



Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a common monogenic condition, with an estimated population prevalence of 1 in 1000 (Refs Reference Ong1, Reference Lanktree2). It is caused by loss-of-function heterozygous germ-line mutations in PKD1 (16p13.3; OMIM 601313; 85% of cases), PKD2 (4q21; OMIM 173910; 15% of cases) or rarely other genes (such as GANAB and DNAJB11) (Refs Reference Rangan3–Reference Cornec-Le Gall5). PKD1 and PKD2 encode, polycystin-1 and -2, respectively, which are transmembrane proteins that localise and regulate primary ciliary function, such that ADPKD is classified as a renal ‘ciliopathy’ (Refs Reference Rangan3, Reference Rangan6). ADPKD is characterised by the bilateral, progressive formation and growth of numerous microscopic, fluid-filled kidney cysts and associated with the deregulation of multiple intracellular signalling pathways (Refs Reference Rangan3, Reference Chatterjee, Verma and Pandey7). Kidney cyst formation has been hypothesised to begin in utero (or during early life) because of reduced intracellular levels of polycystins (Refs Reference Grantham, Geiser and Evan8, Reference Grantham9). Throughout life, kidney cysts continually grow by ~5% per year because of cystic epithelial cell proliferation and fluid secretion, resulting in the gradual loss of healthy kidney tissue (Ref. Reference Rangan3). The severity of renal dysfunction is determined by the number of kidney cysts that develop during life (Ref. Reference Bae10), and ~50% of patients develop end-stage kidney disease (ESKD) before the age of 60 (Refs Reference Ong1, Reference Rangan3). Other disease complications are variable and include hypertension, polycystic liver disease and intracranial aneurysms (Ref. Reference Rangan3).
Despite significant progress in understanding the pathogenesis of ADPKD, the mechanisms that ‘trigger’ kidney cyst formation during life remain unclear, and there are no disease-modifying interventions that specifically target the prevention of cystogenesis (Ref. Reference Rangan3). Several studies have suggested a link between DNA damage, oxidative stress and kidney cyst formation, and therefore the aims of this review were to: (i) critically analyse these data and determine their applicability to ADPKD; (ii) evaluate methods that can be used to assess DNA damage in PKD and (iii) determine the potential for current therapeutic approaches to alter DNA damage in ADPKD.
DNA damage and the DNA damage response (DDR) in normal health and in chronic kidney disease
In normal health, the DNA in each of the 1013 cells that make up the human body encounters tens of thousands of potentially damaging agents and processes per day, which can result in loss of fidelity in the DNA code (Ref. Reference Lindahl and Barnes11). This injury can stall DNA replication and transcription, and if not repaired correctly, can lead to permanent mutations that threaten genome integrity and cause disease (Refs Reference Hoeijmakers12, Reference Jackson and Bartek13). DNA damage is categorised into two types: (1) endogenous DNA damage, which is caused by metabolic processes within the cell itself, such as spontaneous reactions (e.g. hydrolysis) or reactive oxygen and nitrogen species (ROS and RNS); and (2) exogenous DNA damage, which is potentially preventable and occurs when cells are exposed to physical damage or chemical agents such as ultraviolet (UV) radiation (Refs Reference Hoeijmakers12, Reference Kuo and Yang14). Oxidative stress, which refers to a state of imbalance where the production of ROS exceeds the regulatory capacity of antioxidants, is an important cause of endogenous DNA damage (Ref. Reference Jena15). Excess ROS react with different components of DNA and causes a variety of DNA lesions including base modification, inter- and intra-strand crosslinks and DNA breaks (Ref. Reference Jena15). Together, spontaneous DNA damage from endogenous sources can cause up to 105 lesions per cell per day (Refs Reference Hoeijmakers12, Reference Lindahl16). The relative contributions by endogenous and exogenous DNA damage to disease is unknown, however, it has been suggested that exogenous factors are causal in 75–80% of cancer cases (Refs Reference Friedberg, McDaniel and Schultz17, Reference De Bont and van Larebeke18).
To defend against oxidative stress, genes that encode antioxidant enzymes, transcription factors and proteins are activated or silenced in an attempt to maintain redox balance (Ref. Reference Birben19). For example, glutathione (a key antioxidant) will donate its electron to H2O2 to form water and oxygen, reducing the incidence of free radicals (Ref. Reference Birben19). If antioxidant defence is unsuccessful, undesirable DNA modifications may occur (Ref. Reference Birben19). DDR is a series of cellular signalling processes and enzymatic activities that is initiated by DNA damage (Ref. Reference O'Connor20). It is facilitated by ~450 proteins that function to: (1) identify the site of DNA damage, (2) recruit DNA repair factors to the site of damage and (3) repair the physical DNA lesion (Refs Reference O'Connor20, Reference Lord and Ashworth21). In the presence of DNA damage, the histone variant, H2AX, is phosphorylated on the 139th serine residue to form γ-H2AX, and this acts as a signal to facilitate DNA repair (Ref. Reference Kuo and Yang14). The DDR is primarily mediated by Ataxia Telangiectasia and Rad3-related kinase (ATR), Ataxia-Telangiectasia Mutated (ATM) and DNA-dependent protein kinase (DNA-PK), which trigger alternative DNA repair pathways, dependent on the type of DNA lesion that has occurred (Ref. Reference O'Connor20). As part of the DDR, ATR and ATM phosphorylate checkpoint kinase 1 (Chk1) and 2 (Chk2), respectively, which maintains cells in an inactive state and prevents entry into mitosis to allow repair to occur (Ref. Reference O'Connor20).
