Introduction
Safflower (Carthamus tinctorius L., Compositae: Asteraceae) (2n= 24) is a multi-purpose oilseed crop grown for vegetable oil, animal feed, natural dyes and medicinal uses globally (Li and Mündel, Reference Li and Mündel1996). The oil in safflower contains highest polyunsaturated fatty acid (>80%) among edible oils (Kostik et al., Reference Kostik, Memeti and Bauer2012). Genetic variability for agro-morphological and biochemical traits has been well documented in safflower (Fernández-Martinez et al., Reference Fernández-Martinez, del Rio and de Haro1993; Pascual-Villalobos and Alburquerque, Reference Pascual-Villalobos and Albuquerque1996; Johnson et al., Reference Johnson, Bergman and Flynn1999; Dwivedi et al., Reference Dwivedi, Upadhyaya and Hegde2005; Amini et al., Reference Amini, Saeidi and Arzani2008; Mahasi et al., Reference Mahasi, Wachira, Pathak and Riungu2009; Elfadl et al., Reference Elfadl, Reinbrecht and Claupein2010; Safavi et al., Reference Safavi, Pourdad and Safavi2012; Yeilaghi et al., Reference Yeilaghi, Arzani, Ghaderian, Fotovat, Feizi and Pourdad2012; Majidi and Zadhoush, Reference Majidi and Zadhoush2014). It is widely recognized that DNA markers in combination with phenotypic descriptors provide better understanding of plant genetic resources. The multi-locus DNA markers namely amplified fragment length polymorphism (AFLP) (Johnson et al., Reference Johnson, Kisha and Evans2007; Kumar et al., Reference Kumar, Ambreen, Murali, Bali, Agarwal, Kumar, Goel and Jagannath2014), randomly amplified polymorphic DNA (Khan et al., Reference Khan, Witzke-Ehbrecht, Maass and Becker2009), inter-SSR (Yang et al., Reference Yang, Wu, Zheng, Chen, Liu and Huang2007; Golkar et al., Reference Golkar, Arzani and Rezaei2011; Panahi and Neghab, Reference Panahi and Neghab2013; Majidi and Zadhoush, Reference Majidi and Zadhoush2014; Yaman et al., Reference Yaman, Tarıkahya-Hacıoğlu, Arslan and Subaşi2014) and sequence-related amplified polymorphism (Talebi et al., Reference Talebi, Mokhtari, Rahimmalek and Sahhafi2012) have been used extensively to elucidate genetic diversity and species relationships in safflower.
Microsatellites or SSRs are considered ideal genetic markers for assessment of diversity in germplasm collections due to their desirable properties: abundant, locus specific, co-dominant, multi-allelic, high polymorphism and reproducibility. Substantial efforts have been made to develop Expressed Sequence Tag (EST) and genome-based SSR markers (Chapman et al., Reference Chapman, Hvala, Strever, Matvienko, Kozik, Michelmore, Tang, Knapp and Burke2009; Hamdan et al., Reference Hamdan, Garcia-Moreno, Redondo-Nevado, Velasco and Perez-Vich2011; Yamini et al., Reference Yamini, Ramesh, Naresh, Rajendrakumar, Anjani and Dinesh Kumar2013; Lee et al., Reference Lee, Sung, Lee, Chung, Yi, Kim and Lee2014) and their application for germplasm characterization in safflower (Chapman et al., Reference Chapman, Hvala, Strever and Burke2010; Barati and Arzani, Reference Barati and Arzani2012; Derakhshan et al., Reference Derakhshan, Majidi, Sharafi and Mirlohi2014). However, the use of SSR markers to analyze genetic diversity in large collections of safflower core accessions has not yet been reported.
