


Introduction
Cognitive deficits are a core feature of depression in adults of all ages, consistently found in the domains of memory, executive functioning and processing speed (Thomas & O'Brien, Reference Thomas and O'Brien2008). Previously, such deficits were thought to be transient, in its most severe forms called ‘depressive pseudodementia’ (Bulbena & Berrios, Reference Bulbena and Berrios1986), but mounting evidence shows cognitive deficits persist despite remission of depressive symptoms (Abas et al. Reference Abas, Sahakian and Levy1990; Beats et al. Reference Beats, Sahakian and Levy1996; Nebes et al. Reference Nebes, Butters, Mulsant, Pollock, Zmuda, Houck and Reynolds2000; Devanand et al. Reference Devanand, Pelton, Marston, Camacho, Roose, Stern and Sackeim2003; Portella et al. Reference Portella, Marcos, Rami, Navarro, Gastó and Salamero2003; Adler et al. Reference Adler, Chwalek and Jajcevic2004; Neu et al. Reference Neu, Bajbouj, Schilling, Godemann, Berman and Schlattmann2005; Bhalla et al. Reference Bhalla, Butters, Mulsant, Begley, Zmuda, Schoderbek, Pollock, Reynolds and Becker2006; Lee et al. Reference Lee, Potter, Wagner, Welsh-Bohmer and Steffens2007). These persisting deficits may be related to underlying neurobiological changes, including brain atrophy and an increased prevalence of white matter hyperintensities (Schweitzer et al. Reference Schweitzer, Tuckwell, Ames and O'Brien2001; Herrmann et al. Reference Herrmann, Le Masurier and Ebmeier2008).
Although cognitive impairment is nowadays thought to be stable for the group of patients as a whole, recent studies have been short term (⩽12 months) and longer-term outcome has not been determined. There might also be differences between patients with specific clinical characteristics. For example, younger patients show a similar cognitive profile, but impairment is generally found to be more severe in older individuals (Gualtieri & Johnson, Reference Gualtieri and Johnson2008; Thomas et al. Reference Thomas, Gallagher, Robinson, Porter, Young, Ferrier and O'Brien2009) and might be related to a late onset of depressive disorder (⩾60 years) in particular (Herrmann et al. Reference Herrmann, Goodwin and Ebmeier2007). Although modest improvement of cognition may occur in patients who were selected based on good response to antidepressant (AD) treatment (Butters et al. Reference Butters, Becker, Nebes, Zmuda, Mulsant, Pollock and Reynolds2000; Gallassi et al. Reference Gallassi, Di Sarro, Morreale and Amore2006; Mandelli et al. Reference Mandelli, Serretti, Colombo, Florita, Santoro, Rossini, Zanardi and Smeraldi2006), it is largely unknown whether current AD treatment impacts on patients' cognition compared to healthy subjects. Furthermore, controversy remains as to whether cognitive impairment affects all cognitive domains or whether apparently multi-modal deficits in fact reflect a deficit in a single core neuropsychological function. Although the most suitable candidate, processing speed, has indeed been found to be a strong mediator of other cognitive deficits (Nebes et al. Reference Nebes, Butters, Mulsant, Pollock, Zmuda, Houck and Reynolds2000; Butters et al. Reference Butters, Whyte, Nebes, Begley, Dew, Mulsant, Zmuda, Bhalla, Meltzer, Pollock, Reynolds and Becker2004), its effect might be greater for executive functioning than for episodic memory (Delaloye et al. Reference Delaloye, Baudois, de Bilbao, Dubois Remund, Hofer, Lamon, Ragno Paquier, Weber, Herrmann, Giardini and Giannakopoulos2008).
To address these questions we examined the pattern of cognitive deficits in healthy subjects and individuals with late-life major depression over time. We report differences between patients due to current symptom severity, remission status, age of depression onset and AD treatment. We hypothesized that (i) current symptom severity would only marginally affect cognitive deficits, (ii) remitted patients would therefore show some amelioration of deficits but remain impaired, (iii) later age of onset would be associated with more severe deficits without differences in the domains affected and (iv) those treated with ADs would not differ from those not treated. In addition, we addressed the question whether processing speed mediates deficits in other cognitive domains.
Method
Case ascertainment
Sixty-seven patients aged ⩾60 years who fulfilled DSM-IV criteria for major depression were recruited from clinical old age psychiatry services covering geographically based catchment areas and including referrals from day hospitals, in-patient units and out-patient clinics. A control group (n=36) of similar aged older people (also all ⩾60 years) with no past history of depression or current depression were recruited from community sources such as The Royal British Legion and spouses of patients attending the same hospital units. The baseline neuropsychological profile of this group has been reported previously (O'Brien et al. Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier2004). We excluded both subjects and controls with a history of prior cognitive impairment, a history or evidence of stroke or transient ischaemic attack, severe or unstable physical illness (e.g. insulin-dependent diabetes mellitus, untreated hypothyroidism, uncontrolled heart failure, cancer) or a Cambridge Cognitive Examination (CAMCOG; Roth et al. Reference Roth, Huppert, Mountjoy and Tym1999) score of <75 (patients) or <80 (controls). Additional exclusion criteria were: history or current substance/alcohol abuse; long-term use (>2 months) of steroids during lifetime; use of steroid or other medication within the past 3 months thought to interfere with the hypothalamic–pituitary–adrenal (HPA) axis; electroconvulsive therapy (ECT) in the past 3 months; use of medication thought to affect cognition (e.g. non-hypnotic benzodiazepines, antipsychotics or anticholinergic medication); the presence of other neurological diagnosis. Use of newer ADs [e.g. selective serotonin reuptake inhibitors (SSRIs) and venlafaxine] and lithium was permitted, and only seven patients were taking tricyclic ADs (one dothiepin, six lofepramine). The study was approved by the local ethics committee and all patients and controls gave written informed consent.