ADPKD also shares some common histological features with other forms of chronic kidney disease (CKD), such as tubulointerstitial fibrosis and inflammation (Ref. Reference Rangan6). Published data over the last 30 years have described an association between DNA damage and progression of CKD (Refs Reference Christensson22–Reference Akagi31). Epidemiological data indicate that poor kidney function is correlated with an increased risk of cancer, and this has been attributed to genomic instability, increased cellular DNA damage and impaired DNA repair (Refs Reference Christensson22, Reference Wong23). Cengiz et al. were the first to demonstrate that uraemia was associated with abnormalities in chromosome structure and an increased rate of sister chromatid exchange (an indicator of carcinogenic or mutagenic potential) in lymphocytes (Ref. Reference Cengiz24). Subsequent studies have also shown that increasing genetic damage is associated with decline in kidney function (Refs Reference Sandoval25, Reference Sandoval26). Furthermore, in CKD, DNA repair is inhibited (Refs Reference Vamvakas27, Reference Zevin28), and higher rates of oxidative stress are reported (Refs Reference Small29, Reference Sung30). In this regard, elevated serum 8-hydroxy-2′-deoxyguanosine (8-OHdG; a biomarker of oxidative stress) also correlated the progression of kidney function decline in CKD patients (Ref. Reference Akagi31). These data suggest that DNA damage may have a pathogenic role in mediating the progression of CKD.
Genetic and cellular mechanisms underlying focal kidney cyst formation in ADPKD
One of the long-standing conundrums of ADPKD is that kidney cyst formation is focal and arises from only 1–2% of nephrons even though all cells from an affected individual carry one copy of the mutated PKD gene (Refs Reference Grantham, Geiser and Evan8, Reference Martinez and Grantham32). Previous studies suggest that further postnatal reductions of PKD1 (or PKD2) precedes, and is necessary, for initiation of kidney cysts (Refs Reference Lantinga-van Leeuwen33–Reference Hopp36). The gene dose-dependent model of cystogenesis in ADPKD was described by Rossetti et al., who demonstrated that incompletely penetrant PKD1 alleles were associated with mild disease severity in humans (Ref. Reference Rossetti35). Subsequently, Hopp et al. demonstrated that the hypomorphic PKD1 p.R3277C (RC) allele is also associated with a milder ADPKD phenotype, where Pkd1+/null mice are do not develop kidney cysts, Pkd1RC/null mice exhibit rapidly progressive disease, and Pkd1RC/RC animals develop kidney cysts gradually (Ref. Reference Hopp36). It is believed the healthy PKD gene allele inherited from the parent without ADPKD provides sufficient levels of polycystin protein, but when this allele is inactivated within an individual renal tubular cell by a somatic ‘second hit’ (mutation or by stochastic mechanisms, as discussed further below), a kidney cyst develops (Ref. Reference Grantham, Chapman and Torres37). As the cyst enlarges, it separates itself from the tubule and becomes an isolated, self-contained structure (Ref. Reference Calvet38). The kidney cyst then undergoes aberrant proliferation, increasing in cell number and size and enlarges by fluid secretion into the cyst lumen (Ref. Reference Calvet38). Over time, thousands of cysts burden the kidney, varying in diameter from one to several centimetres and the kidney can weigh up to 5 kg (Ref. Reference Rangan6).
Thus, the ‘two-hit’ model for cyst formation suggests that loss of heterozygosity because of a somatic or ‘second-hit’ mutation in the healthy allele causes cyst formation (Ref. Reference Harris39). This hypothesis is supported by extensive in vitro and in vivo studies (Refs Reference Qian40–Reference Tan47), and is described in Figure 1a. Evidence of loss of heterozygosity was initially demonstrated by genetic analysis of cystic epithelium, which revealed loss of the wild-type copy of PKD1 (Refs Reference Qian40, Reference Brasier and Henske41). Moreover, somatic inactivation of PKD2 by homologous recombination in adult mice was causative in renal and hepatic cyst formation (Ref. Reference Wu42). Most recently in support of this hypothesis, it was found that in a cohort of nine ADPKD patients, somatic mutations of PKD1 or PKD2 were present in all and occurred in 90% of kidney cysts (Ref. Reference Tan47).
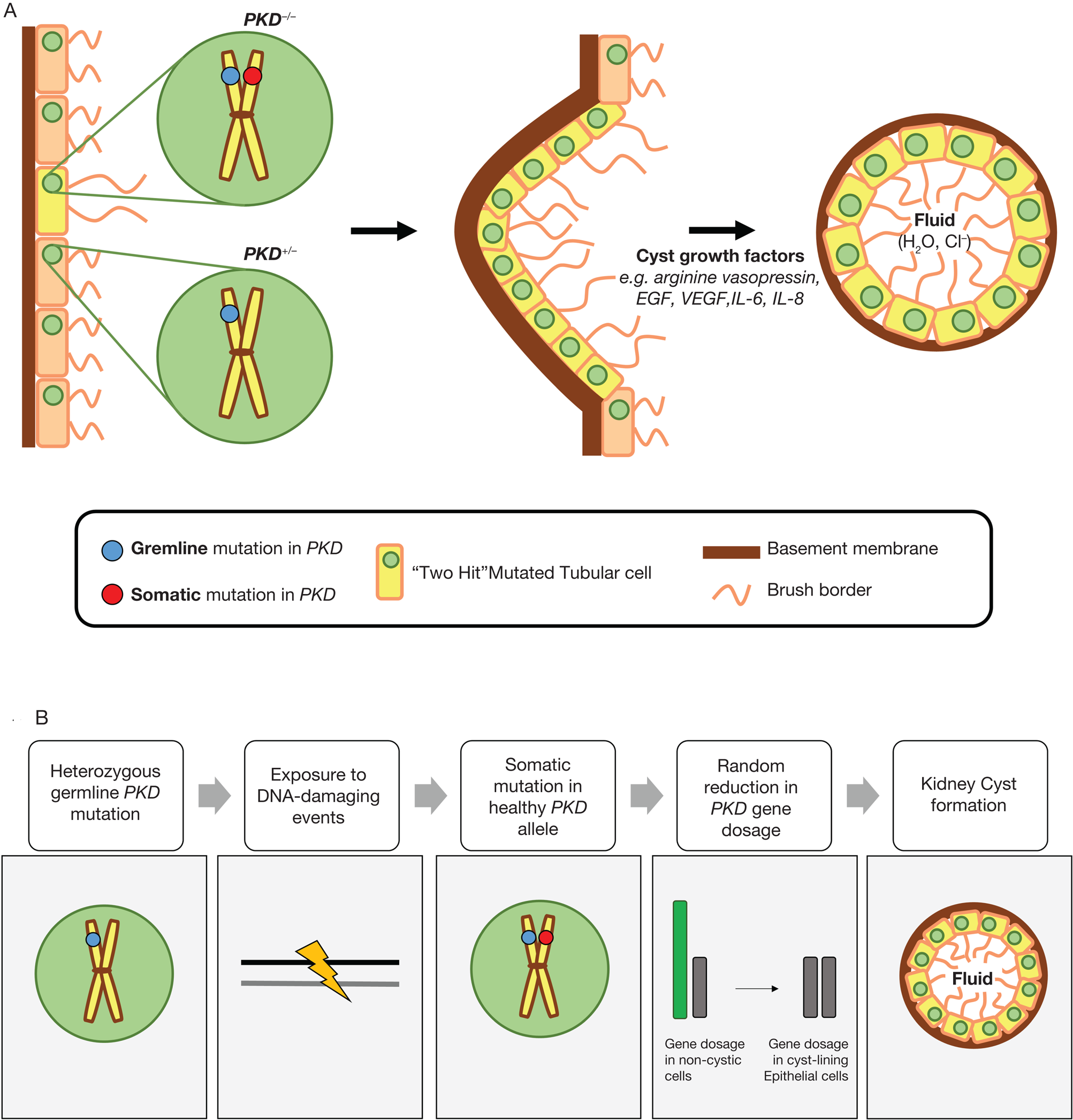
