INTRODUCTION
Cysteine proteases are one of the main catalytic groups of peptide hydrolases, together with serine, threonine, aspartate and metallo-proteases (McKerrow, 1989; Coombs & Mottram, 1997; Tort et al. 1999). Also referred to as thiol or sulfhydryl proteases (Barrett, 1994), cysteine proteases have been identified in plants (Glazer & Smith, 1971; Kumar Dubey & Jagannadham, 2003), animals (Barrett & McDonald, 1980; Bania et al. 2003), viruses (Bazan & Fletterick, 1988; Ziebuhr et al. 2003), bacteria (Morihara, 1974; Svensson et al. 2000) and eukaryotic microorganisms (North, 1982; Nesterenko et al. 1995). Cysteine proteases of parasitic organisms are divided into two main groups, referred to as clans CA and CD according to sequence similarity, possession of inserted peptide loops and biochemical specificity to small peptide substrates (Rawlings & Barrett, 1993; Barrett, 1994). The majority of parasite cysteine proteases belong to the family C1 within clan CA, and are further divided into cathepsin B and cathepsin L-like subfamilies.
Most of the human cathepsins have an acidic pH optimum which allows full activity within the lysosomal compartment (Barrett & Kirschke, 1981). In contrast, many parasitic cysteine proteases are more active at neutral or slightly alkaline pH (Eakin et al. 1992; Caffrey et al. 2001; Sajid & McKerrow, 2002). Neutral or alkaline pH optima are in accordance with the extracellular activity observed for these proteases. Roles of parasitic cysteine proteases in nutrition, tissue and cell invasion, ex/encystement, hatching and immunoevasion have been recently discussed in detail (Sajid & McKerrow, 2002).
Because of the ubiquity of cysteine proteases in both protozoan and helminth parasites, they represent attractive targets for anti-parasitic drug development. Most of this work has focused to date on the papain family of proteases (cathepsin L and B-like proteases) (Li et al. 1994; Du et al. 2002; Rosenthal et al. 2002). In addition, it has been extensively demonstrated that many cysteine proteases are immunogenic and this has been exploited in their use as convenient immunological diagnostic markers for infectious diseases, including infections of Ancylostoma caninum (see Loukas et al. 2000), Fasciola hepatica (see Neyra, Chavarry & Espinoza, 2002), Fasciola gigantica (see Dixit, Yadav & Sharma, 2002) and Clonorchis sinensis (see Na et al. 2002). Antibodies directed against cysteine proteases can have an inhibitory effect on their proteolytic activity. A number of encouraging studies to verify the application of an anti-cysteine protease vaccine against parasitic organisms has been carried out in Trypanosoma congolense (see Authie et al. 2001), F. hepatica (see Dalton et al. 1996), Ostertagia ostertagi (see Geldhof et al. 2002) and H. contortus (see Skuce et al. 1999). These observations may explain the increasing attention addressed to cysteine proteases of parasites.
One of the best characterized families of cathepsin B-like proteases have been described in the abomasal nematode H. contortus (see Pratt et al. 1990, 1992; Rehman & Jasmer, 1998; Skuce et al. 1999; Jasmer, Roth & Myler, 2001). Inter- and intrageographical variation of cysteine proteases has been demonstrated among different strains of H. contortus in the protease profile using gelatin-containing SDS–PAGE gels (Karanu et al. 1993, 1997) and at the genetic level (Rehman & Jasmer, 1998; Skuce et al. 1999). However, to date, the magnitude of this variability has not been assessed within and among different strains of H. contortus from different species of host for a same set of genes under similar laboratory conditions. Using PCR coupled Single-Strand Conformation Polymorphism (SSCP) and sequencing methodologies, we have estimated the genetic polymorphism in five previously reported cysteine protease genes of H. contortus from sheep (North America) and goats (Spain).
MATERIALS AND METHODS
Parasites
Individual worms from 2 strains of H. contortus were used in this study. One strain (NA) corresponded to H. contortus from North America and was maintained in experimentally infected sheep at the Institute of Parasitology, McGill University. The second strain (SP) of H. contortus was originally isolated from naturally infected goats from the Canary Islands (Spain) and maintained experimentally in goats at the Faculty of Veterinary of the University of Las Palmas de Gran Canaria.
