INTRODUCTION
Infectious diseases such as malaria, tuberculosis and HIV are amongst the highest causes of morbidity and mortality worldwide (Wenzel and Edmond, Reference Wenzel and Edmond2000). Many of the gold-standard treatments for these diseases are under threat due to resistance factors and new medicines are urgently required to reduce this burden. Despite this, active anti-infective drug discovery programmes within the pharmaceutical industry have been on the decline since the 1990s.
Since the determination of the complete bacterial and eukaryotic genomes in the mid-1990s, drug discovery used a genomics-based approach whereby essential and conserved genes for the survival of a pathogen were identified and their corresponding proteins subjected to HTS to identify inhibitors. Unfortunately, this target-based genomic approach has failed to deliver the number of anti-infective drugs expected by the pharmaceutical industry (Livermore, Reference Livermore2011). Phenotypic screening has been at the forefront of anti-infective drug discovery for many years (Calderón et al. Reference Calderón, Barros, Bueno, Coterón, Fernández, Gamo, Lavandera, León, Macdonald, Mallo, Manzano, Porras, Fiandor and Castro2011) and the screening of large compound libraries is a long established method for the development of novel chemotherapeutics. However, this method of drug discovery only allows the investigation of a relatively small area of chemical space (López-Vallejo et al. Reference López-Vallejo, Giulianotti, Houghten and Medina-Franco2012).
Structure-based drug design (SBDD) utilizes both the knowledge of three-dimensional protein structures and in silico techniques to identify putative small molecules with biological activity against desired protein targets (Stahl et al. Reference Stahl, Guba and Kansy2006; Simmons et al. Reference Simmons, Chopra and Fishwick2010). SBDD allows greater access to diverse areas of chemical space than compared to HTS, since in silico compound libraries can contain compounds not yet synthesized. Additionally, de novo design can allow access to all possible regions of chemical space (Simmons et al. Reference Simmons, Chopra and Fishwick2010). Each method allows the development of novel chemotherapeutics outside the constraints of classical HTS.
Our research group uses the three main methods of SBDD (1) substrate-inspired design, (2) virtual high throughput screening (vHTS) and (3) de novo design of inhibitors. After the synthesis or purchase of the desired compounds they are subjected to biological evaluation to establish their level of inhibition against the target protein. Once hit compounds have been identified, further iterative rounds of design and optimization occur to improve binding affinity and physicochemical properties (Fig. 1). We will review the main computational tools for our in silico structure-based approach to novel anti-infectives and give examples of our successes in this therapeutic area.

Fig. 1. From X-ray crystallography to lead compounds: an overview of the SBDD process.
COMPUTATIONAL TOOLS FOR SBDD
SPROUT (Gillet et al. Reference Gillet, Newell, Mata, Myatt, Sike, Zsoldos and Johnson1994) is the main de novo design tool for the generation of ligands used by our research group, developed in-house by the Institute for Computer Applications in Molecular Science (ICAMS, University of Leeds, UK). The programme utilizes the principles of shape complementarity – the favourable fit of the three-dimensional shape of a molecule into the volume of a protein cavity coupled with optimal creation of binding interactions between the binding ligand and protein. Structure generation (Fig. 2) is achieved by firstly placing small fragments at chosen sites within the targeted binding region, each of which is placed to make a specific interaction with the protein. These molecular fragments are then joined in a stepwise fashion which satisfies the steric requirements, as well as the electrostatic and hydrophobic properties imposed by the protein to create the complete molecular scaffold. SPROUT can score the generated molecular scaffolds based on predicted binding affinity (calculated pKi of all the binding interactions) and/or molecular complexity (measure of synthetic accessibility).
Fig. 2. Skeleton generation in SPROUT. First the primary constraints are defined by the boundary of the receptor site. Skeleton generation then places a template at the target sites until all sites are satisfied and no boundary violations have occurred. Finally, atoms are substituted in each skeleton to satisfy secondary constraints such as electrostatics and hydrophobicity.