Fig. 1. (a) The ‘two-hit’ model of cystogenesis in autosomal dominant polycystic kidney disease (ADPKD). In ADPKD, all tubular epithelial cells exhibit a heterozygous germline mutation in either PKD1 or PKD2 (shown by the blue circle), and this is the ‘first-hit’. A loss of heterozygosity by an acquired or somatic mutation of the normal PKD allele in an individual cell is required to initiate cyst formation (shown by the red circle), and this is the ‘second-hit’. This epithelial cell then undergoes proliferation, potentiated by cyst growth factors such as arginine vasopressin epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), interleukin-6 (IL-6) and interleukin-8 (IL-8). The cyst eventually detaches from the original nephron and progressively increases in size because of epithelial cell proliferation, dedifferentiation and abnormal fluid secretion. (b) A hypothetical model to explain the role of DNA damage in autosomal dominant polycystic kidney disease (ADPKD). Exposure to DNA-damaging events is causal in the somatic mutation of the healthy PKD allele, resulting in a random reduction in gene dosage and resulting in kidney cyst formation. These DNA-damaging events may be because of endogenous factors (ageing, replication stress from PKD1 duplication, increased oxidative stress, epigenetic modifications and inflammation) and/or exogenous sources (elevated NaCl concentration in the inner medulla).
Other hypotheses to explain the reduction in PKD dosage include an age-related decline (Ref. Reference Rossetti35), random fluctuations during development and/or influence of environmental factors (Ref. Reference Rangan6). Regarding the latter, a ‘third hit’ hypothesis, namely proliferative signals, also appears to be essential for kidney cyst formation (Refs Reference Takakura46, Reference Takakura48). This ‘third hit’ was proposed to explain the discrepancy between the slower onset of focal cystic disease when PKD1 was inactivated in adult rather than infantile mice (Ref. Reference Takakura46), and the rapid development of kidney cysts in response to a third-hit, such as renal ischaemia-reperfusion injury (Ref. Reference Takakura48).
Finally, and more recently, the ‘snowball effect’ of cyst formation has also been described. Leonhard et al. observed that cysts formed in clusters, and PKD-related signalling (pSTAT3, pCREB, pAkt, pERK 1/2 and LCN2) was upregulated in tissue surrounding these clusters, suggesting that paracrine influences from a founder cyst may trigger the formation of new cysts (Ref. Reference Leonhard49). In addition, recent in vitro data indicate cyst fluid itself may contain accelerating paracrine trophic factors to induce regional cystic kidney disease (Ref. Reference Kenter50).
Despite some common elements in these hypotheses, the unifying molecular pathways that link them and explain the focal nature of cyst formation in ADPKD has not been clarified. As the severity of ADPKD is directly related to the occurrence frequency of this focal cyst-initiating event, targeting this mechanism is likely to be the key to preventing ESKD.
Potential role of DNA damage in reducing PKD gene dose in ADPKD
Previous studies have shown that alterations in DNA repair proteins are associated with development of human ciliopathies (Refs Reference Johnson and Collis51–Reference Slaats55). For example, Chaki et al. found that genes encoding DNA repair proteins (MRE11, ZNF423 and CEP164) are mutated in nephronophthisis-related ciliopathies (Ref. Reference Chaki54). Moreover, CEP164 deficiency was associated with systemic DNA damage, increased sensitivity to DNA damaging events, and impaired DNA damage signalling (Refs Reference Chaki54, Reference Slaats55). MRE11 and ZNF423 both play a role in the ATM-Chk2 DDR pathway, recruiting ATM to sites of damage, and CEP164 is activated by ATM and ATR, as well as necessary for Chk1 activation. Further experimental and clinical evidence linking DNA damage and cystic kidney disorders, can be summarised as follows:
Preclinical studies
Kidney cysts in ADPKD and ARPKD originate mainly from tubular epithelial cells present in nephrons from the inner medulla (Ref. Reference Torres and Harris56). Dmitrieva et al. demonstrated that murine inner medullary tubular epithelial cells are more susceptible to DNA damage compared with those from the renal cortex (Ref. Reference Dmitrieva, Cai and Burg57). Furthermore, DNA repair in inner medullary cells was impaired in response to total body irradiation, as evidenced by the reduced H2AX phosphorylation and lack of newly synthesised DNA (Ref. Reference Dmitrieva, Cai and Burg57). Therefore, it is possible that the susceptibility of the distal nephron to renal cyst formation could be related, in part, to the more hostile environment causing DNA damage of inner medullary tubules, together with impaired DNA repair.
