INTRODUCTION
The Trypanozoon subgenus plays a very important role in trypanosomiasis that affects a variety of mammalian species including man and domestic animals. Trypanosoma brucei gambiense and T. b. rhodesiense are responsible for sleeping sickness in humans, while T. b. brucei and T. evansi respectively cause nagana and surra in both domestic animals and game. T. equiperdum is believed to cause dourine in equines through sexual contacts.
At its surface, a trypanosome bears a homogenous coat of approximately 107 molecules of variant surface glycoproteins (VSGs) that are highly immunogenic. At any time, a given parasite expresses only 1 type of VSG. This dense layer of VSG dimers acts as a protective umbrella for underlying surface molecules such as the invariant surface glycoproteins (ISGs). Of all the bloodstream-form ISGs that have been characterized so far, including ISG64 (Jackson et al. 1993), ISG65 or ISG70 (Ziegelbauer and Overath, 1992), ISG75 (Ziegelbauer et al. 1992) and ISG100 (Nolan et al. 1997), their functions and structures remain unknown. In a bloodstream-form T. b. brucei, there are approximately 5×104 ISG75 molecules evenly distributed over its surface. The deduced ISG75 polypeptide of 523 amino acids is comprised of 4 main regions: an amino-terminal hydrophobic signal sequence (28 amino acids) that is cleaved off yielding a mature protein starting at E29; a large hydrophilic extracellular domain; a stretch of hydrophobic residues (20 amino acids) close to the carboxyl-terminus forming a single trans-membrane α-helix; and a short hydrophilic domain (29 amino acids) exposed on the cytoplasmic face of the plasma membrane (Ziegelbauer et al. 1995). Three ISG75 sequences of only T. b. brucei were revealed by Ziegelbauer and colleagues, and 5 ISG75 open reading frames have been reported by the T. brucei Genome Project (Berriman et al. 2005).
This study was designed to extend these observations with the isolation and sequence analysis of ISG75 gDNA and cDNA in all the pathogenic species and subspecies of Trypanozoon, including T. b. brucei, T. b. gambiense, T. b. rhodesiense, T. evansi and T. equiperdum.
MATERIALS AND METHODS
Trypanosomes and preparation of trypanosomal DNA
A total of 8 trypanosome strains, of which 5 are cloned populations, were used in this study. Their origins and hosts from which they were isolated are described in Table 1. Trypanosome pellets and total DNA were prepared as described by Claes et al. (2003). Quality control of the extracted gDNA was performed by PCR using primer set ESAG6/7 to amplify the transferrin receptor of trypanosomes (Holland et al. 2001).

Primer design
A primer set ISG75-1F (TTC TAC GGC CAA GGT AAC GTG), ISG75-2R (GTT CGG GCA CCT GTG ATA GT) was derived from the published 5′-untranslated region and the 3′-untranslated region respectively of the ISG75 sequence of T. b. brucei (Ziegelbauer et al. 1995). This primer set was designed to amplify a 1679-bp DNA region including the whole open reading frame of ISG75 (1572 bp) flanked by the 5′-untranslated region (84 bp) and 3′-untranslated region (22 bp). A second primer set ISG75-3F (AAG GCT GAG AAG GCA AAG GAG), ISG75-2R (GTT CGG GCA CCT GTG ATA G) targeted the 3′-end fragment of ISG75 (550 bp). The designed primers lacked homology with any other known sequence present in the databases.
PCR assays
To check for the presence of ISG75, PCR assays were performed using primer set ISG75-1F, ISG75-2R and ISG75-3F, ISG75-2R with Hotstart Taq polymerase (Qiagen, Germany). The PCR mix contained 1·5 units of Hotstart Taq polymerase, 0·8 μM of each primer, 0·2 mM dNTPs and 1×Q solution. Conditions were as follows: initial denaturation for 15 min at 95 °C, followed by 35 amplification cycles of 1 min, denaturation at 95 °C, 1 min, primer-template annealing at 55 °C and 1 min elongation at 72 °C. A final extension step was carried out for 5 min at 72 °C. For cloning of full-length ISG75, high fidelity Platinum® Pfx DNA polymerase (Invitrogen, UK) was employed to isolate the ISG75 gDNA. The PCR mix contained 1 unit of Pfx, 0·4 μM of each primer, 0·3 mM dTNPs, 1×PCR buffer, 1 mM Mg2+. Cycling conditions were as follows: denaturation for 2 min at 94 °C, followed by 35 amplification cycles of 30 sec, denaturation at 94 °C for 30 sec, primer-template annealing at 68 °C and 2 min polymerization at 68 °C. A final extension step was carried out for 10 min at 68 °C.