SPROUT-HitOpt (Heikkila et al. Reference Heikkila, Ramsey, Davies, Galtier, Stead, Johnson, Fishwick, Boa and McConkey2007; Simmons et al. Reference Simmons, Chopra and Fishwick2010) can be utilized once a hit compound has been identified in order to help guide the optimization of its binding interactions with the target protein. SPROUT-HitOpt operates via core extension whereby additional fragments or groups are added to structures representing the previously identified hit compounds so as to fill additional space and make additional interactions within the protein cavity. This software uses a retrosynthetic approach, utilizing a reaction database and a library of commercially available compounds from which alternative ‘monomers’ can be selected in order to extend the structure of the hit in ways which are compatible with its actual structure.
eHiTS (Zsoldos et al. Reference Zsoldos, Reid, Simon, Sadjad and Johnson2007) is a flexible ligand docking and vHTS software package developed by SimBioSys Inc. This software uses an innovative exhaustive docking algorithm and consistently performs well in pose-prediction studies, where various docking programs are asked to reproduce co-crystal protein-ligand binding conformations. The docking algorithm searches all possible areas of space within the binding site. To achieve this, eHiTS splits the ligand(s) into smaller units consisting of rigid and flexible fragments, which are then docked into the three-dimensional space of the protein receptor in all possible configurations. The program then generates ‘solutions’ from the docked fragments that satisfy the steric constraints of the receptor surface (Fig. 3), which are ranked using the eHiTS scoring function.
Fig. 3. The workflow of the eHiTS docking program.
AutoDock (Morris et al. Reference Morris, Huey, Lindstrom, Sanner, Belew, Goodsell and Olson2009) is a docking software suite which predicts the ability of a small molecule to bind to a receptor. The AutoDock suite is a combination of three programs; AutoGrid, which calculates grid maps of interaction energies for the atom types present in ligands being docked, AutoTors, to identify and define rotatable bonds within the ligand, and AutoDock which utilizes a Lamarckian-Genetic algorithm to dock the ligands into the protein (Huey et al. Reference Huey, Morris, Olson and Goodsell2007). Each module is manipulated through AutoDockTools which is the GUI for AutoDock, AutoTors and AutoGrid (Morris et al. Reference Morris, Huey, Lindstrom, Sanner, Belew, Goodsell and Olson2009). AutoDock searches all possible ligand conformations within the binding site (Fig. 4). This technique should identify the lowest energy ‘pose’ a ligand can take within the binding site. AutoDock achieves this via a series of small conformational changes and larger conformational ‘mutations’ to the ligand to escape local energy minima in-order to find the most stable binding pose for a given ligand. It is important to note that this technique is more computationally intensive than the approach utilized by eHiTS resulting in much smaller numbers of compound structures being screened within a comparable time period.
Fig. 4. Schematic of the AutoDock force field evaluating the free energy of binding in two steps. First, AutoDock calculates the intramolecular energetics of the transition of the ligand and protein from its unbound to bond conformation. Secondly, AutoDock evaluates the intermolecular energetics of combing the ligand and protein in their bound conformations. Adapted from (Huey et al. Reference Huey, Morris, Olson and Goodsell2007).
Rapid Overlay of Chemical Structure (ROCS) (Grant et al. Reference Grant, Gallardo and Pickup1996) uses shape similarity to overlay a target molecule to a compound library to search for molecules that may have similar chemical and biological properties. ROCs operate under the assumption that compounds with similar three-dimensional shape and chemical properties to a known hit compound could show similar biological activity. ROCS compares molecular structures based upon three-dimensional shape where the software tries to maximize shared volume between the molecules, while an additional option allows the overlap of similar chemical properties/ electrostatics such as hydrogen bond donors or acceptors, cationic, anionic or hydrophobic groups.
