulation of sequences implicated in histone acetylation or the down-regulation of HISTONE DEACETYLASE 14 (-16.92), which was also within the up-regulated DEGs (35.97). These contradictions might be explained either by the functions of the genes they control (positive or negative regulators of dormancy), or by variations in the acetylation patterns found between different areas or parts of the seeds when imbibed in water in darkness at 30°C, as described previously (Gomez-Cabellos et al., Reference Gomez-Cabellos, Toorop, Cañal, Iannetta, Fernández-Pascual, Pritchard and Visscher2021). In that work, the immunolocalizations showed that after different periods of imbibition in darkness at 30°C, certain parts of the seed presented higher H4Ac signals than others, which would be in accordance with the apparent contradictions described here.
With respect to histone (de)methylation, we observed both up- and down-regulation of genes involved, although the expression patterns were generally in accordance with the difference in secondary dormancy depth between the accessions (Table 3). However, this was not always the case, such as for the LYSINE-SPECIFIC HISTONE DEMETHYLASE 1 HOMOLOG 1 (LDL1), which presented both down- (-4.43) and up-regulation (4.06). LDL1 acts redundantly with LDL2 in repressing primary seed dormancy (Zhao et al., Reference Zhao, Yang, Liu and Wu2015). The ldl1 ldl2 double mutant displays increased seed dormancy, whereas overexpression of LDL1 or LDL2 in Arabidopsis causes reduced dormancy. Our results show that LDL1 may not play a significant role in secondary dormancy depth between accessions. Regarding DNA methylation, the fact that only one DNA methylase was differentially regulated seems to be in accordance with results from previous work that showed elevated global DNA methylation levels after 3 d in darkness at 30°C in seeds from both the deep and non-deep dormant accessions studied here (Gomez-Cabellos et al., Reference Gomez-Cabellos, Toorop, Cañal, Iannetta, Fernández-Pascual, Pritchard and Visscher2021).
Thus, these results indicate an active involvement of epigenetic regulation in the establishment of different secondary seed dormancy depths. Moreover, it seems that histone (de)acetylation and (de)methylation play a larger role in establishing differences in secondary seed dormancy depth than DNA methylation. However, the effect of specific epigenetic processes on the level of secondary seed dormancy depth could potentially depend on the function of the genes they regulate (negative or positive regulators of dormancy), the area or part of the seed, or the time period since secondary dormancy induction. Future studies could focus on particular genes, sub-sections of seeds or different time points to further elucidate the epigenetic mechanisms underlying differences in secondary seed dormancy depth.
Conclusion
The HDAC inhibitors TSA and valproic acid caused a delay of t50 for germination but not of t50 for testa rupture in comparison with their respective controls, demonstrating they were lowering the speed of germination. In addition, valproic acid exposure led to an increase in secondary dormancy depth. Transcriptome analysis of non-deep dormant seeds exposed to valproic acid or water revealed that even though sequences related to ABA showed complex regulation in seeds treated with valproic acid, the synthesis of GAs (GA20ox1), BRs (CYP708A2) and auxins (CYP79B3, SNL1) was negatively regulated (Table 2), thereby providing a potential mechanistic explanation for the observed delay in t50 for germination in seeds treated with HDAC inhibitors. Among those genes implicated in epigenetic regulation, SNL1 could be involved in the enhanced secondary seed dormancy observed.
Transcriptome analysis of the deep dormant versus non-deep dormant seeds comparison showed that differential regulation of phytohormone-related genes may play an important role in secondary dormancy depth of C. bursa-pastoris (Table 3). Taking into account all the ABA-related DEGs, most of the up-regulated genes were promoters of its synthesis or signalling (e.g. NCED6, NCED2, ABCG40 and ABI3), while in the down-regulated set, the most abundant were repressors or sequences related to its catabolism (e.g. AFP4, ARAC7, ARAC10, ZFP8, ZFP3, ABI1, CYP707A3 and CYP707A1). In contrast, several key (regulators of) genes related to the biosynthesis of gibberellins were down-regulated, such as GA20ox1, GA20ox2 and bHLH93. Down-regulation was also observed for important genes involved in the synthesis and signalling pathways of ethylene (ACO1, ERF4-LIKE, ERF105, ERF109-LIKE), BRs (BIA1, CYP708A2-LIKE, probable WRKY46, BAK1, BEN1, BES1 and BRI1) and auxin (GH3.3, GH3.6, ABCB19, TGG4, AUX1, PIN6 and WAT1). Future research could confirm whether the differential regulation of a potential SNL1, several histone deacetylases and associated genes (HDA14, HDA6-LIKE, HDA-LIKE, ING2, JMJ30), sequences linked to acetylases (bZIP11, ARID1A-LIKE), or to gene silencing through histone methylation (SUVH7, SUVH9, CLF), may be responsible for the observed differential expression of genes related to phytohormones between accessions of C. bursa-pastoris varying in secondary dormancy depth.
Author contributions
S.G.-C.: conceptualization, methodology, investigation, formal analysis, visualization, writing (original draft), writing (review and editing); P.E.T.: supervision, project administration, conceptualization, methodology, investigation, writing (review and editing); E.F.-P.: formal analysis, visualization, writing (original draft), writing (review and editing); P.P.M.I.: resources (ecotypes), writing (review and editing); H.W.P.: project administration, resources, supervision, writing (review and editing); A.M.V.: supervision, conceptualization, methodology, formal analysis, writing (original draft), writing (review and editing).
Supplementary material
To view supplementary material for this article, please visit: https://doi.org/10.1017/S0960258522000265.
Acknowledgements
We would like to thank STAB VIDA for the sequencing of our mRNA samples and the analysis of differentially expressed genes. In addition, we thank Mauro Maver for critical reading of the manuscript.
Financial support
This work was supported by grant-in-aid from the Department for Environment, Food and Rural Affairs to the Royal Botanic Gardens, Kew. E.F.P. was supported by the Jardín Botánico Atlántico (SV-20-GIJON-JBA). P.P.M.I. and The James Hutton Institute were supported by the Rural and Environmental Science and Analytical Services (RESAS), a division of the Scottish Government. The funders had no role in study design, report writing and article submission.
Conflicts of interest
The authors declare that they have no conflict of interest.