Assessment
All depressed cases underwent a comprehensive psychiatric assessment including history, mental state, physical examination and a test of general cognitive functioning (CAMCOG). The CAMCOG is part of the Cambridge Mental Disorders of the Elderly Examination (CAMDEX; Roth et al. Reference Roth, Huppert, Mountjoy and Tym1999) and assesses general cognitive functioning and is used frequently in research and clinical practice.
Depression was diagnosed according to DSM-IV criteria (APA, 1994) and symptom severity was rated using the Montgomery–Åsberg Depression Rating Scale (MADRS; Montgomery & Åsberg, Reference Montgomery and Åsberg1979). In the present study, remission was defined as a MADRS score ⩽9 (Hawley et al. Reference Hawley, Gale and Sivakumaran2002; Zimmerman et al. Reference Zimmerman, Chelminski and Posternak2004). Demographic information (including past and current medical and psychiatric history, medication taken, family history, education and social class) and psychiatric history of past episodes of depression were collected from multiple sources to validate or enrich information from face-to-face interviews with subjects and informants [e.g. case-notes, general practitioner (GP) records and informant accounts to determine number of previous episodes, age of onset and total lifetime duration of depression]. An extensive neuropsychological test battery was administered to controls and all patients who consented to it.
Neuropsychological assessment
The test battery was designed primarily to measure memory, processing speed and executive function as they represent core neuropsychological deficits in late-life depression (Thomas & O'Brien, Reference Thomas and O'Brien2008). Tests used in the present study included both traditional pen-and-paper and computerized tasks:
(1) The Rey Auditory Verbal Learning Test (RAVLT; Rey, Reference Rey1964), a test of episodic memory. The three measures immediate recall, delayed recall and delayed recognition (number of correct items) were used.
(2) The FAS verbal fluency test (Lezak et al. Reference Lezak, Howieson and Loring2004), a task sensitive to frontal lobe impairment.
(3) The Trail Making Test (TMT; Lezak et al. Reference Lezak, Howieson and Loring2004), a test of mental flexibility and divided attention.
(4) The Stroop Color Word Test (SCWT; Stroop, Reference Stroop1935), a test for response inhibition and selective attention.
(5) A computerized continuous performance task (VIGIL; Cegalis & Bowlin, Reference Cegalis and Bowlin1991). Over 8 min, subjects have to press a button to a complex target stimulus (letter K when preceded by the letter A), presented 100 times within a total of 480 stimuli (displayed serially in a pseudo-random fashion). Errors of omission and commission can be used as a measure of vigilance and inhibition but in the present study only response latencies (in ms) were used as a measure of processing speed.
Definition of generalized cognitive impairment (GCI)
There is no universally accepted definition of a suitable cut-off to denote significant cognitive impairment and 1, 1.5 and 2 standard deviations (s.d.) have all been used. In their definition of ageing-associated cognitive decline, Levy et al. (Reference Levy1994) chose 1 s.d. The narrower, and more universally accepted, concept of mild cognitive impairment (Petersen et al. Reference Petersen, Smith, Waring, Ivnik, Tangalos and Kokmen1999) used 1.5 s.d. Consistent with this, we defined GCI as a score of >1.5 s.d. below the healthy control groups' mean on the CAMCOG at each assessment.
Follow-up
Patients and controls were reassessed 6 and 18 months and again 4 years after baseline. At each time point, a psychiatric assessment, administration of rating scales and neuropsychological tests were repeated. At 6 months, 93 (90%) participants of the baseline sample were reassessed and 78 (76%) at 18 months. At 4 years, only 36 (35%) individuals, including 15 patients, were available for follow-up. Our analysis therefore focuses on the 6 and 18 months follow-up data, but because longer-term follow-up cognitive data on such patients are rarely available, we have also included the 4-year data. Although all patients had undergone clinical examination and CAMCOG testing at baseline, only 34 out of 67 of them were tested with the extended neuropsychological battery. Since more subjects could be tested at 6 (51 out of 57) and 18 (41 out of 45) months, this means that samples at different time points are not perfectly comparable. We thus decided to look at the associations cross-sectionally only.
Statistical analysis
For ease of comparison, neuropsychological test scores were standardized using the control group's mean and s.d. at baseline. An overall memory z score was created by adding up the three z scores of the RAVLT (immediate recall, delayed recall, delayed recognition) and this ‘compound score’ was again standardized to a z score using the control group's mean and s.d. at baseline. Similarly, an overall executive functioning z score was created by adding up the z score of verbal fluency, TMT difference A–B and SCWT correct responses. By this, we had three cognitive domains with higher scores indicating better performance: memory, executive functioning and processing speed (inverted VIGIL latencies). The risk of having GCI at follow-up was assessed with logistic regression analyses yielding odds ratios (ORs) and 95% confidence intervals (CIs). We then used multiple linear regression analyses to test associations within cognitive domains. The impact of key clinical variables was investigated by comparing remitters and non-remitters, early onset and late onset, and AD users and non-users to healthy controls. In patients we also tested whether current MADRS scores (symptom severity), continuous age of onset and lifetime duration of AD intake predicted neuropsychological performance. All comparisons were adjusted for age, gender and years of education. The α level for statistical significance was fixed at p⩽0.05. All tests were performed with Stata 9.2 (StataCorp, 2006).