RESULTS AND DISCUSSION
Example 1 – bacterial infections
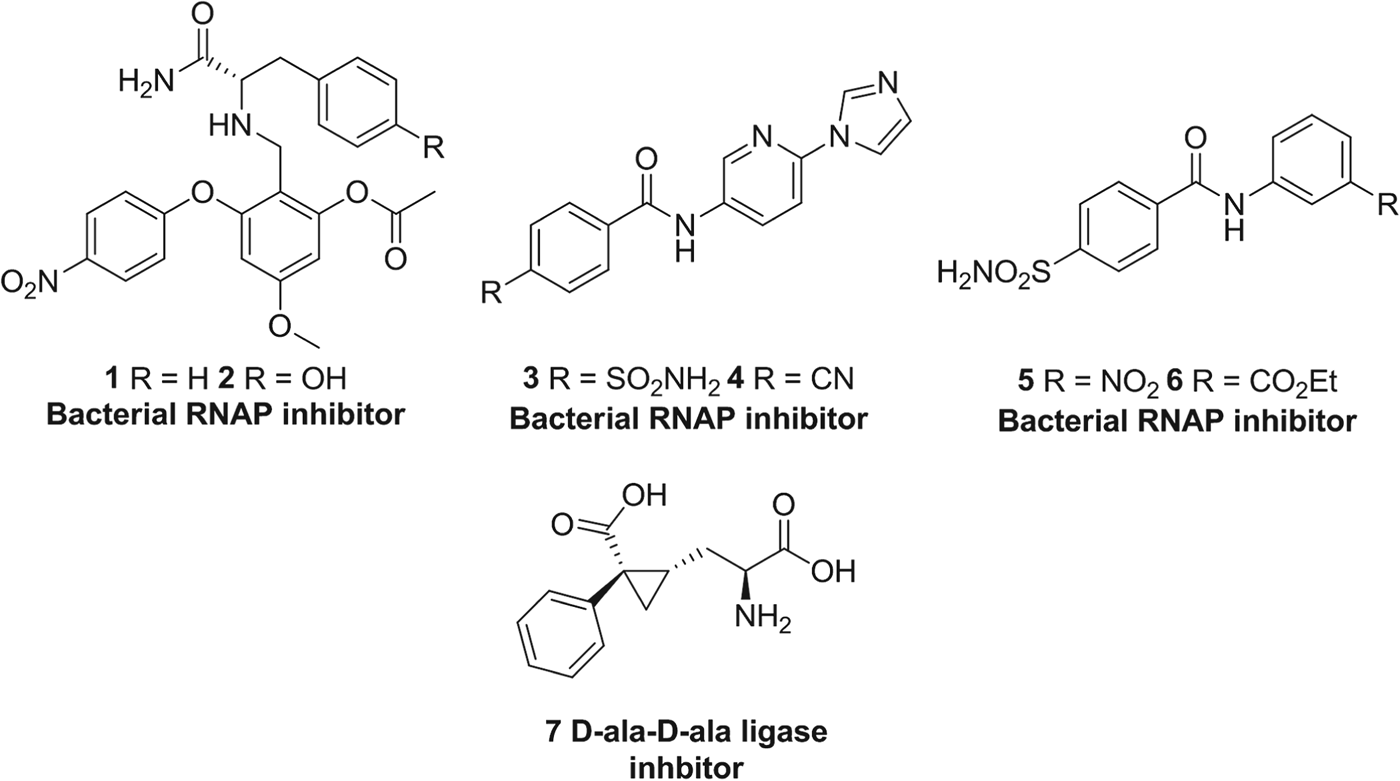
During the golden age of anti-bacterial drug discovery between 1940 and 1962, over 20 distinct antibiotic classes were introduced to the market, but only three classes have been approved in the following 45 years: linezolid in 2000, daptomycin in 2003 and retapamulin in 2007 (Coates et al. Reference Coates, Halls and Hu2011). This decline in productivity is in part due to dereplication of natural products and a failure to deliver new agents using HTS. Novel inhibitor classes are now being sought which have alternative modes of action to existing antibiotic classes. Our interest in combating bacterial resistance is focused on the bacterial targets, D-ala-D-ala ligase and RNA polymerase, respectively and here we summarize our work that has identified several series of new small molecule inhibitors (e.g. Fig. 5, 1–7) of these enzymes.
Fig. 5. Inhibitors of various bacterial enzymes designed using in silico methods by our research group.
RNAP polymerase
DNA-dependent RNA polymerase (RNAP) is a nucleotidyl transferase enzyme, essential for gene expression in all living organisms. It is the central enzyme in the transcription cycle, catalysing the production of RNA from a DNA template (Cramer et al. Reference Cramer, Bushnell and Kornberg2001). Despite its conserved function, RNAP does not share extensive structural homology amongst bacterial, archaeal, eukaryotic and viral RNAPs so enabling the design of selective therapies. This makes RNAP a very attractive drug target for anti-bacterial agents (Chopra, Reference Chopra2007), especially considering that its large size offers many distinct binding sites for small molecular inhibitors. Our research efforts have focused on targeting the rifamycin and myxopyronin B inhibitor binding sites, two natural products with discrete binding cavities within RNAP (Fig. 6).
Fig. 6. (A) Thermos thermophilus RNAP with inserts of the rifamycin binding site (blue) and switch region (green), highlighting the binding modes of rifampicin and myxopyronin B respectively. (B) De novo designed inhibitor 2 targeting the rifamycin binding site, showing contacts to RNAP. (C) Switch region inhibitor 4 showing contacts to RNAP.