Southern blot and densitometry
Single restriction endonuclease digestion was performed for each trypanosome strain. Thirty μl of genomic DNA (approximately 150 ngċμl−1) was digested overnight at 37 °C with 40U of HindIII or EcoRI (NEB, UK). Twenty μl of the digested sample and λ DNA Molecular Weight Marker II (Eurogentec, Belgium) were resolved by electrophoresis in a 1·5% agarose gel for 17 h at 20 V. Subsequently, DNA from the agarose gel was transferred to a nitrocellulose membrane according to published methods (Sambrook and Russell, 2001).
The 3′-end fragment of T. b. brucei AnTat 2.2 obtained via the primer set ISG75-3F, ISG75-2R was used as DNA probe. α-32P-dCTP labelling of the probe was performed using DecaLabelTM DNA Labeling Kit (MBI Fermentas, Germany). The ProbeQuantTM G-50 Micro Column was used for removal of unincorporated labelled nucleotides (Amersham Biosciences, UK). Hybridization and subsequent washing steps was performed according to the protocols of Sambrook and Russell (2001). The intensity and area of the autogradiographic bands obtained from Southern blotting were quantified by densitometry using Image Master 1D Elite version 3.01 (Pharmacia, UK). Autoradiograms were selected which corresponded closely to an idealized linearity of the signal (Victoir et al. 1995).
RNA extraction
Seven Trypanosoma strains as described in Table 1 (except T. b. rhodesiense AnTat 12.1 S) were used for RNA extraction. Total trypanosome RNA was extracted using the RNAqueous™-Midi Kit (Ambion, UK). Concentration and quality of the isolated RNA were analysed by capillary electrophoresis RNA 6000 Nano Assay, using a RNA 6000 Nano LapChips® (Agilent, UK). To assure that the total RNA in each sample was free of gDNA, a PCR (primer set ISG75-1F, ISG75-2R) was performed using the total RNA as template. One or 2 μg of the total RNA was mixed with a PCR cocktail containing the components as described for amplification of the full-length ISG75.
RT-PCR
The synthesis of first-strand cDNA was carried out using the Omniscript™ Reverse Transcriptase (Qiagen, Germany) according to the manufacturer's manual. The reverse transcription reaction was performed in a Biometra® Trio-block thermocycler at 37 °C for 60 min. For isolation of full-length ISG75 cDNA, 10 μl of the RT product were subjected to PCR assay using Platinum® Pfx DNA polymerase as described earlier.
gDNA and cDNA cloning
Cloning of the (RT-) PCR products was performed using the Zero Blunt® Topo® PCR Cloning Kit for Sequencing (Invitrogen, UK) according to the manufacturer's manual. To screen for the presence of the desired insert (i.e. 1679-bp ISG75) in the cells, PCR was performed using vector T3 and T7 primers flanking the insert. The plasmids containing the desired insert were extracted from the cells using the QIAprep® Miniprep Kits (Qiagen, Germany).
DNA sequencing
The extracted plasmids were sequenced with T3 and T7 primers. The obtained nucleotide sequences were assembled, from which a set of internal primers was designed for the second strand sequencing. The obtained sequences by internal-primer sequencing were assembled and confirmed with the sequences that were acquired by the T3–T7 sequencing. Sequence analysis was performed using DNAman (Lynnon Soft, Australia) and publicly available softwares, including InterProScan for topology prediction (http://www.ebi.ac.uk/InterProScan/), SignalP for signal peptide prediction (http://www.cbs.dtu.dk/services/SignalP/), and NetNGlyc for N-linked glycosylation prediction (http://www.cbs.dtu.dk/services/NetNGlyc/).