India is among major producers of safflower seed in the world (0.15 million tonnes in 2012) (FAOSTAT, 2012, http://faostat.fao.org). Its cultivation continues to decline in India since 1988, which is mainly attributed to low productivity caused by low oil yield potential of the cultivars and susceptibility to biotic (wilt and aphid) and abiotic (moisture and nutrient deficiency) stresses (Nimbkar, Reference Nimbkar, Knights and Potter2008). The situation demands broadening genetic base of breeding materials by exploiting diverse genetic resources available in the country and elsewhere. India maintains the largest safflower germplasm collection (>6000 accessions) (Mukta, Reference Mukta, Knights and Potter2008) and an extensive network of safflower breeding programmes. Identification of a subset of accessions that represents maximum possible genetic diversity of the whole collection would facilitate better exploitation of genetic resources for breeding as well as mining of useful genes (Reeves et al., Reference Reeves, Panella and Richards2012). At the Indian Council of Agricultural Research (ICAR)-Indian Institute of Oilseeds Research (IIOR), a core set of safflower accessions has been identified based on variability in agronomic traits (Alivelu and Mukta, Reference Alivelu and Mukta2012). In this study, our aim was to characterize a subset of 148 accessions of the core collection for genetic diversity, population structure and linkage disequilibrium (LD) using SSR markers to enable its utilization in breeding as well as trait mapping purposes.
Materials and methods
Plant material
A core subset of 148 safflower accessions were used in this study (Table S1, available online). The accessions represented 15 countries namely India (105), Mexico (13), USA (8), Italy (4), Iran (3), Turkey (2), Afghanistan (2), Sudan (2), Pakistan (1), Israel (1), Australia (1), Hungary (1), Portugal (1), Belgium (1) and China (1). The origin of two accessions was uncertain. The accessions were maintained at the germplasm management unit (GMU) of IIOR. The core subset of accessions was chosen from a core of 620 accessions, which was developed earlier by Alivelu and Mukta (Reference Alivelu and Mukta2012) using the procedure briefly described here. A total of 6201 accessions were arrayed into 20 distinct clusters based on 13 quantitative descriptors using Ward's minimum variance method (Ward, Reference Ward1963). From each cluster, stratified sampling was carried out to establish the core set of 620 (10% of total) accessions. The statistical parameters namely mean difference (MD) percentage, variance difference (VD) percentage, coincidence rate (CR) of range and variable rate (VR) of coefficient of variation (Wang et al., Reference Wang, Guan, Wang, Zhu, Wang, Hu and Hu2014) were used to evaluate the representativeness of the core with the original collection. A core collection is considered to be the representative of the initial collection under the following situations: (1) when no more than 20% of the traits have different means (significant at a= 0.05) between the core collection and the initial collection; (2) the CR retained by the core collection is no less than 80%; (3) higher VD and VR. The values of MD, VD, CR and VR of the core were 0, 60.1, 87.8 and 98.4, respectively, which indicted the representativeness of the core with the original collection.
Trait evaluation
The accessions (15 plants per accession) were grown on a single row basis (3 m length; 45 cm × 20 cm spacing) for phenotypic evaluation. The quantitative traits namely days to 50% flowering, days to physiological maturity, plant height (cm), number of primary branches per plant, number of capitula per plant, diameter of main capitula (cm), 100-seed weight (g) and oil content (%) were measured based on the standard procedures of trait evaluation prescribed by Bioversity International (IBPGR, 1983). Data on days to 50% flowering and days to physiological maturity were collected on row basis. Days to physiological maturity was recorded when bracts of the first formed capitula turned brown and only little green remained on the latest formed capitula in more than 75% of the plants. Data on plant height, number of primary branches per plant, number of capitula per plant and diameter of main capitula were collected from five plants. Data on 100-seed weight and oil content were collected from a pooled seed sample. The estimation of oil content (%) was done in nuclear magnetic resonance analyser using about 20 g of seed samples with the moisture content of about 7–8%. The evaluation trials were conducted in alfisol at the research farm of IIOR during October to February months of two consecutive years, 2012–13 and 2013–14.