Results
Descriptive analyses
Patients and their comparison subjects were well matched for age (p=0.609) and gender (p=0.633), but patients had higher MADRS scores (t=−12.2, df=101, p<0.001) and fewer years of formal education (t=2.06, df=101, p=0.042) (Table 1).
Table 1. Baseline demographic characteristics for depressed and control subjects
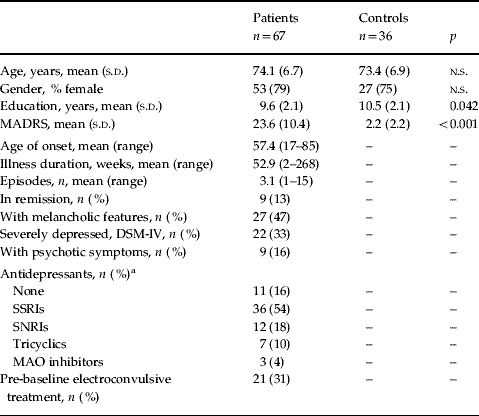
MADRS, Montgomery–Åsberg Depression Rating Scale; SSRI, selective serotonin reuptake inhibitor; SNRI, serotonin-norepinephrine reuptake inhibitor; MAO, monoamine oxidase; s.d., standard deviation; n.s., not significant.
a Percentages do not add up to 100 because two depressed subjects were taking SSRI (citalopram) and tricyclic (one dothiepin, one lofepramine) antidepressant medication.
Loss to follow-up
At 18 months, 22 (21%) participants were lost to follow-up (LTFU), all within the patient group. Of these, 19 refused participation and three had died. Three control subjects had no data on CAMCOG or other neuropsychological testing. Among the patients, being LTFU was not related to age (t=0.10, df=65, p=0.919), gender (χ2=0.07, df=65, p=0.797), years of education (t=−0.27, df=65, p=0.785), age of onset (t=−0.04, df=65, p=0.972), MADRS score (baseline: t=−1.47, df=65, p=0.147; 6 months: t=0.34, df=55, p=0.739), remission status (baseline: χ2=0.53, df=65, p=0.466; 6 months: χ2=1.04, df=55, p=0.308), baseline AD use (χ2=0.04, df=65, p=0.836) or weeks on medication (baseline: t=0.10, df=64, p=0.919; 6 months: t=1.10, df=43, p=0.277). In addition, there were no significant differences between groups in total CAMCOG (baseline: t=0.18, df=64, p=0.857; 6 months: t=1.74, df=55, p=0.087), memory (baseline: t=1.12, df=32, p=0.270; 6 months: t=1.11, df=49, p=0.274), executive functions (baseline: t=1.19, df=35, p=0.241; 6 months: t=1.87, df=49, p=0.067) and processing speed (baseline: t=0.83, df=28, p=0.414; 6 months: t=0.72, df=44, p=0.478). However, all patients LTFU were on medication at the 6-month follow-up, resulting in a significant difference with patients not LTFU (χ2=4.03, df=55, p=0.045).
Depression and persistent generalized cognitive impairment (GCI)
One patient with missing CAMCOG scores was excluded from this analysis. Of the remaining 66 patients, 33 (50%) showed GCI defined as 1.5 s.d. below the control group's CAMCOG mean (Fig. 1). Having GCI at baseline was highly predictive of having persistent GCI at 6 months (OR 6.0, 95% CI 1.86–19.40, p=0.003) and at 18 months (OR 5.2, 95% CI 1.41–19.18, p=0.011). The risk increment remained robust after adjustment for age, gender, years of education, age of onset, remission status and current AD use (6 months: OR 5.85, 95% CI 1.43–23.97, p=0.014; 18 months: OR 5.91, 95% CI 1.12–31.23, p=0.036).