The X-ray co-crystal structure of Thermus aquaticus RNAP containing rifampicin (PDB ID: 1I6V) (Campbell et al. Reference Campbell, Korzheva, Mustaev, Murakami, Nair, Goldfarb and Darst2001) was used to evaluate the RNAP-rifamycin binding site, revealing a solvent-exposed cavity, approximately 12 Å away from RNAP's active site. Rifampicin makes several specific hydrogen bonds to RNAP (Fig. 6A) via the hydroxyl groups on the naphthalene ring and ansa bridge. When designing novel inhibitors for this site, we wished to utilize these RNAP residues, as well as additional regions of hydrophobicity located near the residues L391, L413 and F394. Application of the SPROUT software produced a synthetically attractive molecular skeleton based on a 1,2,4,6-tetrasubstituted aromatic core (Fig. 6B) (Agarwal et al. Reference Agarwal, Johnson and Fishwick2008). Several examples were synthesized and assayed for their ability to inhibit Escherichia coli RNAP using a SYBR Green assay (Daubendiek et al. Reference Daubendiek, Ryan and Kool1995). Compounds 1 and 2 (Fig. 5) had IC50 values of 70 μ m and 60 μ m respectively, and represent the first examples of non-macrocyclic inhibitors that may inhibit RNAP at the rifamycin binding site. Further optimization of the scaffold is ongoing, as is an investigation into the mode of action of these compounds.
We have also applied in silico methods to the switch region of bacterial RNAP (Srivastava et al. Reference Srivastava, Talaue, Liu, Degen, Ebright, Sineva, Chakraborty, Druzhinin, Chatterjee, Mukhopadhyay, Ebright, Zozula, Shen, Sengupta, Niedfeldt, Xin, Kaneko, Irschik, Jansen, Donadio, Connell and Ebright2011) in order to design new RNAP inhibitors. The switch region is a highly mobile unit of RNAP controlling the movement of the β’ subunit and allowing double-stranded DNA to enter the active centre. Ebright et al. (Mukhopadhyay et al. Reference Mukhopadhyay, Das, Ismail, Koppstein, Jang, Hudson, Sarafianos, Tuske, Patel, Jansen, Irschik, Arnold and Ebright2008) showed that the structurally related antibiotics myxopyronin B and corallopyronin A, and the macrocyclic antibiotic ripostatin B bind to the switch region and inhibit bacterial RNAP with IC50 values less than 10 μ m. At present, these antibiotics do not represent attractive lead candidates due to their unfavourable physicochemical properties (high molecular weights and logP values) and narrow spectrum of antibacterial activity. After applying the SPROUT software to this cavity, and utilizing the hydrogen bond interactions made by myxopyronin B (Fig. 6B) to the protein, several new scaffolds were produced based on a benzamide core (McPhillie et al. Reference McPhillie, Trowbridge, Mariner, O'Neill, Johnson, Chopra and Fishwick2011). Using the heteroatom substitution tool within SPROUT, a pyridyl-benzamide compound 3 (Fig. 5) was designed and following synthesis and biological screening, showed promising inhibition of E. coli RNAP (33% inhibition at 100 μ m). Iterative rounds of optimization, guided by molecular docking, improved binding affinity for this scaffold, giving compound 4 (Fig. 6C) with an IC50 value of 7·2 μ m.
The fragment-like compounds 5 and 6 (Fig. 5) also showed promising activity against RNAP at the micromolar level of inhibition. A structure-activity relationship study showed that meta-electron withdrawing groups on the N-phenyl ring maximized potency of the compounds (5: IC50 23·3 μ m, 6: IC50 5·6 μ m). Indeed, both this class and the pyridyl-benzamide compounds proved to be selective for bacterial RNAP when assayed for inhibition of S. cerevisiae RNAP and the structurally unrelated enzymes, malate dehydrogenase and chymotrypsin (Seidler et al. Reference Seidler, McGovern, Doman and Shoichet2003). Unfortunately, both series of compounds suffer from a lack of antibacterial activity against both Gram-positive and Gram-negative bacteria which we attributed to poor cell membrane penetration.