SSR analysis
DNA was extracted from the pooled leaf samples (ten plants per accession) using the protocol developed by Doyle and Doyle (Reference Doyle and Doyle1987). Forty-four SSR primer pairs representing 11 out of 12 linkage groups (LG) were chosen based on their map position published by Mayerhofer et al. (Reference Mayerhofer, Archibald, Bowles and Good2010). A number of SSR loci on each LG were 7 (LG-1), 3 (LG-2), 3 (LG-3), 5 (LG-4), 8 (LG-5), 5 (LG-6), 3 (LG-7), 3 (LG-8), 3 (LG-9), 3 (LG-10) and 1 (LG-11). The details of SSR primer pairs are provided in Table S2 (available online). The PCR was carried out with the following conditions: 94°C for 5 min for initial denaturation, 35 cycles of 94°C for 30 s for denaturation, 55°C for 30 s for annealing, and 72°C for 30 s for extension followed by 72°C for 5 min for final extension in a thermocycler (BioRad Laboratories, Inc.). Different annealing temperatures were maintained depending upon the requirement for a specific primer pair. The PCR products were resolved in 6% polyacrylamide gel electrophoresis (BioRad Laboratories, Inc.) and the polymorphisms were visualized after silver staining. The polymorphic SSR alleles were scored as co-dominant markers using different characters such as 1, 2, 3 etc.
Data analysis
Trait variation
The basic statistics namely mean, range and standard deviation (SD) values for each trait were obtained using Cropstat version 7.2 (IRRI, 2009).
Genetic diversity estimates
The number of alleles (N A), observed heterozygosity (H o), gene diversity (expected heterozygosity, H e) and polymorphism information content (PIC) were obtained using the software program PowerMarker version 3.25 (Liu and Muse, Reference Liu and Muse2005).
Genetic relatedness
Neighbour-joining (NJ) tree was constructed based on pair-wise simple matching coefficients as implemented in Dissimilarity Analysis and Representation for windows V.5.0.158 (Perrier and Jacquemoud-Collet, Reference Perrier and Jacquemoud-Collet2006) to depict genetic relationships among accessions. Bootstrap analysis was carried out with 1000 replications.
Population structure
STRUCTURE 2.3.3 program (Pritchard et al., Reference Pritchard, Stephens and Donnelly2000) was used to detect the number of populations (K) within the collection of 148 accessions. The possible number of K was assumed from 1 to 10 in order to determine the optimal K. The mean posterior probability (LnP(D)) values per K were obtained based on ten replications. The delta K measure (Evanno et al., Reference Evanno, Regnaut and Goudet2005) was used to determine the K as implemented in the online version of STRUCTURE HARVESTER (http://tayloro.biologyucla.edu/Struct_harvest) (Earl and VonHoldt, Reference Earl and von Holdt2012). The membership coefficient of each genotype in the putative number of populations was obtained using the admixture model with a burn-in period of 200,000 and replications of 500,000. The accessions with membership coefficient of more than 0.75 were assigned to the respective population and less than 0.75 were assigned to the admixture group. The STRUCTURE analysis was repeated for each population to determine subpopulations within them. Pair-wise F st estimates among populations were obtained and hierarchical analysis of molecular variance (AMOVA) was carried out to determine the extent of genetic differentiation among populations using the software Arlequin 3.11 with 1000 permutations (Excoffier et al., Reference Excoffier, Laval and Schneider2005).
LD between SSR loci
The pair-wise LD was determined based on squared allele-frequency correlations (r 2) among 44 SSR loci as implemented in the software program PowerMarker version 3.25 (Liu and Muse, Reference Liu and Muse2005) with 1000 permutations. The LD between SSR loci was considered significant when r 2 values were ≥ 0.1 (Abdurakhmonov and Abdukarimov, Reference Abdurakhmonov and Abdukarimov2008).
Results
Trait variation
The variation observed in the core subset of 148 safflower accessions for agronomic traits is presented in Table 1. Days to 50% flowering and physiological maturity showed narrow range of variation with about 3 week difference from the early to late accessions. Maximum oil content of 33% was observed in the collection. However, considerable differences were observed for seed yield-related traits namely number of primary branches per plant, number of capitula per plant, diameter of main capitula and 100-seed weight.