DNA isolation
H. contortus adult males from both the NA and the SP strains were taken from the abomasum of the corresponding single host (sheep or goats) experimentally infected with 3rd-stage larvae (L3) of the parasite. Only adult males were used to avoid the possibility of DNA contamination from the eggs or sperm present in females resulting in more than a single genotype in each sample. The worms were washed in RPMI medium (Sigma–Aldrich) at 37 °C and then frozen at −80 °C until DNA isolation was performed. The DNA was isolated from 55 individual males of the SP strain and 97 males of the NA strain. Each worm was transferred to a tube containing 200 μl of STE (0·1 M NaCl, 10 mM Tris–HCl, pH 8·0, 1 mM EDTA, pH 8·0), 0·6 M β-mercaptoethanol, 0·5% SDS and 200 μg/ml proteinase K (Sambrook, Fritsch & Maniatis, 1989) and incubated overnight at 55 °C. Two DNA extractions, one with phenol and another with phenol/chloroform, were performed. The DNA was then precipitated with 2·5 M ammonium acetate and 50% ethanol, following the addition of 10 μg of linear acrylamide as co-precipitant (Gaillard & Strauss, 1990). The DNA pellet was air-dried and redissolved in 50 μl of 1×TE (10 mM Tris–HCl, 1 mM EDTA, pH 8·0).
Polymerase chain reaction (PCR) amplification
PCR reactions were performed in a PTC-200 Peltier Thermal Cycler (MJ Research Inc.) using reagents and Taq polymerase provided by Gibco BRL. Forward and reverse primers (Table 1) were based on reported sequences from H. contortus encoding cysteine protease genes AC-1 (Pratt et al. 1990), AC-3, AC-4 and AC-5 (Pratt et al. 1992) and GCP-7 (Rehman & Jasmer, 1998), and the annealing temperatures for the PCR amplifications were: 59 °C for the set of primers AC-5, 54 °C for the primers AC-1 and AC-3, and 53 °C for the primers AC-4 and GCP-7. The other conditions for the amplifications were the same for all pairs of primers: 94 °C for 2 min to denature the template DNA, followed by 35 cycles of 15 sec at 95 °C, 30 sec at the corresponding annealing temperature and 1 min at 70 °C. A final step of 2 min at 15 °C was included. For the PCR reactions ~2 ng (1–2 μl) of DNA from individual male worms were used. In addition, the reaction mixture for all pairs of primers contained 0·5 U Taq polymerase, 0·8 μM corresponding forward and reverse primers, 0·2 mM dNTPs, 1 mM MgCl2, 10% (v/v) 10× buffer reaction and H2O to a total volume of 25 μl. Finally, the PCR products were subjected to electrophoresis in 1·2% agarose gels and subsequent staining with ethidium bromide.
Single Strand Conformation Polymorphism (SSCP)
One μl of PCR product was mixed with 15 μl of SSCP loading buffer (95% formamide, 10 mM NaOH, 0·25% xylene cyanol, 0·25% bromophenol blue). The mixture was denatured at 95 °C for 2 min and cooled immediately on iced water before being loaded onto a non-denaturing polyacrylamide gel. Electrophoresis was performed in a Hoefer SE600 (Pharmacia Biotech, San Francisco, CA). Electrophoresis conditions were optimized for each gene (AC-1, AC-3, AC-4, AC-5 and GCP-7) and strain (SP and NA), in order to produce unique migration and separation patterns for the single strands of the different alleles. The parameters optimized in the electrophoresis were the running time (ranging from 12 to 23 h), the power (from 75 to 110 volts) and the percentage of acrylamide/bis-acrylamide (from 10 to 15%). The gels were made using different percentages of a 49[ratio ]1 proportion of acrylamide[ratio ]bis-acrylamide, buffer TBE (1·11 M Tris, 1·11 M boric acid, 0·003 M EDTA, pH 8·0), 0·09% (v/v) N,N,N,N′-tetra-methyl-ethylenediamine and 0·07% (w/v) ammonium persulphate (the solutions are expressed as final concentrations). Gels were run in 1×TBE buffer (0·89 M Tris, 0·89 M boric acid, 0·002 M EDTA, pH 8·0) at room temperature (22–24 °C). The gels were stained with ethidium bromide, scanned using a Bio-Rad Molecular Imager® FX, and the patterns recorded with the corresponding Quantity One Software (Version 4.2.1) for subsequent analysis.