Nek8 is a mitotic kinase that regulates intracellular levels of polycystins and is mutated in the jck mouse model of PKD and human nephronophthisis (a cystic kidney disease) (Refs Reference Smith58, Reference Sohara59). Choi et al. reported that knockdown of Nek8 in vitro causes the accumulation of double-strand breaks (DSBs) in DNA, irregular origin firing (unscheduled DNA replication), and a higher likelihood of replication fork collapse (Ref. Reference Choi60). In vivo, kidney DNA damage, measured by γ-H2AX, was increased in jck mice compared with the wild-type group (Ref. Reference Choi60). Activation of DNA repair signalling has also been associated with mutation in Sdccag8 (gene mutation causing nephronophthisis), characterised by increased γ-H2AX levels and ATM activity both in vitro and in vivo (Ref. Reference Airik61). Finally, kidney tubular cells extracted from 8-week-old Pkd1 knockout mice revealed an increase in DNA breaks, as determined by the alkaline comet assay (Ref. Reference Cassini62). However, analysis of γ-H2AX by western blotting in kidney lysates of these mice showed no differences when compared with wild-type (Ref. Reference Cassini62).
Human studies
Evidence that DNA damage is increased in human ADPKD is lacking. Interestingly, Cengiz et al. reported the highest levels of sister chromatid exchange in patients with PKD (n = 3) compared with other causes of uraemia, providing preliminary clinical evidence for a link between DNA damage and ADPKD (Ref. Reference Cengiz24). Ta et al. showed that DNA damage, measured by γ-H2AX positive immunostaining, is increased in human ADPKD kidney cyst-lining epithelial cells (Ref. Reference Ta63). Thus, further investigation is required to determine whether DNA damage has a direct role in human ADPKD.
Potential reasons for increased DNA damage in ADPKD
Evidence from preclinical and clinical studies indicate that DNA damage is increased in renal ciliopathies and renal cystic diseases. Most of these studies attribute this increase to defective DDR, resulting in impaired DNA repair and DNA damage accumulation. For example, Choi et al. hypothesised that Nek8 may play an integral role in preventing the accumulation of DNA damage through its interaction with the DNA repair proteins, ATR and Chk1 (Ref. Reference Choi60). However, the mechanisms for increased DNA damage in PKD are not clear and could include several other possibilities.
PKD1 gene is prone to replication stress and DNA damage because of large mirror repeat sequences
Replication stress is a term that describes barriers to DNA replication, including variations in DNA, complex structures that are difficult to replicate and/or exhaustion of the nucleotide pool (Ref. Reference Lecona and Fernández-Capetillo64). PKD1 contains a polypurine-polypyrimidine (Pu-Py) tract made up of large mirror repeat sequences that form complex, ‘difficult to replicate’ structures (Ref. Reference Liu65). Liu et al. found evidence that these non-B secondary DNA structures from the PKD1 gene lead to replication fork stalling, followed by DSBs and subsequent activation of DNA repair signalling (Ref. Reference Liu65). Thus, the observed DNA damage may, in part, be because of replication stress associated with PKD1 replication. Further investigation is required to examine whether this could be the cause of the ‘second hit’ mutation to the healthy allele in ADPKD. Lea et al. reported that the presence of the long polypyrimidine tract led to aberrant splicing in human PKD1, resulting in 61.5% of PKD1 transcripts having premature stop codons (Ref. Reference Lea66). This, in turn, resulted in decreased levels of full-length mRNAs, and formation of a lower molecular weight, truncated form of polycystin-1 (Ref. Reference Lea66). Lea et al. hypothesised that the decreased full-length polycystin-1 from normal alleles causes reduced polycystin signalling below ‘threshold’ levels, resulting in cyst initiation and development (Ref. Reference Lea66).
Genetic mutations associated with cystic kidney diseases increase susceptibility to DNA damage
Cells isolated from ADPKD patients are more susceptible to DNA damage after irradiation compared with healthy controls (Ref. Reference Li67). Battini et al. provided in vivo evidence that conditional knock-out (KO) of PKD1 was associated with significant centrosome amplification, and the loss of polycystin-1 resulted in mitotic catastrophe and genomic instability during disease progression in this model (Ref. Reference Battini68). Both Li et al. and Battini et al. proposed that the ‘second hit’ or somatic mutation to the PKD gene is required for dysregulated polycystin-1 expression and provides an explanation for increased genomic instability (Refs Reference Li67, Reference Battini68). The increased genomic instability may also explain the phenotypic variability in ADPKD and sporadic cyst formation in few nephrons (Ref. Reference Battini68). In addition, a cross-species meta-analysis of conserved biological pathways in ADPKD also suggested that transcriptomic alterations related to genomic instability were associated with regulating cyst formation (Ref. Reference Chatterjee, Verma and Pandey7). Finally, a cohort study from Taiwan suggested that patients with ADPKD have higher risk of developing liver, colon and renal cancer compared with those without the disease (Ref. Reference Yu69). However, this has not been confirmed by all studies, and overall, the relationship between ADPKD and cancer incidence remains conflicting (Refs Reference Cachat and Renella70, Reference Chapman and Wong71). For example, in contrast, Wetmore et al. reported that renal transplant recipients with PKD were 16% less likely to develop cancer compared with non-PKD patients (Ref. Reference Wetmore72).
Increased oxidative stress results in subsequent endogenous DNA damage
Kidney cystic disorders are characterised by an increase in oxidative stress, which may contribute to endogenous DNA damage (Refs Reference Cassini62, Reference Apeland, Holdaas and Mansoor73–Reference Nowak75). A case-control study showed that ADPKD patients had higher levels of aminothiols (marker of oxidative stress) compared with healthy controls (Ref. Reference Tariq74). The redox imbalance in CKD may be because of reduced estimated glomerular filtration rate (eGFR) and/or hypertension (Refs Reference Apeland, Holdaas and Mansoor73, Reference Tariq74). In particular, elevated levels of plasma total homocysteine (a component of the antioxidant defence system) are associated with reduced eGFR, and may provide one explanation for increased oxidative stress in ADPKD (Ref. Reference Apeland, Holdaas and Mansoor73). More direct evidence was provided by Cassini et al., who demonstrated that 8-OHdG was expressed in cyst-lining cells of Pkd1 mutant mice (Ref. Reference Cassini62). Finally, Nowak et al. demonstrated that acute intravenous infusion with ascorbic acid (an inhibitor of free radicals) improved brachial artery flow-mediated dilation (a measure of endothelial dysfunction) in humans with ADPKD, but had no effect on controls, indicating the presence of vascular endothelial oxidative stress (Ref. Reference Nowak75).