RESULTS
Sequence analysis of full-length ISG75 gDNA and cDNA clones
As there were 3 ISG75 sequences of only T. b. brucei reported, we intended to investigate the presence and transcription of ISG75 in all species and subspecies of the Trypanozoon. A 1679-bp PCR product of the full-length ISG75 was isolated in all the tested strains of the Trypanozoon subgenus with either Hotstart Taq or Pfx DNA polymerase. The same result was obtained with RT-PCR in all these strains. Therefore, it shows that ISG75 is present and transcribed in all species and subspecies of the Trypanozoon.
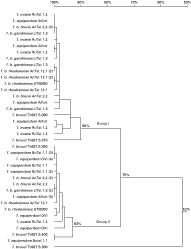
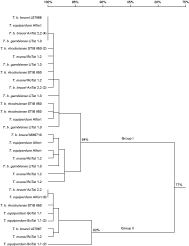
In total, 10–14 gDNA and cDNA clones isolated from each strain were sequenced (Table 1). The isolated gDNA and cDNA sequences show that multiple copies of ISG75 are present and transcribed in the genomes of all the strains. A nucleotide similarity tree constructed from 83 sequences divides the ISG75 cDNA and gDNA of the examined strains into 2 groups (Group I and Group II) with 77% and 75% identity respectively. For clarity purposes, here we present the sequence alignment of all the gDNA as 1 similarity tree (Fig. 1), and of all the cDNA as another (Fig. 2). For either gDNA or cDNA, highly conserved sequences ([ges ]90% or [ges ]94% identity) in Group I are present across different (sub)species of T. b. brucei AnTat 12.1, T. b. gambiense LiTat 1.3, T. b. rhodesiense AnTat 12.1 S and STIB 850 HR, T. evansi RoTat 1.2 and T. equiperdum Alfort. Likewise, Group II harbours highly similar gDNA and cDNA sequences among all strains ([ges ]93% or [ges ]92% identity). Out of 5 DNA sequences of T. b. brucei reported by the genome project, 2 (Tb927.5.390, Tb927.5.370) are clustered in Group I and 2 (Tb927.5.350, Tb927.5.400) belong to Group II in our analysis. However, Tb927.5.380 does not fall into either groups, and has 52% identity with Group I and Group II (Fig. 1). Of the 3 ISG75 sequences identified by Ziegelbauer and colleagues, M86710 and L07866 belong to Group I; and L07867 belongs to Group II in this gene family (Fig. 2). Furthermore, in silico and experimental resstriction analysis of the iolated sequences with MluI revealed that all the gDNA and cDNA sequences in Group I contain a unique MluI site, that is absent in Group II. The RT-PCR of T. equiperdum OVI was negative; hence no ISG75 cDNA of T. equiperdum OVI were obtained. cDNA sequences of only Group II were obtained in T. equiperdum BoTat 1.1. However, PCR of T. equiperdum Botat 1.1 and OVI gDNA using Group I- and Group II-specific primers and MluI restriction digestion were positive for both groups indicating that ISG75 gDNA sequences of Group I and Group II are also present in these two strains.

Fig. 1. ISG75 gDNA similarity tree, constructed with DNAman. Numbers in parentheses indicate the number of identical sequences that were isolated from the E. coli clones of the particular PCR product.

Fig. 2. ISG75 cDNA similarity tree, constructed with DNAman. Numbers in parentheses indicate the number of identical sequences that were isolated from the E. coli clones of the particular RT-PCR product.
All the cDNA and gDNA were translated into amino acid sequences and multiple alignment of the putative amino acid sequences distinguished Group I and Group II based on the following. Firstly, all the sequences in Group II lack 1 amino acid residue at position 157 as compared to Group I (S or L). As a result, Group I sequences have 523 residues (except 2 gDNA sequences of T. b. gambiense that had 520 residues, lacking residues A147E148K149); Group II sequences have 522. Secondly, all Group II sequences have a potential N-linked glycosylation site at position 134 (tripeptide NSS) and most of the Group II sequences have a second site at position 115 (tripeptide NRT); whereas all Group I sequences have only 1 potential N-linked glycosylation site at position 134 (tripeptide NAS) (Fig. 3).