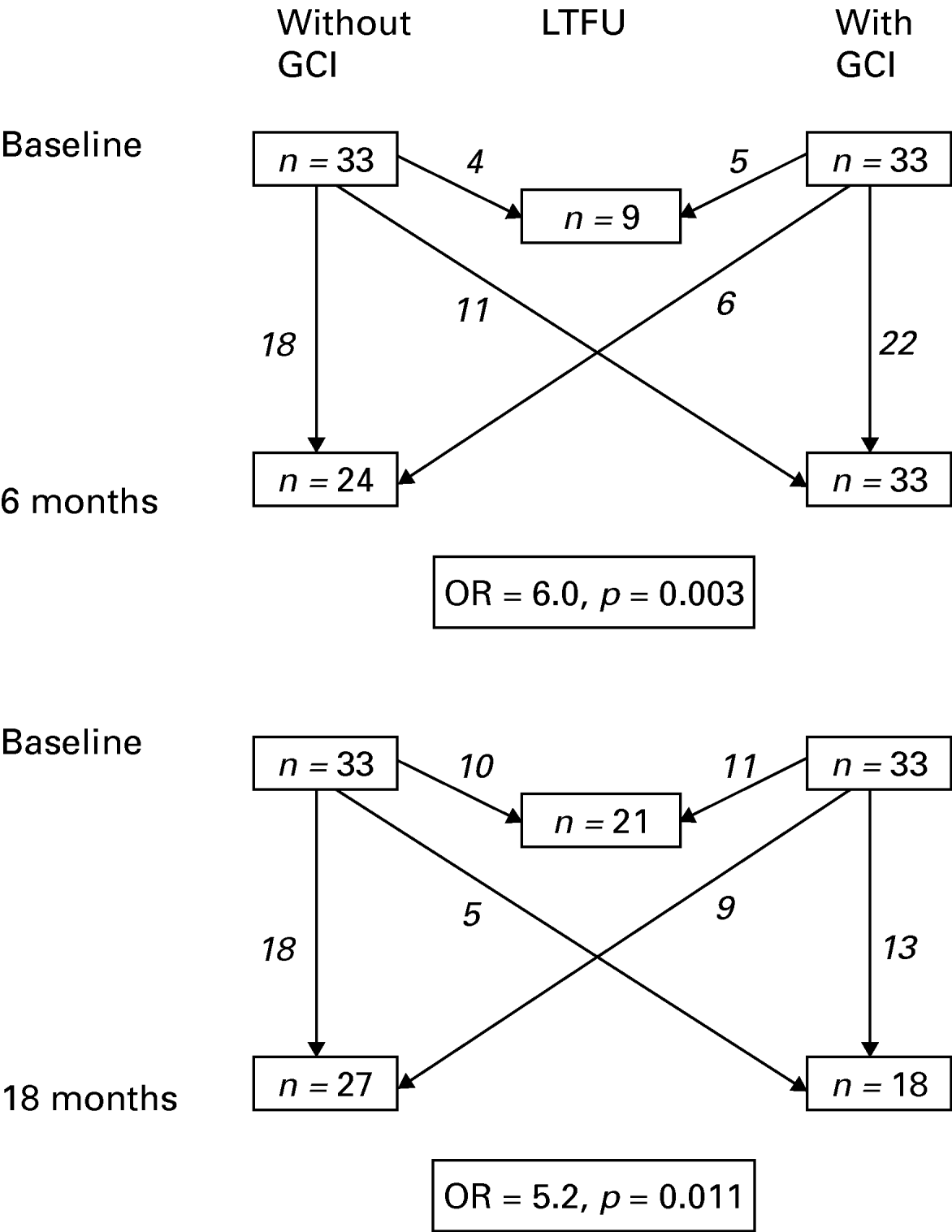
Fig. 1. Diagram showing numbers of depressed patients with and without generalized cognitive impairment (GCI) at each assessment. Arrows indicate how many patients were lost to follow-up (LTFU), remained with or without GCI, or made transitions between GCI groups from baseline to follow-up. The odds ratio (OR) and p value for having GCI at follow-up given GCI at baseline is also shown.
Single-domain or multiple-domain cognitive impairment
We wanted to test whether cognitive impairment is domain specific or affects multiple cognitive domains. Separate linear regression analyses adjusted for age, gender and years of education showed that patients did significantly worse at all time points and in all domains (Table 2). Figure 2 illustrates this by showing little deviation from parallel running lines representing both groups' unadjusted mean z scores up to 18 months.
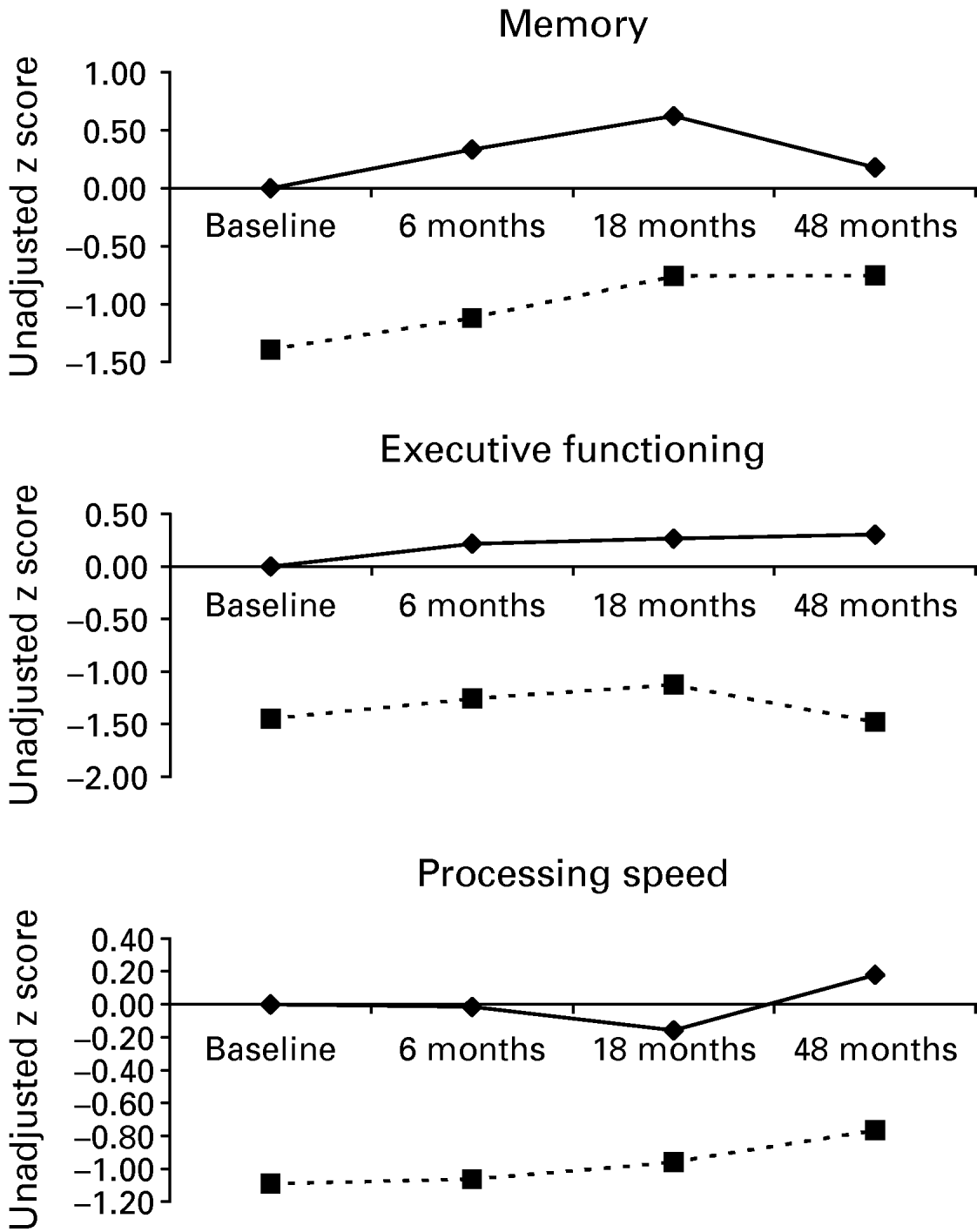
Fig. 2. Plotted unadjusted z score means illustrating cognitive trajectories over time for control (–♦–) and depressed (- -▪- -) subjects in individual cognitive domains.
Table 2. Difference in mean z scores of individual cognitive domains between depressed subjects and controls at baseline and follow-up

CI, Confidence interval.
*** p⩽0.001, ** p⩽0.01, * p⩽0.05.
Do deficits in processing speed drive the impairment in patients?
To test the mediating role of processing speed, analyses were repeated but controlled for VIGIL latency z scores. Adjusted for group, age, gender and education, processing speed was positively and significantly associated with memory (baseline: b=0.31, 95% CI 0.12–0.50, p=0.002; 6 months: b=0.43, 95% CI 0.19–0.67, p=0.001; 18 months: b=0.34, 95% CI 0.08–0.60, p=0.012) and executive functioning (baseline: b=0.42, 0.19–0.65, p=0.001; 6 months: b=0.40, 95% CI 0.13–0.68, p=0.004; 18 months: b=0.46, 95% CI 0.23–0.70, p<0.001). As can be seen in Table 2, adding processing speed to the regression model explained another 6–8% of the variance in memory scores. For executive functioning, this rose to 7–16%. However, differences between groups remained significant in both domains at all time points.