D-alaine-D-alanineligase
D-alanine-D-alanine ligase (Ddl) participates in peptidoglycan biosynthesis and catalyses the conversion of D-alanine to the D-ala-D-ala dipeptide, a substrate for the MurF enzyme. Inhibition of Ddl leads to a loss in cell rigidity and integrity, followed by cell death (Green, Reference Green2002). Therefore Ddl, as well as the peptidoglycan synthesis pathway, is an excellent target for novel anti-microbial agents. We used an in silico approach in order to identify new inhibitors of Ddl using SPROUT in conjugation with the E. coli Ddl crystal structure (pdb code: 2DLN) (Fan et al. Reference Fan, Moews, Walsh and Knox1994). A number of key residues involved in binding the phosphonate-based transition-state isostere were chosen as target sites, as well as an additional small hydrophobic pocket not utilized by the phosphonate inhibitor (Besong et al. Reference Besong, Bostock, Stubbings, Chopra, Roper, Lloyd, Fishwick and Johnson2005). A cyclopropyl-based amino acid 7 was selected for chemical synthesis as it satisfied the hydrogen-bond and steric requirements of the cavity (Fig. 7). Although 7 was synthesized as a 1:1 mixture of disastereomers, it displayed a K i value of 12·5±0·1 μ m against Dlb. The K i value of cycloserine, a known inhibitor of Dlb, was 1·4 μ m, and therefore compound 7 is an excellent starting point for further optimization.
Fig. 7. (A) D-ala-D-ala ligase inhibitor 7 as designed using SPROUT in the enzyme active site. (B) Schematic diagram showing the inhibitor-protein interactions of inhibitor 7.
Example 2 – Malaria
Malaria is a global problem. Approximately 50% of the world's population is at risk of infection from the parasites which cause malaria (WHO, 2011), five of which are able to infect humans under normal conditions. The two parasites responsible for the greatest malaria burden are Plasmodium falciparum and Plasmodium vivax (White, Reference White1999; WHO, 2011). Although there are myriads of current drugs used to treat malaria, unfortunately resistance to all current treatments has been observed. The development of resistance to all first-in-line treatments for malaria, coupled with the fact that the anti-malarial pipeline contains a very small number of novel chemical entities with novel modes of action (Olliaro and Wells, Reference Olliaro and Wells2009), highlights an urgent requirement for the development of novel chemotherapeutics to combat this disease, particularly against strains which have previously developed resistance to earlier therapies.
Dihydroorotate dehydrogenase
Dihydroorotate dehydrogenase catalyses the fourth and only redox step of pyrimidine biosynthesis; the oxidation of dihydroorotate to orotate. This is achieved through the reduction of the resident enzyme co-factor FMN coupled with the oxidation of a respiratory quinolone such as co-enzyme Q10 (Christopherson et al. Reference Christopherson, Lyons and Wilson2002). Interestingly, DHODH has been linked with the electron transport chain and it has been shown that its loss leads to mitochondrial dysfunction (Fang et al. Reference Fang, Uchiumi, Yagi, Matsumoto, Amaoto, Takazaki, Yamaza, Nonaka and Kang2012). Although humans also have an homologous DHODH enzyme, it has a low (26%) sequence similarity with PfDHODH, and they are not dependent on this method for acquiring pyrimidine nucleosides. However, inhibition of the human homologue has been shown to have an immunosuppressive effect (Christopherson et al. Reference Christopherson, Lyons and Wilson2002; Haque et al. Reference Haque, Tadesse, Marcinkeviciene, Rogers, Sizemore, Kopcho, Amsler, Ecret, Zhan, Hobbs, Slee, Trainor, Stern, Copeland and Combs2002) and therefore it is important to avoid this undesirable effect in patients with an infection such as malaria.