Sequence analysis
DNA fragments that displayed different electrophoretic patterns in the SSCP analyses were selected for sequencing. PCR products were purified using the Nucleospin extraction kit (Clontech) and ligated into the plasmid vector pCR®2.1 (Gibco BRL). Transformation into One Shot® competent Escherichia coli cells was then carried out according to the manufacturer's instructions (TA Cloning® Kit, Gibco BRL). The plasmid DNA was isolated using a Qiaprep® Spin Miniprep Kit (Qiagen) and sequencing was carried out by the ABI Big Dye cycle sequencing kit and an ABI Prism 377 automated sequencer. Before sequencing, the Miniprep products were subjected to PCR–SSCP analysis to confirm the electrophoretic patterns of inserts from recombinant clones. This analysis proved that, for every individual male used in the sequencing, the SSCP profile obtained in the first screening of the whole population (North American or Spanish populations) was identical to that observed after cloning. According to the reproducibility of the SSCP results in all the assays it is quite unlikely that the observed diversity causing the SSCP patterns was a consequence of base misincorporation during PCR.
Data analysis
Genotype frequencies for each gene were tested for Hardy–Weinberg equilibrium for an excess of homozygotes, calculating from a binomial distribution based on the observed allele frequencies (Sokal & Rohlf, 1981). Differences in allele frequencies between strains were tested for significance using a G test for heterogeneity, pooling allele classes if necessary to ensure a minimum expected number of at least 5 individuals (Sokal & Rohlf, 1981). Significance was taken at the 5% level. To further analyse the allelic frequencies, a set of intra- and inter-population genetic statistics were estimated, and corrected for small sample size (Nei, 1978) and small number of populations (Nei, 1986) using the GeneStat-pc 3.3 computer program (Lewis, 1994). Some of the statistics included the percentage of polymorphic loci (P) (95% criterion), the mean number of alleles per locus (A), expected heterozygosity (He), total genetic diversity (Ht), genetic diversity within populations (Hs), genetic diversity among populations (Dst) and the relative magnitude of genetic differentiation among populations (Gst=Dst/Ht) (Nei, 1978).
In order to analyse the nucleotide variability and the phylogenic relations among the different genes in both the SP and the NA strains of H. contortus, the sequences obtained were initially aligned using CLUSTAL W (1.81) (Higgins et al. 1994) and then treated with the computer program MEGA version 2.1 (Kumar et al. 2001). Jukes-Cantor's genetic distances (Jukes & Cantor, 1969) for each locus were calculated for all pairwise combinations. The mean distance within a subpopulation, the mean inter-populational distance and the mean diversity for the entire population were also determined for all loci using the same distance method. A dendrogram was constructed based on the matrix of the distances using the Neighbour-Joining Tree method (NJ) (Saitou & Nei, 1987). In all cases the standard error was estimated by a bootstrap procedure with a total of 500 replications. Further estimations of the variability within the two strains of H. contortus, including the number of polymorphic sites (S), the nucleotide diversity (π) and the heterozygosity per nucleotide site (θ) were performed using the computer program DnaSP version 3.51 (Rozas & Rozas, 1999).
RESULTS
SSCP analysis
The amplification of the loci AC-1, AC-3, AC-4 and AC-5 resulted in PCR products of ~350 bp. A PCR product of ~625 bp was identified when the locus GCP-7 was amplified. While no variation in size was detectable among the PCR products from individual males on agarose gels, SSCP results revealed distinct profiles among some of the samples for all the genes analysed. Examples of SSCP gels are shown in Figs 1 and 2. The frequencies of the different SSCP patterns determined for each locus are displayed in Table 2. Based on the SSCP results, a total of 20 different alleles were identified for the 5 loci assessed. Subsequent sequencing of PCR products samples for each allele confirmed that the differences in banding patterns (homozygosity and heterozygosity) detected in the SSCP results were due to nucleotide variation. The allelic frequencies for each locus are shown in Table 3. These frequencies did not differ significantly from the Hardy–Weinberg equilibrium for any of the loci assessed for both the SP and the NA strain.