Stability of cognitive impairment: depression severity
We tested whether cognitive impairments, despite being relatively stable for the group of patients as a whole, showed some variability due to differential associations with a priori identified clinical factors (see Appendix). To test the influence of symptom severity in patients, we tested whether MADRS scores at the relevant follow-up point predicted cognition and found that they did not: memory (baseline: b=0.02, 95% CI −0.06 to 0.11, p=0.587; 6 months: b=−0.01, 95% CI −0.05 to 0.03, p=0.593; 18 months: b=−0.02, 95% CI −0.06 to 0.03, p=0.415); executive functioning (baseline: b=0.01, 95% CI −0.08 to 0.10, p=0.814; 6 months: b=−0.00, 95% CI −0.04 to 0.04, p=0.870; 18 months: b=−0.02, 95% CI −0.07 to 0.03, p=0.444); processing speed (baseline: b=0.08, 95% CI −0.03 to 0.18, p=0.153; 6 months: b=−0.02, 95% CI −0.06 to 0.01, p=0.179; 18 months: b=−0.02, 95% CI −0.06 to 0.02, p=0.366).
Stability of cognitive impairment: remitted versus persistently depressed patients
Whether remission of depression went together with an amelioration of cognitive deficits was analysed in a subsample from which patients already in remission at baseline (MADRS <10, n=9) had been removed. At 6 months, 21 out of 48 (44%) available formerly depressed subjects were in remission, with another 14 out of 38 (37%) available patients in remission at 18 months. At both follow-ups, remitting patients performed closer to healthy controls than depressed patients but both groups were still considerably impaired in memory and executive functioning (Table 3). For processing speed, both groups showed impairment at 6 months but no significant difference from healthy controls at the 18-month follow-up.
Table 3. Association between categorical measures of remission status, age of onset and antidepressant use on cognitionFootnote a

AD, Antidepressant; CI, confidence interval.
a Values represent unadjusted z score differences with healthy comparison subjects.
b Patients in remission at baseline were excluded in this analysis.
c Current antidepressant use at time of testing.