De novo design of DHODH inhibitors
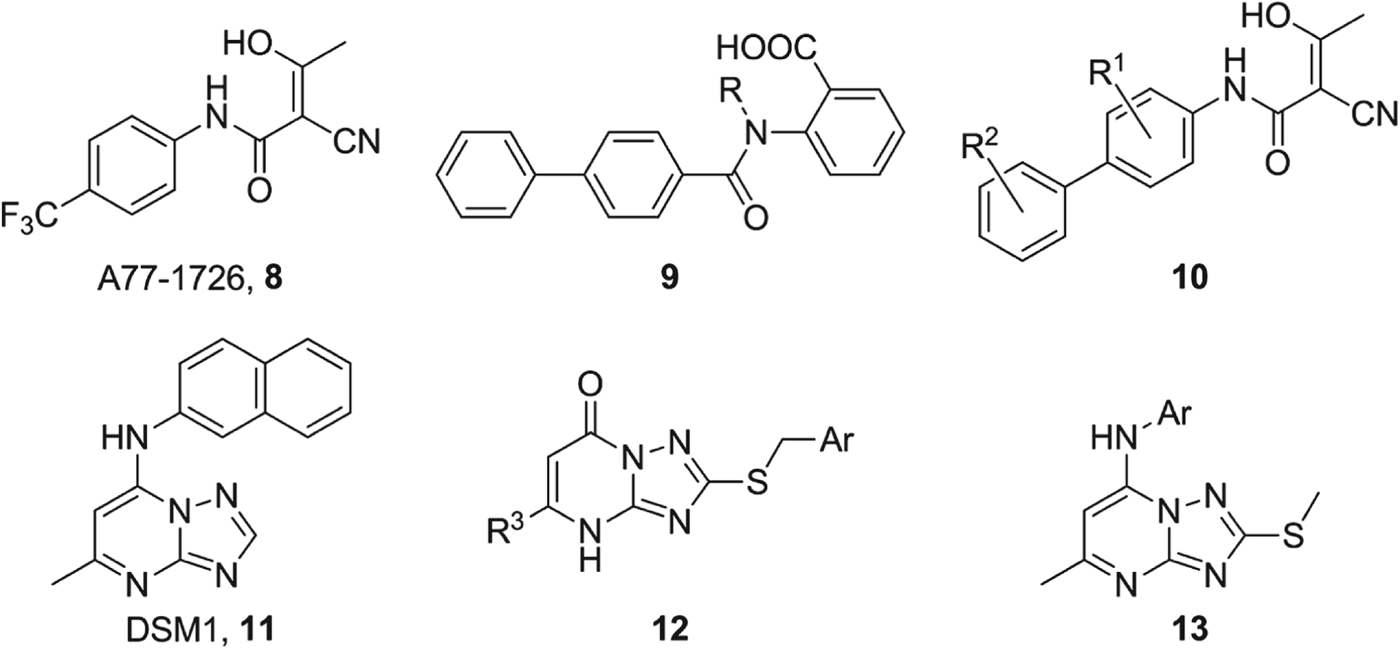
A detailed comparison of the active sites within the human and Plasmodial DHODH enzymes (from co-crystal structures 1D3H and 1TV5 respectively) (Liu et al. Reference Liu, Neidhardt, Grossman, Ocain and Clardy2000; Hurt et al. Reference Hurt, Widom and Clardy2006), containing the same bound inhibitor A77-1726 8 (Fig. 8), revealed that the binding site in the human enzyme was ‘flattened’ compared to the less congested plasmodial binding site by the protrusion of AlaHs59 into the cavity (Fig. 9A) (Heikkila et al. Reference Heikkila, Thirumalairajan, Davies, Parsons, McConkey, Fishwick and Johnson2006).
Fig. 8. Structures of the previously identified inhibitors of PfDHODH A77-1726 8 and DSM1 11, and the novel DHODH inhibitors developed by our group.
Fig. 9. (A) PfDHODH binding site (top) with bound inhibitor A77-1726 8, showing the less congested nature of the PfDHODH binding site. HsDHODH binding site (bottom) with Ala59 protruding into the cavity and closing the hydrophobic binding site. (B) Overlay of the DHODH crystal structures 1TV5 (orange) and 1D3H (blue), showing the key residues in each binding site.
To exploit this structural difference we applied our de novo molecular design program SPROUT (Gillet et al. Reference Gillet, Newell, Mata, Myatt, Sike, Zsoldos and Johnson1994) to the binding site of the co-crystal structure of PfDHODH (1TV5, (Hurt et al. Reference Hurt, Widom and Clardy2006)). In order to design structurally simple inhibitors, only two direct hydrogen bonding interactions were specified (ArgPf265 and HisPf185), resulting in an anthranilic acid scaffold 9 (Fig. 8). A series of inhibitors were synthesized and tested against both Pf and HsDHODH (Heikkila et al. Reference Heikkila, Thirumalairajan, Davies, Parsons, McConkey, Fishwick and Johnson2006) in a colorimetric DCIP assay (Hines et al. Reference Hines, Keys and Johnston1986), the most potent of which, compound 9 (R=Me), was selective for PfDHODH (IC50 value of 42·6±4·6 μ m) over HsDHODH (IC50 value of 200 μ m). It was assumed that this selectivity could be attributed to the non-planar conformation of the molecule induced by the presence of the methyl group on the amide nitrogen (Fig. 10B). This hypothesis was reinforced by the reduced activity of unsubstituted analogue compound 9 (R=H) against PfDHODH (153·5±13·2 μ m) and its preferred binding to HsDHODH (5·0±1·6 μ m).
Fig. 10. In silico dockings for the designed inhibitors in PfDHODH synthesized by our research group. (A) PfDHODH co-crystal structure (1TV5), indicating the location of the substrate binding site (red box). (B) anthranilic acid derivative 9 (R=Me) bound in PfDHODH (1TV5). (C) A77-1726 inspired compound 10 (R 1 & R 2=H) bound in PfDHODH (1TV5). (D) S-benzyl series 12 identified via shape similarity screens based on DMS1 11 bound in PfDHODH, from co-crystal structure 3I68.