Epigenetic changes results in DNA damage
A limited number of studies have examined the functional significance of epigenetics in ADPKD (Refs Reference Woo76–Reference Woo78). Woo et al. observed that PKD1 was hypermethylated, and inhibiting DNA methylation ameliorated cyst formation in vitro (Ref. Reference Woo76). Moreover, the non-coding RNA, miR-182-5p, was found to be novel regulator of progression in conditional PKD1/2-deficient mice (Ref. Reference Woo78). Genes that have undergone epigenetic modifications, such as methylation of the DNA base, cytosine, may be more prone to oxidative stress later in life, as well as exhibiting reduced ability to repair oxidised DNA bases (Ref. Reference Zawia, Lahiri and Cardozo-Pelaez79). In the context of Alzheimer's disease, hypomethylation of disease-related genes led to overproduction of ROS-producing proteins, and hypermethylation led to diminished capacity to repair 8-OHdG (a product of oxidative stress) (Ref. Reference Zawia, Lahiri and Cardozo-Pelaez79). Therefore, this observed DNA damage may be a consequence of epigenetic modifications to the PKD gene, as hypermethylation may result in increased oxidative stress, reduced base excision repair capacity and subsequent DNA lesions.
Increased renal interstitial inflammation causes DNA damage
There is clear interplay between inflammation, DNA damage and DNA repair (Ref. Reference Pálmai-Pallag and Bachrati80), and inflammation is also a key pathological feature of PKD, as reviewed previously (Ref. Reference Ta, Harris and Rangan81). In both acute and chronic inflammation in the kidney, neutrophils, eosinophils and macrophages are activated and as a defence mechanism, large volumes of ROS and RNS are produced, which may have genotoxic effects as well as inhibiting the DDR response (Ref. Reference Pálmai-Pallag and Bachrati80). Cassini et al. investigated this hypothesis and found reduced DNA breaks in PKD1−/−/MCP1−/− mice compared with PKD1−/−/MCP1+/+ mice (Ref. Reference Cassini62). Thus, observed DNA damage may be a result of interstitial pro-inflammatory signalling around kidney cysts.
Exposure to endogenous and exogenous factors results in DNA damage
Both endogenous and exogenous DNA-damaging events may also contribute to DNA damage observed in both CKD and cystic kidney disease. Ageing is associated with genome instability because of endogenous factors, as DNA undergoes time-dependent deterioration from spontaneous biological reactions (Ref. Reference Hoeijmakers12). Moreover, previous in vitro studies have demonstrated that acute increases in NaCl inhibits the DDR (evidenced by lack of H2AX and Chk1 phosphorylation), resulting in the accumulation of DNA breaks and impaired DNA repair (Ref. Reference Dmitrieva, Bulavin and Burg82). In addition, both in vitro and in vivo studies have demonstrated that angiotensin II can also trigger DNA damage (Refs Reference Schupp83–Reference Brand, Amann and Schupp85). The increase in damage could be attenuated by administration of an angiotensin II type-1 receptor blocker (candesartan) and antioxidants (N-acetylcysteine and α-tocopherol), highlighting that angiotensin II causes DNA damage via this receptor and subsequent formation of oxidative stress (Refs Reference Schupp83, Reference Brand, Amann and Schupp85).
Exogenous events such as renal ischaemia-reperfusion stimulate phosphorylation of DDR proteins, ATM, H2AX, Chk2 and p53 in vivo, which is likely because of increased ROS (Ref. Reference Ma86). As mentioned earlier, ischaemia and nephrotoxic injury is associated with exacerbated cyst burden in PKD (Refs Reference Takakura48, Reference Prasad87, Reference Happé88). Therefore, DNA lesions resulting from oxidative damage because of ageing, high sodium content of the inner renal medulla, high intra-renal levels of angiotensin II, ischaemia or nephrotoxic injury are other potential explanations for increased DNA damage in ADPKD. The role of chronic exposure to endogenous and exogenous DNA-damaging events in disease pathogenesis is important to consider because of the incidence of somatic mutations in ADPKD pathogenesis. Alexandrov et al. demonstrated that ‘clock-like’ mutational signatures present in clear cell and papillary renal cancers originate from continuous exposure and reabsorption of mutagens in the proximal tubular epithelium of the kidney (Ref. Reference Alexandrov89), highlighting a causal role for endogenous and exogenous factors in the development of mutations and disease.
It is unlikely that any of these possible explanations occur in isolation in the complex microenvironment of the polycystic kidney. Therefore, a combination of replication stress associated with the PKD gene, oxidative stress, exposure to endogenous and exogenous factors, inflammation, genetic mutations and epigenetic changes, most likely explains the observed increase in DNA damage in ADPKD.
Biomarkers that could be used to assess DNA damage in ADPKD
Multiple techniques can be used for the detection, analysis and quantification of DNA damage, in both preclinical and clinical settings. These techniques vary in specificity, sensitivity and ease of administration, and each have their own advantages and disadvantages. The key methods for measuring DNA damage in the current literature are: (i) the comet assay, (ii) γ-H2AX and (iii) 8-OHdG, and these are summarised in Table 1 and below.
Table 1. A summary of methods that could be used to assess DNA damage in cystic kidney diseases

ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3 related; DNA-PK, DNA-dependent protein kinase; DSB, double-stranded break; ELISA, enzyme-linked immunosorbent assay; MRN, MRE11-RAD50-NBS1; SSB, single-stranded break; 8-OHdG, 8-hydroxy-2′-deoxyguanosine; γ-H2AX, gamma-H2AX.
Preclinical biomarkers
Comet assay: Overall, the comet assay is widely used and provides a reliable assessment of DNA damage levels. The benefit of the comet assay is that it can be performed in cells isolated non-invasively from humans (e.g. peripheral blood lymphocytes), however it is unclear whether DNA damage detected in these cells is an accurate reflection of damage in tissues of interest (Ref. Reference Collins90). Previous studies have used the comet assay to examine DNA damage in healthy individuals compared with those on dialysis (Ref. Reference Schupp, Stopper and Heidland91), and these demonstrated that DNA damage was increased in the latter when performed on peripheral blood lymphocyte cells (Ref. Reference Schupp, Stopper and Heidland91). Choi et al. also observed that an increased tail moment, representative of increased DNA damage, when neutral comet assays were performed on HeLa cells with Nek8 knockdown, indicating increased DSB (Ref. Reference Choi60).