*** p⩽0.001, ** p⩽0.01, * p⩽0.05.
Stability of cognitive impairment: early versus late onset depression
When defined on a continuous scale, age of onset was not significantly associated with executive functioning (baseline: b=0.00, 95% CI −0.03 to 0.03, p=0.915; 6 months: b=−0.01, 95% CI −0.03 to 0.01, p=0.423; 18 months: b=−0.02, 95% CI −0.05 to 0.01, p=0.226) or processing speed (baseline: b=0.02, 95% CI −0.01 to 0.05, p=0.227; 6 months: b=−0.00, 95% CI −0.02 to 0.02, p=0.955; 18 months: b=0.00, 95% CI −0.02 to 0.03, p=0.796), but increasing age of onset was negatively related to episodic memory (baseline: b=−0.03, 95% CI −0.06 to −0.00, p=0.049; 6 months: b=−0.03, 95% CI −0.05 to −0.01, p=0.010; 18 months: b=−0.03, 95% CI −0.05 to −0.00, p=0.043). Thirty (45%) patients had an onset before age 60 (early onset depression, EOD) and 37 (55%) thereafter (late onset depression, LOD). Both groups were impaired relative to controls at all time points in memory and executive functioning and at baseline and 6 months testing of processing speed, but processing speed was not significantly impaired in either onset group at the 18 months follow-up (Table 3). Mean z score differences with controls (as displayed in Table 3) suggest some improvement in EOD for all domains, whereas LOD showed signs of deterioration in memory and executive functioning relative to controls. Paired t tests on the longitudinal association between 6 and 18 months cognition confirmed this by showing improved memory scores in controls (t=−3.24, df=31, p=0.003) and EOD (t=−2.63, df=18, p=0.017) but not LOD (t=−1.50, df=19, p=0.150) and stable executive functioning scores in controls (t=0.22, df=31, p=0.828) and EOD (t=−1.70, df=18, p=0.106), but a decline in LOD (t=2.33, df=19, p=0.031).
Stability of cognitive impairment: influence of AD use
Both the acute effects of current AD use at time of testing (yes, no) and the possible long-term effects due to (cumulative) lifetime duration of AD intake (in weeks) were analysed. Duration of lifetime AD intake was not significantly associated with memory (b=0.001, 95% CI −0.001 to 0.004, p=0.299; 6 months: b=0.001, 95% CI −0.002 to 0.003, p=0.486; 18 months: b=0.001, 95% CI −0.002 to 0.003, p=0.533), executive functioning (b=0.000, 95% CI −0.003 to 0.003, p=0.942; 6 months: b=−0.002, 95% CI −0.003 to 0.002, p=0.863; 18 months: b=0.002, 95% CI −0.001 to 0.007, p=0.178) or processing speed (b=−0.001, 95% CI −0.004 to 0.002, p=0.503; 6 months: b=0.001, 95% CI −0.001 to 0.003, p=0.247; 18 months: b=0.000, 95% CI −0.002 to 0.002, p=0.958). At baseline, 57 (85%) patients were on medication. Of those available at follow-up, 46 (81%) were on AD at 6 months and 11 (19%) were not, and 34 (79%) were on AD at 18 months whereas nine (21%) were not. Both current AD users and non-users displayed significant memory impairment at all time points and impaired executive functioning and processing speed at baseline and 6 months (Table 3). Overall, non-users had lower mean z scores in all domains at baseline and 6 months, but at 18 months they did not differ significantly from controls in executive functioning and processing speed.
Exploratory analyses of 4-year follow-up data
LTFU from baseline to 4-year follow-up was high with 67 (65%) of baseline participants dropping out of the study [52 (78%) patients, 15 (41%) controls]. Reasons for LTFU were refusal (n=41, 61%), death (n=10, 15%), being too late for follow-up (n=6, 9%), physical health (n=6, 9%), and other (n=4, 6%). One patient had developed possible dementia. Being LTFU at 4 years was independent of age (t=−1.41, df=101, p=0.160), gender (χ2=2.27, df=101, p=0.132), remission status at 18 months (χ2=0.83, df=43, p=0.362) and baseline executive functioning (p=0.112) and processing speed scores (p=0.589), but was significantly associated with fewer years of education (t=−2.06, df=101, p=0.042) and worse baseline episodic memory (t=−2.72, df=68, p=0.008). In addition, patients with a later onset (t=−2.53, df=65, p=0.014) were more likely to be LTFU. Taken together, this pattern reflects the higher attrition in the patient group than in the controls.
Of the 15 patients followed up, six had a GCI at baseline and three of them had persistent GCI after 4 years, but statistical testing failed to reach significance (OR 8.0, 95% CI 0.58–110.27, p=0.120). A fairly wide 95% CI indicated that this was probably because of the small sample size, and it is notable that the OR was similar in magnitude to that found at 6 and 18 months. Regarding domain-specific impairment, t tests suggest a pattern that is consistent with the 6 and 18 months follow-up, but tests lacked power and were therefore not always conclusive. Thus, patients' impairment seemed to persist relative to controls in executive functioning (t=3.00, df=34, p=0.005) and processing speed (t=2.36, df=29, p=0.025), with a trend in the same direction for episodic memory (t=1.90, df=34, p=0.065).
Discussion
Main findings
We found that cognitive impairment persists in many depressed subjects, affects multiple cognitive domains and is not significantly influenced by illness factors such as current mood, remission status or current AD use. Persistence was only partially explained by information processing speed. Patients with a later age of onset displayed worse episodic memory functioning.