Despite significant research we were unable to improve the potency of this compound series further, although the discovery that the selectivity between species could be achieved via the use of conformational staggering between adjacent aryl rings would be applied to later compound series.
Substrate-inspired structure-based drug design
A77-1726 8 (Fig. 8) (Bruneau et al. Reference Bruneau, Yea, Spinella-Jaegle, Fudali, Woodward, Robson, Sautes, Westwood, Kuo, Williamson and Ruuth1998; Heikkila et al. Reference Heikkila, Ramsey, Davies, Galtier, Stead, Johnson, Fishwick, Boa and McConkey2007; Davies et al. Reference Davies, Heikkila, McConkey, Fishwick, Parsons and Johnson2009) is a weak inhibitor of PfDHODH but the co-crystal structure of this compound with PfDHODH offered a good starting point for substrate inspired structure-based drug design of inhibitors (Hurt et al. Reference Hurt, Widom and Clardy2006; Heikkila, Reference Heikkila2007). Further examination of the human and Plasmodium co-crystal structures containing this inhibitor revealed several additional differences to those previously discussed. The hydrogen bonding network for the bound inhibitors is different in both enzymes; in PfDHODH there are three distinct hydrogen bonds to HisPf185, ArgPf265 and TyrPf528 whereas in the HsDHODH complex there are two water-mediated hydrogen bonds to ArgHs136 and GlnHs47 in addition to a direct interaction with TyrHs356 (Fig. 9B).
The SPROUT-HitOpt program (Simmons et al. Reference Simmons, Chopra and Fishwick2010) was utilized to generate variants of the bound inhibitor 8. We opted to retain the hydrogen bonding network and ‘head group’ of 8, whilst optimizing the aromatic ‘tail’ as it was reasoned that the difference in shape and hydrogen bonding networks of the binding site in each enzyme would allow for the development of species-selective inhibitors (Heikkila et al. Reference Heikkila, Ramsey, Davies, Galtier, Stead, Johnson, Fishwick, Boa and McConkey2007). A series of inhibitors utilizing substituted biphenyls as extensions of the hydrophobic ‘tail’ of 8 was synthesized (Fig. 8, 10). As we observed in our previous work that the PfDHODH binding site was better suited to non-planar groups in the hydrophobic pocket of the binding region, substituents were placed in the ortho-positions of the rings to ‘lock’ them into an orthogonal conformation. Unfortunately, these compounds were only weakly active against the PfDHODH enzyme (4·0 μ m, R 1 & R 2=Cl, 10). It is important to note that some of these inhibitors displayed potent inhibition of the HsDHODH with IC50 values as low as 22 nm (R 1 & R 2=Cl, 10).
In silico docking studies of these compounds in the PfDHODH binding site indicated that the residue MetPf536 at the entrance to the binding cavity would clash with the phenyl ring of the inhibitors (Fig. 10B). The human enzyme has the much smaller ProHs364 residue at this position which is small enough not to sterically clash with the inhibitors, consistent with the good observed inhibition of this enzyme (Hurt et al. Reference Hurt, Widom and Clardy2006).
We were subsequently able to obtain several X-ray co-crystal structures of these inhibitors bound within HsDHODH which helped confirm these predictions. Additionally, it was observed that the residue HisHs56 (homologous to HisPf185) was unable to hydrogen bond to these inhibitors, even when they adopt a similar orientation to that observed for A77-1726 8 bound within PfDHODH (Fig. 9B). Instead, HisHs56 is seen to hydrogen bond to TyrHs147. The homologous residue to TyrHs147 in PfDHODH, CysPf276, is unable to form this hydrogen bond resulting in HisPf185 and HisHs56 adopting different conformations in each enzyme respectively and more importantly, removing the possibility of HisHs56 to form hydrogen bonds with a bound inhibitor (Fig. 9B). This observation coupled with previously published mutagenesis studies, where mutation of HisPf185 resulted in the loss of tight binding for some selective inhibitors of PfDHODH, showed that in order for a compound to be selective for PfDHODH over HsDHODH, a hydrogen bond to HisPf185 is required (Heikkila et al. Reference Heikkila, Ramsey, Davies, Galtier, Stead, Johnson, Fishwick, Boa and McConkey2007). Since publishing our findings, a number of other studies have highlighted the importance of HisPf185 for selective binding to PfDHODH (Bedingfield et al. Reference Bedingfield, Cowen, Acklam, Cunningham, Parsons, McConkey, Fishwick and Johnson2012).