γ-H2AX: The measurement of γ-H2AX is also limited to a preclinical setting, using in vitro and in vivo models of disease, as it requires tissue and cannot be measured from routine specimens collected from human patients such as blood and urine. The advantage of γ-H2AX is that it provides insight into potential molecular pathways involved in the body's response to DNA damage, and its upstream effectors or downstream targets may be related to disease causality. Overall, immunoblotting is less sensitive and is not informative of the location or nature of γ-H2AX foci (Ref. Reference Sharma, Singh, Almasan and Bjergbæk92). This technique would be useful in the examination of ADPKD tissue as the localisation of γ-H2AX in cyst-lining cells compared with non-cyst-lining cells would be of interest. However, the quantification of immunostaining can be challenging as there is variable background level of γ-H2AX associated with DNA replication occurring in S-phase cells (Ref. Reference Bonner93). To overcome these difficulties, detection of other proteins involved in DNA repair could act as a surrogate measure of DNA damage (Ref. Reference Bonner93). However, immunostaining for these other proteins (e.g. p53, ATR and ATM) is not equivalent to γ-H2AX, as most DNA repair proteins already exist in the nucleus. Therefore, their rates of accumulation may not correspond directly to onset of DNA damage, as background protein levels may be present (Ref. Reference Bonner93). Alternatively, if the protein of interest is also phosphorylated de novo (in a similar nature to γ-H2AX), using an antibody to its phosphorylated form may yield more accurate results (Ref. Reference Bonner93).
Clinical biomarkers
8-Hydroxy-2′-deoxyguanosine (8-OHdG): The measurement of 8-OHdG could be easily incorporated into clinical research studies as it can be measured from routinely collected specimens, and the availability of enzyme-linked immunosorbent assay (ELISA) provides a simple, high-throughput technique. However, 8-OHdG lacks sensitivity, as it is an indicator of DNA damage caused by oxidative stress exclusively, excluding other types of DNA damage. Furthermore, the antibodies available for 8-OHdG measurement have been previously criticised for the lack of specificity and tendency for overestimation, highlighting the importance of controls and careful data interpretation (Ref. Reference Santella100). Overall, the measurement of 8-OHdG would primarily demonstrate the contribution of endogenous and exogenous factors to DNA damage in ADPKD. Similar to the comet assay, 8-OHdG levels have also been compared in healthy and dialysed individuals, and a review of all studies revealed higher oxidative stress in individuals undergoing haemodialysis (Ref. Reference Schupp, Stopper and Heidland91).
In summary, there is no perfect method for assessing DNA damage in either experimental or clinical context. The comet assay is regarded as the ‘gold-standard’ for assessment of DNA damage (Ref. Reference Figueroa-Gonzalez and Perez-Plasencia101). In preclinical studies, the comet assay provides an indication of the presence or absence of DNA damage, encompassing all the different types of DNA damage, and can be quantified using specific software. Despite this, γ-H2AX tends to be routinely used in preclinical studies (instead of the comet assay), probably because it can be measured using routine laboratory techniques such as immunoblotting and immunohistochemistry. In contrast, in clinical research studies, urinary or serum 8-OHdG, is a practical method to use as it can quantified using ELISA. Therefore, based on our analysis of the methodological data, our recommendation is for investigators to use a combination of methods to verify the hypothesis.
Clinical implications and applications
It is possible that current therapies for ADPKD could, in part, be protective because of their effect on DNA damage and further studies might be helpful in addressing this hypothesis. In this regard, current therapies in clinical practice, as well as those under investigation, and their potential link to DNA damage are summarised below and in Table 2.
Table 2. A summary of therapies that influence cyst growth in ADPKD and their effect on DNA damage

ACEi, angiotensin converting enzyme inhibitors; ARBs, angiotensin II receptor blockers.
a Denotes this effect was demonstrated in human studies.
b Denotes this effect was demonstrated in vitro.
c Denotes this effect was demonstrated in animal studies.
Angiotensin converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs)
Previous in vitro and in vivo studies have demonstrated that angiotensin II can trigger DNA damage. Schmid et al. found that increased levels of angiotensin II caused a significant increase in DNA strand breaks (measured by the comet assay) in isolated perfused mouse kidneys (Ref. Reference Schmid84). In addition, γ-H2AX and 8-OHdG were detected in angiotensin II-treated cells (Ref. Reference Schmid84). Treatment of pig kidney cells with angiotensin-II led to an increase of DNA damage (up to 15-fold measured by the comet assay) and enhanced formation of ROS (Ref. Reference Schupp83). This increased damage could be attenuated by administration of an ARB (candesartan) and antioxidants (N-acetylcysteine and α-tocopherol) (Ref. Reference Schupp83). These findings were consistent in vivo, where treatment with angiotensin II led to increased oxidative stress (measured by ROS formation), elevated DSBs in the kidney (measured by γ-H2AX) and increased poly (ADP-ribose) polymerase activity (PARP; a DNA repair protein) (Ref. Reference Brand103). All these effects were reversed with administration of candesartan (Ref. Reference Brand103). Therefore, the small benefit of lower blood pressure targets in reducing cyst growth in ADPKD mediated by ACE inhibitors and ARBs (Ref. Reference Schrier102), may be related to reduced angiotensin levels and DNA damage inhibition.
Arginine vasopressin (AVP) signalling
The functional role of AVP in renal cyst progression has been confirmed in human ADPKD, where administration of a V2 receptor antagonist (tolvaptan) resulted in modest reductions in total kidney volume increase and eGFR decline (Refs Reference Torres104–Reference Wyatt and Le Meur106). There is limited evidence to suggest that AVP levels directly influence DNA damage or DNA repair. In male Wistar rats, vasopressin infusion exhibited antioxidant properties, reducing oxidative stress and demonstrating a cardioprotective effect against ischaemia-reperfusion injury (Ref. Reference Nazari107). Furthermore, increased cAMP levels, which AVP is known to upregulate, have demonstrated a protective effect against the DNA damaging agents (cisplatin and etoposide) in an in vitro model of chronic myeloid leukaemia (Ref. Reference Xiao and Kan108). However, dehydration and elevated vasopressin levels have been associated with increased oxidative stress, contributing to neurovascular and cognitive defects in adult mice (Ref. Reference Faraco109). It is possible that DNA damage is involved in the earlier stages of disease as a trigger for initial kidney cyst formation, whereas AVP acts as the driver for continued proliferation.