The 18 months findings augment earlier reports of shorter follow-up duration (Adler et al. Reference Adler, Chwalek and Jajcevic2004; Bhalla et al. Reference Bhalla, Butters, Mulsant, Begley, Zmuda, Schoderbek, Pollock, Reynolds and Becker2006; Lee et al. Reference Lee, Potter, Wagner, Welsh-Bohmer and Steffens2007) and studies in younger cohorts (Weiland-Fiedler et al. Reference Weiland-Fiedler, Erickson, Waldeck, Luckenbaugh, Pike, Bonne, Charney and Neumeister2004; Airaksinen et al. Reference Airaksinen, Wahlin, Larsson and Forsell2006; Reppermund et al. Reference Reppermund, Zihl, Lucae, Horstmann, Kloiber, Holsboer and Ising2007) showing that cognitive deficits are highly persistent in depressive disorder. Furthermore, our findings show that incident cognitive impairment can develop in people with prevalent depression whereas (some) amelioration of deficits occurs in some individuals with initial deficits. However, the most common outcome is that of no change at all: either persistent impairment or persistent absence of it.
State or trait effects?
Patients' impairments in single cognitive domains were not related to state effects such as current symptom severity. Likewise, remitting patients showed similar cognitive impairments as depressed patients, albeit milder. Persistent deficits have been reported frequently (Abas et al. Reference Abas, Sahakian and Levy1990; Beats et al. Reference Beats, Sahakian and Levy1996; Nebes et al. Reference Nebes, Butters, Mulsant, Pollock, Zmuda, Houck and Reynolds2000; Devanand et al. Reference Devanand, Pelton, Marston, Camacho, Roose, Stern and Sackeim2003; Portella et al. Reference Portella, Marcos, Rami, Navarro, Gastó and Salamero2003; Adler et al. Reference Adler, Chwalek and Jajcevic2004; Neu et al. Reference Neu, Bajbouj, Schilling, Godemann, Berman and Schlattmann2005; Bhalla et al. Reference Bhalla, Butters, Mulsant, Begley, Zmuda, Schoderbek, Pollock, Reynolds and Becker2006; Lee et al. Reference Lee, Potter, Wagner, Welsh-Bohmer and Steffens2007) and are a core feature of the disorder itself. We found mixed results for the influence of AD treatment on cognition, but overall, there were only small differences between those who were and were not on medication. If anything, patients taking ADs performed slightly better in all cognitive domains at baseline and 6 months, which does not imply that medication affected cognition negatively in this sample. Inconsistent with this was the finding that those who did not take ADs did not differ from controls at 18 months follow-up testing of executive functioning and processing speed. Although, at least for executive functioning, this might have been due to lack of power (the mean score of the eight patients tested and currently not on AD was still 1 s.d. below controls), the lack of a consistent effect of AD treatment can be seen as evidence that it was not a major factor mediating cognitive deficits in our sample. In addition, lifetime AD treatment had no major effects on cognitive functioning at any time point. Taken together, these findings imply a trait effect on neurocognition, most probably caused by structural cerebral changes, which have been consistently reported in depression, especially LOD (Schweitzer et al. Reference Schweitzer, Tuckwell, Ames and O'Brien2001; Herrmann et al. Reference Herrmann, Le Masurier and Ebmeier2008).
The role of processing speed
Consistent with the literature, impairment was found to affect multiple cognitive domains, including episodic memory, executive functioning and processing speed (Thomas & O'Brien, Reference Thomas and O'Brien2008). As in earlier reports, deficient processing speed made major contributions to cognitive deficits in other domains (Nebes et al. Reference Nebes, Butters, Mulsant, Pollock, Zmuda, Houck and Reynolds2000; Butters et al. Reference Butters, Whyte, Nebes, Begley, Dew, Mulsant, Zmuda, Bhalla, Meltzer, Pollock, Reynolds and Becker2004). In the present study, however, its effect on executive functioning deficits was greater than on memory deficits, confirming one earlier report (Delaloye et al. Reference Delaloye, Baudois, de Bilbao, Dubois Remund, Hofer, Lamon, Ragno Paquier, Weber, Herrmann, Giardini and Giannakopoulos2008). However, it was insufficient to fully explain the differences between patients and controls, indicating that other deficits exist in parallel. These may stem from structural brain changes, including hippocampal atrophy (Sapolsky, Reference Sapolsky2000; Steffens et al. Reference Steffens, Byrum, McQuoid, Greenberg, Payne, Blitchington, MacFall and Krishnan2000; O'Brien et al. Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier2004; Hickie et al. Reference Hickie, Naismith, Ward, Turner, Scott, Mitchell, Wilhelm and Parker2005), frontal lobe atrophy/volume reduction (Schweitzer et al. Reference Schweitzer, Tuckwell, Ames and O'Brien2001; Almeida et al. Reference Almeida, Burton, Ferrier, McKeith and O'Brien2003; Lavretsky et al. Reference Lavretsky, Kurbanyan, Ballmaier, Mintz, Toga and Kumar2004) and (mainly frontal) deep white matter lesions (Herrmann et al. Reference Herrmann, Le Masurier and Ebmeier2008), which, in their diversity, do not suggest single-domain impairment.