Table 1 Phenotypic variation in the core subset of 148 safflower accessions based on the evaluation trial for two consecutive years

Genetic diversity
Forty-four polymorphic SSR primer pairs produced a total of 148 alleles across 148 safflower accessions. The number of SSR alleles ranged from 2 to 15 (ct-47) with an average of 3.6 per locus. Majority of primer pairs (19) produced only two alleles. The major allele frequency ranged from 0.244 to 1.000 with an average of 0.824. About 12 rare alleles (frequency of ≤ 0.0074) were found. The H o values ranged from 0.000 to 0.041 with an average of 0.003. The H e values ranged from 0.013 to 0.866 with an average of 0.314. The PIC values of SSR primer pairs ranged from 0.013 to 0.853 with an average of 0.284. The locus-wise details are provided in Table 2. Gel profile showing the allelic variation of SSR locus ct-246 across safflower accessions is provided in Fig. 1.
Table 2 Mean and range values of genetic diversity measures in the safflower core subset of 148 accessions based on 44 polymorphic simple sequence repeat (SSR) loci

PIC, polymorphism information content.

Fig. 1 Gel profile showing the allelic variation of SSR locus ct-246 across safflower accessions.
Genetic relationships
NJ tree showed five major genotypic clusters within the collection of 148 accessions with very low bootstrap support (Fig. 2). Cluster 1 (named as G1) included 27 accessions and cluster 2 (G2) included 25 accessions. Cluster 3 was a major group consisting of 79 accessions with several subgroups. Cluster 4 (G4) and cluster 5 (G5) consisted of eight and nine accessions, respectively. The details of genotypic clusters and geographical distribution of accessions within the clusters are provided in Table S1 (available online). Overall, pair-wise simple matching coefficients ranged from 0.02 (EC-181956-EC-181526) to 0.6 (EC-181267-IC-545045) with an average of 0.2682. The pair-wise simple matching coefficients of G1 ranged from 0.0957 (EC-337879-IC-442753) to 0.450 (EC-151819-EC-159643), G2 ranged from 0.14 (EC-210538-1-EC566072) to 0.573 (EC-181513-IC-338427), G3 ranged from 0.02 (EC-181956-EC-181526) to 0.48 (EC-181267-GMU 4773), G4 ranged from 0.0426 (EC-566012-EC-337155) to 0.14 (IC-337789-IC-443009) and G5 ranged from 0.0625 (EC-337318-EC-337999) to 0.36 (EC-337943-EC-338179). The accessions originated from India, Mexico and USA were distributed across clusters. The accessions from Pakistan, Israel, Australia, Hungary, Portugal, Belgium and China grouped in different clusters.

Fig. 2 NJ tree-based dendrogram depicting genetic relationships among 148 accessions of the safflower core set. Bootstrap percentage values (>30 %) are shown. G1, G2, G3, G4 and G5 indicate major genotypic clusters.