Fig. 3. Multiple alignment of some of the putative amino acid sequences, constructed with DNAman, highlighting the variable and conserved regions between Group I and group II ISG75.
Multiple alignment and hydrophobicity analysis of the putative amino acid sequences demonstrate that ISG75 contains 2 conserved hydrophobic regions: one 28-amino acid segment at the amino terminus indicates a potential signal peptide; and one 21-amino acid region close to the carboxy end, suggests a single α-helix trans-membrane domain of the protein (Fig. 3). The 34-residue segment at the carboxy-terminus is predicted to be a cytoplasmic domain. The signal peptide cleavage site is predicted to be between residue 28 and 29, hence the mature protein potentially starts at residue E29 for most of the Group I sequences or S29 for most of the Group II sequences. A single α-helix trans-membrane domain is predicted at aa 469–491 for Group I sequences, and at aa 468–490 for Group II sequences with 100% sequence identity among all sequences of both groups. A large region between residue 29 and 467 might serve as an extracellular domain of the protein. The region immediately downstream of the signal sequence, from amino acid 29 to approximately amino acid 300, is heterogeneous; whereas roughly 200 amino acids thereafter are highly conserved. The overall predicted topology of the sequences in both groups is consistent with that of T. b. brucei isolated by Ziegelbauer et al. (1992).
In addition to the conserved domains of ISG75 in both Groups I and II, the variable region also shares particularly well-conserved features in all the sequences. These include an N-linked glycosylation site at position 134 as described earlier and 6 conserved cysteines that are located at identical positions in the variable region (Fig. 3). Together with 2 cysteines located in the signal peptide, there are overall 8 conserved cysteines in all the ISG75 (except overall 9 cysteines in 3 of the T. equiperdum BoTat 1.1 sequences).
Estimation of ISG75 copy number
Complementary to the isolation of ISG75 gDNA and cDNA sequences in all species and subspecies of the Trypanozoon, Southern blot and densitometry were performed for estimation of the copy number of the ISG75 in the genomes of these strains. It was verified by restriction digest and sequence analysis that neither HindIII nor EcoRI restriction sites were present in the probe region. Southern blot of gDNA digested with the restriction endonuclease HindIII or EcoRI showed multiple bands in all tested strains. Densitometrical scanning was performed with the bands obtained from the Southern blot for estimation of its copy number. In each lane, the band with lowest intensity was considered as at least 1 gene copy. The results indicate that ISG75 copy number fluctuates among strains of the Trypanozoon, ranging from at least 4 to 16 copies in each strain.
DISCUSSION
This study confirms that all Trypanozoon species and subspecies harbour and transcribe multiple ISG75 sequences. Interestingly, regardless of the strains, all the gDNA and cDNA cluster into 2 groups (Group I and Group II). These sequences have high similarities with the ISG75 sequences of T. b. brucei reported in the databank. All 3 ISG75 sequences identified by Ziegelbauer and colleagues, and 4 out of 5 sequences reported in the T. brucei genome project fall into Group I and Group II in our analysis. Previous observations in terms of glycosylation site and variations/substitutions of residues in the cytoplasmic and signal peptide domains correlate well with our findings in the two groups (Ziegelbauer et al. 1995). Therefore, this study in a sense is an extension to all members of the Trypanozoon subgenus of the previously reported results on T. b. brucei ISG75. Moreover, this study is the first to identify the 2 main groups of the ISG75 gene family in the Trypanozoon subgenus. In agreement with the previous work on T. b. brucei and the high correlation between the Southern blot and gene cloning results in this study, ISG75 is present as multiple copies in the genomes of all the taxa of the Trypanozoon subgenus.
The third-group sequence Tb927.5.380 identified in the T. brucei's genome project was not isolated in our experiment. As our primers were designed based on the untranslated regions of ISG75, this third group sequence either has different untranslated regions or is absent in the tested strains. Furthermore, it has little similarity (approximately 33% identity) with either Group I or Group II at amino acid level. The only similarities between this sequence and the rest of the ISG75 members are a predicted signal peptide, a predicted transmembrane domain and positions of 6 (out of 7) cysteines in the mature putative polypeptide. In addition, it has 3 deletions (total of 15 amino acids) and 4 insertions (total of 44 amino acids) when aligned to the other ISG75 sequences. Therefore, it is approximately 29–30 residues longer than all other ISG75 open reading frames available in the databank. Further research is needed to reveal the general presence and expression of this sequence in the Trypanozoon group and its relationship with Group I and Group II ISG75.