Biological explanations for cognitive impairment in late-life depression
Current explanations of potential mechanisms for these brain changes focus on cerebrovascular pathology (Alexopoulos et al. Reference Alexopoulos, Meyers, Young, Campbell, Silbersweig and Charlson1997) and glucocorticoid action (Sapolsky et al. Reference Sapolsky, Krey and McEwen1986). The ‘vascular hypothesis’ (Alexopoulos, Reference Alexopoulos2006) is based on the consistent finding of white matter hyperintensities (Herrmann et al. Reference Herrmann, Le Masurier and Ebmeier2008), especially in the form of ischaemic lesions (Thomas et al. Reference Thomas, O'Brien, Davis, Ballard, Barber, Kalaria and Perry2002). In normal ageing (Turken et al. Reference Turken, Whitfield-Gabrieli, Bammer, Baldo, Dronkers and Gabrieli2008) and multiple sclerosis (Amato et al. Reference Amato, Zipoli and Portaccio2008), such lesions are associated with reduced processing speed, but have also been related to executive functioning deficits in late-life depression (Sheline et al. Reference Sheline, Price, Vaishnavi, Mintun, Barch, Epstein, Wilkins, Snyder, Couture, Schechtman and McKinstry2008). The ‘glucocorticoid cascade hypothesis’, based on animal models (O'Brien, Reference O'Brien1997; Sapolsky, Reference Sapolsky2000; McEwen, Reference McEwen2005), proposes that the dysregulation of the HPA axis leads to brain atrophy but direct evidence in humans has been inconsistent (O'Brien et al. Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier2004).
Age of onset of depression
Apparently incompatible with the glucocorticoid cascade hypothesis, subjects with an early onset (and thus a longer illness duration) did not display greater memory deficits or increasing memory deficits over time, which may point to different pathogenic pathways between both onset groups. LOD is more strongly related to cerebrovascular changes than EOD (Schweitzer et al. Reference Schweitzer, Tuckwell, Ames and O'Brien2001; Herrmann et al. Reference Herrmann, Le Masurier and Ebmeier2008), and demonstrates greater hippocampal volume reduction (Lloyd et al. Reference Lloyd, Ferrier, Barber, Gholkar, Young and O'Brien2004; Hickie et al. Reference Hickie, Naismith, Ward, Turner, Scott, Mitchell, Wilhelm and Parker2005). These changes might be superimposed on any pathophysiological changes that are shared with EOD (e.g. glucocorticoid action) and so explain the greater cognitive deficits seen in LOD.
Long-term course of late-life depression
During the medium-term course (baseline to 18 months), we found little evidence of further progression of deficits. Patients' change in test scores paralleled that seen in controls, which implies that both profited equally from learning effects and increasing task familiarity. Patients with LOD tended to show worsening of memory and executive functioning relative to controls, due to absence of improvement or to true decline, which was not observed in early onset patients. The exploratory analyses of the 4-year data suggest that patients remain impaired long term, but again without evidence of further decline, and only one patient developed dementia during the study. As patients with more severe impairment tended to drop out of the study, this might, however, give a too favourable picture of the true course.
Methodological considerations
The present study has several strengths, including an age- and gender-matched healthy comparison group tested at the same time points, a relatively long follow-up duration and the administration of a comprehensive neuropsychological test battery tapping into core cognitive domains. However, some methodological shortcomings have also to be considered. First, this study was observational. Hence, we did not manipulate the AD regime. Other studies have found improvement of cognition with AD use, mainly in subgroups of good responding patients (Butters et al. Reference Butters, Becker, Nebes, Zmuda, Mulsant, Pollock and Reynolds2000; Gallassi et al. Reference Gallassi, Di Sarro, Morreale and Amore2006; Mandelli et al. Reference Mandelli, Serretti, Colombo, Florita, Santoro, Rossini, Zanardi and Smeraldi2006). Cognitive functioning might therefore still be a suitable target for AD treatment, especially because the subgroups displayed somewhat better cognitive functioning in the present study, too, despite staying impaired. Second, more subjects could be tested with the extended neuropsychological test battery at follow-up than at baseline, and thus groups at different time points are not perfectly comparable. Therefore, we focused primarily on the cross-sectional analyses and tested longitudinal changes between age of onset groups only from 6 to 18 months but not from baseline. In addition, in contrast to some other reports, we did not control for estimated IQ, but instead used years of education to adjust for pre-morbid level of functioning. LTFU at 18 months among patients was within the normal range and unrelated to differences in variables of interest to the present study, with the exception of a higher drop-out among AD users. However, 75% of those seen at 18 months were on medication and it therefore seems unlikely that bias occurred due to selective drop-out. The coefficients (R 2) suggest that LTFU was preceded by worse cognition at previous assessments, which would explain the apparent convergence of patients and controls at the 18 months assessment (see Table 2). Finally, we reported the 4-year data because of the paucity of longer-term studies in the literature, but at this point we observed high and seemingly non-random drop-out among patients, so we advise interpreting these results with caution. Further studies of comparable follow-up length (and beyond) are clearly needed to verify these findings.
Conclusion
The present study shows that cognitive deficits in late-life depression tend to persist up to at least 4 years without further deterioration, affect multiple domains and seem to be related to trait rather than state effects. Differences in the severity and course of cognitive deficits due to age of onset imply different pathogenic processes between early and late onset depression.
Appendix

Fig. A1. Illustration of the association between remission status (remitted, depressed), age of onset (early onset: before age 60 years, late onset: after 59 years) and antidepressant use (yes, no) on cognition. z scores are expressed as mean differences with the healthy comparison group.
Acknowledgements
We thank the Wellcome Trust and the Stanley Foundation for financial support, L. McGuckin for help with subject recruitment, assessment and database management and J. Allardyce for statistical advice.
Declaration of Interest
None.