Population structure
The mean posterior probability (LnP(D)) value for each given K increased with the increase of K, but the probable K value could not be inferred since there was no abrupt change in LnP(D) (Fig. 3a). However, delta-K (DK) analysis of LnP(D) (Evanno et al., Reference Evanno, Regnaut and Goudet2005), showed a sharp peak of DK at K= 4, suggesting four populations within the collection of 148 accessions (Fig. 3b). Based on the threshold value of membership coefficient ( ≥ 0.75), 77 accessions were assigned to four populations (hereafter referred as P1, P2, P3 and P4) and the remaining 71 accessions to the admixture group. The classification of accessions falling under respective population and admixture group are provided in Table S1 (available online) and are also depicted in the STRUCTURE bar diagram (Fig. 3c). P1 comprised of 36 accessions (32 from India, two from Mexico, one each from Turkey and China). P2 comprised of eight accessions (two from Mexico, one each from India, USA, Turkey, Pakistan, Israel and Sudan). P3 comprised of 28 accessions (23 from India, one each from Mexico, USA, Iran, Afghanistan and Belgium). P4 comprised of only five accessions (three from India and one each from Mexico and Australia). The average gene diversity between individuals in the same cluster was 0.1820, 0.3266, 0.1595 and 0.3110 for P1, P2, P3 and P4, respectively. The mean F st values within P1, P2, P3 and P4 were 0.4024, 0.3023, 0.4678 and 0.2422, respectively. Pair-wise F st values between populations were 0.41 055 (P1 and P2), 0.09 504 (P1 and P3), 0.40 145 (P1 and P4), 0.46 033 (P2 and P3), 0.30 007 (P2 and P4), and 0.48 643 (P3 and P4). P1 and P2, P1 and P4, P2 and P3, and P3 and P4 had higher F st values (>0.3), whereas P1 and P3 had the lowest F st value (0.095). The pair-wise F st value between P1 and admixture group (0.085), and P3 and admixture group (0.084) was also very low. Further subdivision of P1, P3 and admixture group showed four, six and four subpopulations, respectively. Overall, the structure analysis revealed 16 subpopulations. AMOVA partitioned the total genetic variance into two components: among and within populations. Maximum of genetic variation was explained by individuals within the populations (84.99%) and not among populations (15.01%).

Fig. 3 (a) Determination of optimum value of K in the safflower core subset of 148 accessions based on the procedure described by Pritchard et al. (Reference Pritchard, Stephens and Donnelly2000); (b) predicted value of K based on Evanno et al. (Reference Evanno, Regnaut and Goudet2005); (c) model-based clustering of the safflower core subset into four main populations. P1, P2, P3 and P4 indicate the number of populations (K) along with admixture.
LD among SSR loci
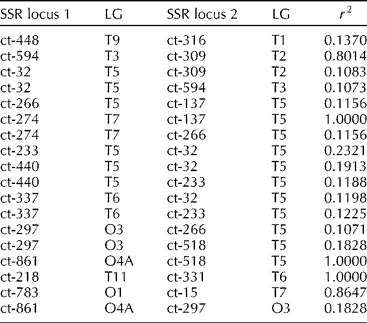
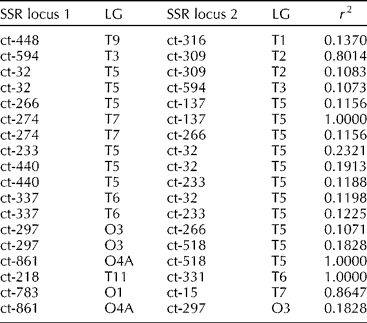
About 1.9% of SSR marker pairs (18 out of 946) showed significant LD (r 2>0.1; Table 3). Strong LD (r 2= 0.2321) was observed between the marker pair, ct-32 and ct-233, which were located very closely (~1.6 cM) on LG-5. Three marker pairs namely [ct-274]-[ct-137], [ct-861]-[ct-518] and [ct-218]-[ct-331] showed complete LD (r 2= 1). Notably, six out of nine markers namely ct-32, ct-266, ct-233, ct-440, ct-137 and ct-518 on LG-5 were in LD with other markers on different LGs.