Shape similarity screening
In 2008 Phillips et al. reported the structure of a series of potent and selective triazolopyrimidine inhibitors (11, Fig. 8) of PfDHODH discovered using HTS (Phillips et al. Reference Phillips, Gujjar, Malmquist, White, El Mazouni, Baldwin and Rathod2008). Based upon this work, we identified compound 12 (Fig. 8, 12, R 3=Me, Ar=4-chlorophenyl) as a moderately selective inhibitor of PfDHODH, with an IC50 value of 1·0 μ m (IC50 value of 39 μ m against HsDHODH) from a shape similarity search, using ROCS with compound 11 as the template and using the Maybridge chemical screening library.
Several analogues were subsequently synthesized and tested against both Hs and PfDHODH. However, no improvement in the inhibition of PfDHODH was found compared with 11. Interestingly, S-benzyl triazolopyrimidine compound 12 (R 3=Me, Ar=2,5-dichlorophenyl) was a potent inhibitor of HsDHODH (51 nm) and was subsequently crystallized with HsDHODH revealing a small hydrophobic pocket adjacent to the pyrimidine methyl group. The extension of the methyl group to an ethyl group maximized these hydrophobic interactions and improved the IC50 value to 13 nm (when R 3=Et, Ar=2,5-dichlorophenyl). Both methyl and ethyl S-benzyl triazolopyrimidine derviatives were also crystallized in HsDHODH revealing a previously unseen conformational change in the two α-helices enclosing the respiratory quinone binding site (Fig. 11) (Bedingfield et al. Reference Bedingfield, Cowen, Acklam, Cunningham, Parsons, McConkey, Fishwick and Johnson2012). In silico studies of the predicted binding of these compounds within PfDHODH suggested that they adopt a similar binding pose to that observed in HsDHODH (Fig. 10). Mutagenesis studies on both PfDHODH and HsDHODH once again confirmed the observation that HisPf185 is a key residue for the binding of selective inhibitors in the PfDHODH binding site (Bedingfield et al. Reference Bedingfield, Cowen, Acklam, Cunningham, Parsons, McConkey, Fishwick and Johnson2012).
Fig. 11. Overlay of all previously published HsDHODH co-crystal structures (grey), and the two newly obtain HsDHODH co-crystal structure (blue and yellow), highlighting the structural shift in the α−1 helices of this enzyme.
Most recently, in silico modelling based upon the S-benzyl series of inhibitors (Fig. 10D), has resulted in the production of a subsequent series of compounds which show excellent inhibitory activity of PfDHODH, and potent anti-plasmodial activity. Full details of this new series will be published elsewhere.
CONCLUSIONS
SBDD utilizes knowledge from three-dimensional protein structures and applies in silico techniques to identify putative small molecules with biological activity against desired protein targets. We have applied these methods to enzymes such as dihydroorotate dehydrogenase, bacterial RNAP and bacterial D-ala-D-ala ligase, and successfully developed compounds which not only inhibit these enzymes but, in the case of dihydroorotate dehydrogenase, are potent against the infecting pathogen. The use of in silico modelling has helped us develop species-selective inhibitors by harnessing subtle differences within the binding site to infer selectivity. This is one of the many advantages of SBDD, that selectivity can be built into a series of molecules from an early stage helping to minimize side effect and undesirable effects in vivo.
Our structure-based approach emphasizes the importance of structural molecular genomics and validates its use within the field of anti-infective drug discovery. It offers rapid progression from target to lead compound at a reduced cost compared to the standard approaches to drug discovery which focus on HTS. Exploiting these tools is essential for anti-infective drug discovery to combat the rise in resistance to front-line therapeutics. Advances in both the development of computational hardware and in the more advanced SBDD algorithms will continue to increase the attractiveness of using this technique to aid drug discovery of anti-infectives. However, the major challenge here is the design of drug-like compounds capable of entering (and remaining within) the cell of the pathogenic organism.
SOFTWARE ACCESSIBILITY
SPROUT, SPROUT-HitOpt and eHiTS software are maintained by SimBioSys Inc. and further information can be obtained via the Symbiosys website; http://www.simbiosys.ca. Autodock is freeware for academics and can be accessed at http://autodock.scripps.edu. ROCS is maintained by OpenEye and is available from http://www.eyesopen.com/rocs.