Modifiable dietary factors
Modifiable dietary factors, namely dietary sodium intake (Refs Reference Torres130–Reference Amro132) and caloric restriction (Refs Reference Warner133–Reference Nowak135), may mediate the progression of ADPKD. In vitro, acute increases in NaCl inhibits the DDR, resulting in the accumulation of DNA breaks and impaired DNA repair (Ref. Reference Dmitrieva, Bulavin and Burg82). Of note, mouse inner medullary collecting duct cells were able to adapt when exposed to a high NaCl environment, with continued rapid proliferation and no evidence of apoptosis (Ref. Reference Dmitrieva, Cai and Burg57). However, the cells did not activate the DNA repair signalling, as evidenced by the lack of H2AX phosphorylation, and had persistent numerous DNA breaks (Ref. Reference Dmitrieva, Cai and Burg57). When NaCl level was decreased, DNA repair signalling was reactivated, indicated by H2AX phosphorylation (Ref. Reference Dmitrieva, Cai and Burg57). Therefore, a clear relationship exists between higher salt, increased DNA damage and impaired DNA repair in vitro, but these data need further verification in vivo.
Recent studies have also suggested a protective role for food restriction in the context of ageing and DNA damage, and this has been reviewed previously (Refs Reference Haley-Zitlin and Richardson136–Reference Subba Rao138). Vermeij et al. observed that in mice deficient in the DNA excision repair gene (Ercc Δ/−), caloric restriction of 30% increased median and maximal lifespans of by three-fold (delaying accelerated ageing), and reduced the number of γ-H2AX foci (Ref. Reference Vermeij139). Therefore, the beneficial effect of food restriction in murine ADPKD may be mediated, at least in part, by reductions in DNA damage.
Nicotinamide
Nicotinamide is the active form of vitamin B3 or niacin, and is a precursor for the synthesis of nicotinamide adenine dinucleotide (Ref. Reference Surjana, Halliday and Damian122). Zhou et al. discovered that administration of nicotinamide drastically reduced cyst formation in Pkd1−/− embryos and Pkd1 KO mice (Ref. Reference Zhou121). However, the mechanism of action proposed for the effect of nicotinamide on delaying cyst growth was via sirtuin 1 inhibition, rather than DNA damage inhibition (Ref. Reference Zhou121). Furthermore, Zhou et al. found that Pkd1 and Sirt1 double KO mice had no further delay in cyst growth compared with just Pkd1 KO mice, suggesting that nicotinamide reduces cyst growth by specifically inhibiting sirtuin 1, and is unlikely to act via other pathways (Ref. Reference Zhou121). Currently, a pilot randomised controlled trial (NIAC-PKD2; ClinicalTrials.gov Identifier: NCT02558595; https://clinicaltrials.gov/ct2/show/NCT02558595) has been completed at the University of Kansas Medical Center to observe the effects of niacinamide (reduced form of niacin) on kidney cyst growth and markers of kidney disease progression. Nicotinamide promotes DNA repair by providing a substrate for PARP-1 activity, an energy reserve for ATP-dependent DNA repair, and preserving the integrity of PARP-1 (Ref. Reference Surjana, Halliday and Damian122). Moreover, the ONTRAC study found that 500 mg of nicotinamide twice daily was associated with a lower rate of new non-melanoma skin cancers in a high-risk population (Ref. Reference Chen123).
Other investigational treatments
Metformin: Recent studies, focused on drug repurposing, have revealed significant therapeutic potential for metformin in the treatment of ADPKD (Refs Reference Takiar110, Reference Chang111). A phase II, double-blinded, randomised placebo-controlled trial (TAME; ClinicalTrials.gov Identifier: NCT02656017; https://clinicaltrials.gov/ct2/show/NCT02656017) is underway to examine efficacy, feasibility, safety and tolerability of metformin use in patients with ADPKD (Ref. Reference Seliger140). In addition, a clinical trial, called Implementation of Metformin therapy to ease decline of kidney function in PKD (IMPEDE-PKD), by the Australasian Kidney Trials Network (AKTN) is in development (https://aktn.org.au/trials/trials-in-development/impede-pkd/). The relationship between metformin and DNA damage remains unclear, where experimental evidence suggests both a harmful and protective role (Refs Reference Ohnishi, Mizutani and Kawanishi112–Reference Othman119). Epidemiological evidence suggests type-2 diabetic patients on metformin have lower cancer incidence (Ref. Reference DeCensi114), and there is in vivo and in vitro evidence that suggests metformin reduces tumour growth rates (Refs Reference Zakikhani115–Reference Zakikhani118). Moreover, metformin reduced insulin-induced DNA damage and oxidative stress in the Zucker diabetic fatty rat (Ref. Reference Othman119). However, some studies have reported elevated oxidative stress and increased DNA damage, associated with metformin treatment (Refs Reference Ohnishi, Mizutani and Kawanishi112, Reference Amador113).