Table 3 Linkage disequilibrium (LD) among simple sequence repeat (SSR) locus pairs from across linkage groups (LG) in the safflower core subset of 148 accessions
Discussion
Genetic diversity in the germplasm collection is the foundation for crop improvement. In this study, a core subset of 148 safflower accessions, mostly of Indian origin, were characterized for the extent of genetic diversity, genetic relatedness among accessions, population structure and LD using 44 genome-wide SSR markers. Microsatellites or SSR markers are considered ideal for characterization of plant genetic resources because they are available in large number, randomly distributed throughout the genome, multi-allelic and highly polymorphic. They are widely used to map genetic diversity in crop germplasm collections (Rao et al., Reference Rao, Kadirvel, Symonds, Geethanjali and Ebert2012). Till recently, the characterization of safflower germplasm using SSR markers has been limited perhaps due to lack of sufficient information. However, Mayerhofer et al. (Reference Mayerhofer, Archibald, Bowles and Good2010), Hamdan et al. (Reference Hamdan, Garcia-Moreno, Redondo-Nevado, Velasco and Perez-Vich2011), Yamini et al. (Reference Yamini, Ramesh, Naresh, Rajendrakumar, Anjani and Dinesh Kumar2013) and Lee et al. (Reference Lee, Sung, Lee, Chung, Yi, Kim and Lee2014) have developed a substantial number of SSR markers in safflower, but most of them have not yet been genetically mapped. For this study, 44 SSR markers were selected randomly based on their position on the linkage maps developed by Mayerhofer et al. (Reference Mayerhofer, Archibald, Bowles and Good2010). The results showed that SSR allelic diversity in the core subset of safflower germplasm was low (N A= 3.6, H e= 0.314 and PIC = 0.284). Only three out of 44 primer pairs (ct-47, ct-50 and ct-246) showed high PIC values (>0.5), which may be more useful for diagnostic applications in safflower. Low SSR polymorphism in safflower is also evident from previous studies. Sehgal and Raina (Reference Sehgal and Raina2005) reported that SSRs were less polymorphic than AFLP markers in the safflower cultivars. Hamdan et al. (Reference Hamdan, Garcia-Moreno, Redondo-Nevado, Velasco and Perez-Vich2011) reported an average of 3.2 alleles per SSR locus among ten safflower cultivars. Barati and Arzani (Reference Barati and Arzani2012) reported that EST–SSR alleles ranged from 2 to 8 with an average of 3.43 per locus in a collection of 48 genotypes belonging to three safflower species (C. lanatus, C. oxyacanthus and C. tinctorius). The PIC values ranged from 0.04 to 0.695 with an average of 0.322. Lee et al. (Reference Lee, Sung, Lee, Chung, Yi, Kim and Lee2014) reported an average of 2.8 alleles per SSR locus, H e= 0.386 and PIC = 0.325 in a collection of 100 safflower accessions from different centres of similarity. Derakhshan et al. (Reference Derakhshan, Majidi, Sharafi and Mirlohi2014) also observed low SSR polymorphism (N A= 3.81 and PIC = 0.30) in a collection that included a few accessions of wild species namely C. oxyacanthus, C. lanatus, C. dentatus, C. boissieri and C. palestinus. The level of polymorphism is influenced by the number of markers, population size and the type of plant material used in the study. In a similar study, Filippi et al. (Reference Filippi, Aguirre, Rivas, Zubrzycki, Puebla, Cordes, Moreno, Fusari, Alvarez, Heinz, Hopp, Paniego and Lia2015) reported an average of N A= 4.95, H e= 0.51 and PIC = 0.50 in a collection of 170 sunflower accessions using 42 SSR markers. Nevertheless, low SSR polymorphism in safflower is a concern because that would limit their use for trait mapping purposes. Our results showed that H o was negligible (0.002) in the germplasm collection, whereas Lee et al. (Reference Lee, Sung, Lee, Chung, Yi, Kim and Lee2014) reported higher H o (0.452). Very low H o suggested that the accessions were highly inbreds perhaps due to no outcrossing during maintenance. The accessions used in this study were maintained under protected net conditions to avoid outcrossing by pollinator bees. Although safflower is predominantly a self-pollinated species, the activity of pollinator bees contribute for outcrossing. Rudolphi et al. (Reference Rudolphi, Becker, Witzke-Ehbrecht, Knights and Potter2008) reported outcrossing of up to 63% under open field conditions. The differences in the maintenance of accessions might probably have contributed for the differences in the estimates of H o in different germplasm collections.