In silico analysis based on preliminary genome sequences of T. congolense and T. vivax (http://www.sanger.ac.uk/Projects) shows that there is no sequence identity between the full-length ISG75 and T. congolense and T. vivax's sequences. It suggests that ISG75 is probably specific for Trypanozoon. Therefore, this may have implications on possible differences in surface architecture or host survival mechanisms between the Trypanozoon and the other subgenera of Trypanosoma.
It is a common feature in Kinetoplastids that genes are transcribed as polycistronic mRNA. The mature mRNA is generated by addition of a miniexon at the 5′-end of the open reading frame and a polyadenylation tail at the 3′-end (Borst, 1986; Agabian, 1990). In Trypanosomatids, several of their characterized genes belong to tandemly arrayed multigene families, including calmodulin (Tschudi et al. 1985), tubulin (Thomashow et al. 1983; Seebeck et al. 1983) and trypanosome hexose transporters (Bringaud and Baltz, 1994). The ISG75 gene family is of no exception and shows an interesting genetic diversity. Highly similar sequence members (at least 92% identity in Group I and 90% identity in Group II at amino acid level) are present across different strains, while the variations between Group I and Group II (67% identity at amino acid level) are mainly situated in discrete segments at the variable regions. In addition, the difference in length of most of the putative amino acid sequences between Groups I and II is by only 1 residue at the same position.
During the course of their evolution, the Trypanozoon have kept the transmembrane and cytoplasmic domain of the ISG75 highly conserved; perhaps because they are well protected in the parasite's membrane and not prone to the host immune system, or perhaps because they have some particular function. However, the parasite allows genetic variability in the extracellular domain that is relatively more exposed to the host environment, hence is able to adapt itself to survival in the mammalian hosts. It is noted that as ISG75 is smaller than the VSG, it is probably hidden in the VSG coat on the surface (Overath et al. 1994). Therefore, it remains unclear why there is a variable extracellular region among all the sequences and why 2 groups of ISG75 sequences are transcribed. It may be hypothesized that different protein sequences from both groups are required for adaptation to different mammalian host species as has been proposed for the transferrin receptor ESAG6/7 that has different affinity for types of transferrins (Isobe et al. 2003). Also it has been reported that the trypanosome hexose transporter THT1 and THT2 that have 6 and 5 direct repeats in tht loci respectively and are developmentally regulated for glucose availability in different hosts (Bringaud and Baltz, 1993, 1994). One could imagine that ISG75, being smaller and less abundant than the VSGs, may play a role as a receptor of some particular ligand for signalling between the host's environment and the parasite or among the parasites themselves. Furthermore, it is possible that 2 molecules of Groups I and II are necessary for the formation of a functional dimeric structure. Since several surface molecules of the trypanosome are known to be dimers, ISG75 might also be either a homodimer formed by either Group I or Group II sequences, like VSG (Freymann et al. 1984); or a heterodimer formed by 2 monomers from Group I and Group II, like the combination ESAG6 and ESAG7 for the transferrin receptor (Salmon et al. 1994).
In conclusion, ISG75 is ‘invariant’ in terms of its presence and transcription in all the Trypanozoon species and subspecies. However, there is a certain degree of genetic variability among different sequences, especially between Group I and Group II, as it is typical of the Trypanosomatids, whose genomes appear to have multiple gene copies that play important roles in their successful parasitic life.
We are grateful to Dr Xu Ying and Dr Jan Van Den Abbeele (Institute of Tropical Medicine, Antwerp) for their practical and scientific assistance. We greatly appreciate the critical comments from Dr Henri De Greve (Free University of Brussels) on the manuscript. This study received financial support from the International Livestock Research Institute (ILRI) in Nairobi, Kenya and a private funding.