Curcumin: Leonhard et al. revealed curcumin treatment was effective at reducing two kidney weight to body weight ratio, cystic index, proliferation and STAT3 activation in the iKsp-Pkd1 del mouse model (Ref. Reference Leonhard124). The clinical efficacy of curcumin as an oral supplement (25 mg/kg per day) in treating vascular dysfunction in ADPKD is currently being investigated in a paediatric and young adult population (6–25 years of age) (ClinicalTrials.gov Identifier: NCT02494141; https://clinicaltrials.gov/ct2/show/study/NCT02494141). A secondary outcome measure for this study is change in urine 8-OHdG after 1 year of curcumin treatment, as a biomarker of oxidative stress. The effect of curcumin in DNA damage and DNA repair proteins has also been examined in vitro and in vivo (Refs Reference Shang125, Reference Cao126, Reference Chen128, Reference Lu141). In vivo, curcumin significantly improved survival rate of tumour-bearing mice (Ref. Reference Chen128). In vitro, curcumin reduced DNA damage induced by carboplatin treatment (chemotherapeutic agent) and increased expression of DNA repair pathway proteins such as BRCA1, BRCA2 and ERCC1 (Ref. Reference Chen128). In contrast, studies in mouse-rate hybrid retina ganglion N18 cells, demonstrated that curcumin treatment induced DNA damage and reduced expression of DNA repair proteins including ATM, ATR, BRCA1 and DNA-PK (Ref. Reference Lu141). Moreover, in human hepatoma G2 cells, curcumin increased 8-OHdG staining in a dose-dependent manner (Ref. Reference Cao126). Owing to this conflicting evidence, Cao et al. proposed a dual, dose-dependent role for curcumin, where low level curcumin treatment exhibits antioxidant properties and high doses cause oxidative stress and DNA damage (Ref. Reference Cao126).
Glycosphingolipid (GSL) metabolism: Altered GSL metabolism has emerged as a pathological feature of hyperplastic or hypertrophic renal diseases, including renal cell carcinoma, diabetic nephropathy and PKD, because of its role in regulating proliferation and apoptosis (Refs Reference Shayman142, Reference Shayman143). Elevated components of GSL metabolism including glucosylceramide, lactosylceramide and ganglioside GM3 have been reported in human ADPKD and the cpk mouse (Refs Reference Deshmukh144, Reference Chatterjee145), and Natoli et al. demonstrated that pharmacological inhibition of glucosylceramide synthase with Genz-123346 (blocks conversion of ceramide to glucosylceramide) reduced cystic disease burden and improved renal function in numerous mouse models of PKD (Pkd1 conditional knockout mice, jck mice and pcy mice) via downregulation of Akt-mTOR pathways and inhibition of cell cycle (Ref. Reference Natoli129). Recruitment is currently underway for a 2-year intervention study (STAGED-PKD) to determine whether venglustat (a glucosylceramide synthase inhibitor) is effective at reducing the rate of total kidney volume growth and eGFR decline in ADPKD patients with rapidly progressive disease (18–50 years of age, eGFR 45–90 mL/min/1.73 m2, Mayo Imaging Classification Class 1C-E; ClinicalTrials.gov Identifier: NCT03523728; https://clinicaltrials.gov/ct2/show/NCT03523728). Studies have demonstrated interaction between sphingolipids and the DDR, as reviewed by Carroll et al. (Ref. Reference Carroll, Donaldson and Obeid146), where elevated ceramides may be a downstream consequence of ATM activation in vitro (Ref. Reference Vit and Rosselli147), as well as associated with conditions of oxidative stress (Refs Reference Couto148, Reference Couto149). However, there is currently no evidence to suggest glucosylceramide synthase inhibition directly influences levels of DNA damage and/or DNA repair mechanisms.
In summary, almost all current therapies in use or under investigation for the treatment of ADPKD, at least in part, suppress DNA damage and/or oxidative stress. However, further studies are required to clarify the extent to which these therapies are mediated, at least in part, by their effect on suppressing DNA damage.
Conclusion and future directions
This review has outlined the current evidence linking DNA damage and kidney cyst formation in ADPKD. The proposed hypothesis is outlined in Figure 1b. Briefly, we propose that exposure to DNA-damaging events is causal in the formation of somatic mutations and reduced gene dosage, resulting in subsequent kidney cyst formation. This hypothesis requires further investigation in future studies, and specific questions that should be addressed include the following:
(i) Verification that DNA damage is increased in ADPKD compared with healthy controls. Future studies are required to examine the in vivo characteristics and kinetics of DNA damage and DDR signalling in genetically orthologous animal models and human ADPKD, in relation to the progression of cystic kidney disease. These studies will determine if the presence of DNA damage precedes or follows cyst formation in ADPKD, and whether DNA damage is increased in early- or late-stage disease.
(ii) Are ADPKD patients more susceptible to DNA damage? Further investigation is required to examine whether PKD1 deficiency results in increased susceptibility to DNA damage, and whether DNA repair is defective in ADPKD. This hypothesis can be examined in vitro and in vivo by exposing PKD models to DNA damaging agents (e.g. UV light, cytotoxic agents) and observing their response compared with healthy controls. Genomic analyses would also be useful to examine whether mutations in PKD1 or PKD2 are also associated with mutations in DDR genes, potentially providing an explanation for a defective DDR in ADPKD.
(iii) Will modifying DNA damage influence disease outcomes in ADPKD? It remains unknown whether reducing or increasing DNA damage will improve disease outcomes in ADPKD. Initial studies are required to confirm the functional significance of DNA damage. If DNA damage proves causal, studies would be required to examine if approaches to reduce DNA damage (e.g. antioxidants) are effective at reducing cyst burden in ADPKD. An examination of mutational signatures and patterns observed in cystic tissue compared with non-cystic tissue may be useful in identifying whether any changes in DDR occur as a cyst-initiating event. Furthermore, the use of a systems biology approach will be most useful to determine the therapeutically potential for modifying DNA damage in ADPKD. An examination of transcriptomics, proteomics and metabolomics can reveal novel pathways and mechanisms involved in disease pathogenesis, allowing for the discovery of new small molecule drug targets and drug re-purposing. Numerous ‘omics’ analyses have been conducted previously in PKD using cell lines, rat or mouse tissue and human samples (Ref. Reference Menezes and Germino150), and further examination of this data from a DNA damage and DDR perspective would be of interest.
ADPKD is a genetic condition that affects approximately 12 million people worldwide, where at least half will experience ESKD during their lifetime. Owing to the economic burden of renal replacement therapy and the increasing longevity of the population, the identification of drug targets and development of treatments to ameliorate kidney cyst formation in ADPKD are vital. Targeting DNA damage may provide a promising path forward towards curative ADPKD therapies.
Financial support
This work was supported by a Research Training Program Stipend from the University of Sydney (J.Z.), the PKD Foundation of Australia and the National Health and Medical Council of Australia (grant number 1164128).
Conflict of interest
None.