NJ tree analysis revealed at least five possible major clusters (G1, G2, G3, G4 and G5) in the accessions. Several sub-clusters within the major clusters were also clearly observable. Indian safflower accessions were distributed across all seven clusters. Similarly, the accessions from Mexico and USA were found in most of the clusters. These results indicated that there was no relationship between genotypic clusters and geographical origin. The accessions EC-181956 (India) and EC-181526 (Afghanistan) were the closest whereas EC-181267 (India) and IC-545045 (India) were the most divergent based on simple matching coefficients. The Indian accessions included some breeding lines or cultivars developed at various safflower research centres (Jalgaon, Varanasi, Akola, Phaltan, Indore and Parbhani). The wide range of simple matching coefficients within G1, G2 and G3 suggested that substantial diversity existed within those clusters. However, it should be noted that the clustering may not be dependable as the bootstrap percentage values were very low.
STRUCTURE is one of the most widely used software for population analysis, which helps to assess the patterns of genetic structure in a subset of samples (Porras-Hurtado et al., Reference Porras-Hurtado, Ruiz, Santos, Phillips, Carracedo and Lareu2013). The structure analysis grouped about 52% of the accessions into four main populations (P1, P2, P3 and P4) with several sub-populations and the rest 48% of the accessions into an admixture group suggesting that recognizable genetic structure existed in the collection. The average distances (gene diversity) (range 0.1595–0.3266) and F st values (range 0.2422–0.4678) within the main populations were high; therefore, further subdivision of them produced three, five and three subpopulations, respectively along with admixture groups. The pair-wise F st values among major genotypic groups, P1, P3 and admixture were low (F st< 0.1) suggesting low to moderate genetic divergence. These three groups together consisted of 91% (135) of the total number of accessions suggesting that the genetic structuring in the collection was only weak. The P2 and P4 were more divergent populations (F st= 0.30–0.48) and together consisted of 13 accessions. The P2 consisted of eight accessions originated from different countries including Turkey, Pakistan, Israel, Sudan, India, USA and Mexico. The P4 consisted of five accessions originated from India, Australia (cultivar Sirothora) and Mexico. However, it is difficult to conclude that P2 and P4 were highly diverse due to different geographical origin because only one or two accessions represented those countries. It may be noted that limited sample size, small number of markers, LD between markers may affect the ability of Bayesian methods in predicting subpopulations reliably (Porras-Hurtado et al., Reference Porras-Hurtado, Ruiz, Santos, Phillips, Carracedo and Lareu2013). Therefore, care was taken to the extent possible to select reasonably large population size and widely spaced (unlinked) markers on linkage maps for this study. Admixture model was followed considering that the collection included substantial number of breeding lines and cultivars originated from different countries. AMOVA showed that 85% of the total genetic variation was explained by the individuals within populations and only 15% was explained between populations, which also did not support for strong genetic structuring in the collection of 148 accessions.
The pattern of classification by NJ tree and STRUCTURE methods were comparable. The most divergent populations P2 and P4 detected by STRUCTURE consisted of 13 accessions, which were also present in the most diverse NJ tree based groups G1 and G2 (Table S1, available online). Notably, these accessions originated from diverse countries.
A large number of the accessions in the core subset (48%) were classified under admixture group suggesting that they have mixed ancestry. Our passport data indicate that some of the Indian accessions imported from USDA were originally collected in India. This is understandable considering that USDA's safflower collection includes a substantial number of Indian accessions, which were collected in India by Professor Knowles and the USDA accessions are used extensively in safflower breeding programmes across the world. Knowles (1969) previously reported that safflower accessions were more morphologically similar within the centres of origin, but substantially differed between centres. However, Ashri (Reference Ashri1975) found no obvious morphological divergence in the safflower collection between centres of origin, which he attributed to lack of details on origin of accessions, inclusion of recently developed varieties, unexpected exchange of seeds, etc. Khan et al. (Reference Khan, Witzke-Ehbrecht, Maass and Becker2009) also found no clear relationship of genetic diversity pattern with geographical origin of the accessions and reasoned that this could have occurred due to duplications in the gene banks. Furthermore, molecular marker data support this view by grouping the accessions randomly across all clusters (Johnson et al., Reference Johnson, Kisha and Evans2007; Khan et al., Reference Khan, Witzke-Ehbrecht, Maass and Becker2009). Chapman et al. (Reference Chapman, Hvala, Strever and Burke2010) found genetic structuring within a collection of safflower accessions from ten proposed geographical centres of similarities across the world, but genetic diversity pattern did not correspond with geographical origin. Majidi and Zadhoush (Reference Majidi and Zadhoush2014) reported that classification of 79 cultivated safflower accessions of the world-wide collection, which represented regional gene pools, was in general agreement with the available information regarding the origins of these genotypes; some accessions did not correlate with the geographic origin. Our results also suggested that the mild genetic structure found within the core subset of 148 accessions did not relate to geographical origin but might probably have occurred by breeding history and mating system. The core subset includes a number of breeding lines and cultivars developed by safflower research centres across the world. Furthermore, being a self-pollinated species, safflower is expected to show differentiated populations, which may arise by limited gene flow by pollen (Hamrick and Godt, Reference Hamrick and Godt1996).
The extent of LD (non-random association of alleles at different loci) is critical information for exploitation of core germplasm collection for genetic studies. To our knowledge, LD has not been examined in a safflower germplasm collection so far. The proportion of SSR marker pairs in significant LD in the core subset was low (1.9%), which might perhaps be underestimated due to less number of markers. Forty-four loci are too small to evaluate the extent of LD across the large genome (~1.4 Gb) of safflower (Garnatje et al., Reference Garnatje, Garcia, Vilatersana and Vallès2006). Therefore, the result of LD was only indicative and a thorough evaluation of LD in the safflower core subset would require genome-wide analysis. Lack of high density SSR linkage map in safflower was a limitation to choose markers; however, care was taken to represent the genome fairly by covering 11 out of 12 LGs as per Mayerhofer et al. (Reference Mayerhofer, Archibald, Bowles and Good2010) and to ensure most of locus pairs were possibly unlinked. The current analysis provided clear indication of LD among linked as well as unlinked SSR loci in the safflower core subset but the proportion of locus pairs in LD appeared to be low. It was interesting to note that more markers on LG-5 were in significant LD with other markers within and across LGs. The long stretched LD or LD between unlinked loci indicate the existence of other LD generating factors than linkage itself in a genome such as selection that generates LD through high frequency sweeping and fixing of alleles flanking a favoured variant (hitchhiking effect) and co-selection of loci (epistatic selection and co-adopted genes) during breeding for multiple traits (Abdurakhmonov and Abdukarimov, Reference Abdurakhmonov and Abdukarimov2008). Therefore, it would be interesting to examine if any evolutionary significance of LD region on LG-5. Pearl et al. (Reference Pearl, Bowers, Reyes-Chin-Wo, Michelmore and Burke2014) reported several quantitative trait loci (QTLs) associated with domestication-related traits in safflower. Unfortunately, the relationship of those QTLs with the LD region of LG-5 could not be analysed due to lack of common markers and linkage map.
Phenotypic diversity, genetic structure and LD in a given population are important considerations for association analysis (Stich et al., Reference Stich, Melchinger, Frisch, Maurer, Heckenberger and Reif2005; Abdurakhmonov and Abdukarimov, Reference Abdurakhmonov and Abdukarimov2008; Zhang et al., Reference Zhang, Liu, Tong, Lu and Li2014). The phenotypic evaluation of core subset of 148 accessions showed considerable variability for important agronomic traits namely number of primary branches per plant, number of capitula per plant and 100-seed weight. The collection also possessed weak genetic structure with low level of LD among unlinked SSR loci. These observations suggest that the core subset of safflower accessions could be a suitable germplasm panel for association mapping to identify genetic loci associated with seed yield components.
Supplementary material
To view supplementary material for this article, please visit http://dx.doi.org/10.1017/S1479262115000295
Acknowledgements
The authors kindly acknowledge Dr. V. Dinesh Kumar for critical review of the manuscript. They also gratefully acknowledge ICAR, New Delhi, India for financial support.