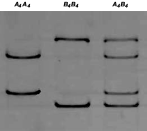
Fig. 1. SSCP profiles for the gene encoding cysteine proteases AC-4 in two strains of Haemonchus contortus (Spanish and North American strains). A4 and B4 represent the alleles detected in the two strains of the parasite. The homozygotes A4A4 and B4B4 and the corresponding heterozygote A4B4 were present in both strains.
Fig. 2. SSCP profiles for the gene encoding cysteine proteases GCP-7 in two strains of Haemonchus contortus. The profiles for the Spanish and the North American strains are represented separately in (A) and (B), respectively. A7, B7, C7 and D7 represent the different alleles identified in the two strains of the parasite. In the Spanish strain, 3 homozygotes (A7A7, B7B7 and C7C7) and the corresponding heterozygotes (A7B7, A7C7 and B7C7) were identified. In the NA strain the homozygotes A7A7 and D7D7 and the heterozygotes A7B7, A7D7 and B7D7 were detected (B).
Table 2. Frequencies of SSCP profiles in 5 genes encoding cysteine proteases of Haemonchus contortus from Spain (SP) and North America (NA)
(Differences in the SSCP profiles between the strains were tested for significance using a G test, pooling SSCP migration patterns when necessary to ensure a minimum of at least five individuals. Significant differences between populations were observed at the 0·1% level (P<0·001*) in genes AC-1, AC-3 and GCP-7, while any statistical difference was detected for genes AC-4 and AC-5. The most abundant SSCP profiles were A1A1, A3B3, B4B4, A5A5 and A7A7–A7C7 (Spanish strain) and A1B1, B3D3, B4B4, A5A5 and A7A7 (North American strain), for genes AC-1, AC-3, AC-4, AC-5 and GCP-7, respectively.)
Table 3. Allelic frequencies estimated by SSCP in genes encoding cysteine proteases of H. contortus from Spain (SP) and North America (NA)
(Differences in the allele frequencies between the strains were tested for significance using a G test, pooling allele classes when necessary to ensure a minimum of at least five individuals. Significant differences between populations were observed at the 0·1% level (P<0·001*) in genes AC-1, AC-3 and GCP-7, while any statistical difference was detected for genes AC-4 and AC-5. The most abundant alleles were A1, B3, B4, A5 and A7 (Spanish strain) and A1, D3, B4, A5 and A7 (North American strain) for genes AC-1, AC-3, AC-4, AC-5 and GCP-7, respectively.)
Five different alleles were detected within the Spanish (SP) and the North American strains (NA) for the locus AC-1 (A1, B1, C1, D1 and E1). The homozygotes A1A1 and B1B1, the corresponding heterozygote (A1B1) and 3 heterozygotes for allele A1 (A1C1, A1D1 and A1E1) were detected for all males analysed. The allelic frequencies ranged from 0·0070 (NA strain E1) to 0·8560 (SP strain A1) and were statistically different between the two populations (Table 3). Except for the allele E1, which was only detected in the NA strain, the other alleles were present in both strains. The most frequent SSCP patterns were A1A1 and A1B1 for the SP and NA strains, respectively (Table 2).
A total of 8 different alleles were detected within the two populations for the locus AC-3 (A3, B3, C3, D3, E3, F3, G3 and H3). In the SP strain, the homozygotes A3A3, B3B3, C3C3 and D3D3, the corresponding heterozygotes (A3B3, A3C3, B3C3 and B3D3) and other heterozygotes for the allele A3 (A3F3 and A3G3) and for the allele B3 (B3H3) were identified. In the NA strain, the homozygotes D3D3, B3B3 and E3E3, the corresponding heterozygotes (D3B3, D3E3 and B3E3) and other heterozygotes for the allele D3 (A3D3) or B3 (A3B3) were also found. The allelic frequencies ranged from 0·0110 (SP strain H3) to 0·4720 (NA strain D3) (Table 3) and were statistically different between the two strains. Alleles C3, F3, G3 and H3 were exclusive to the SP strain. The most frequent SSCP patterns were A3B3 and B3D3 for the SP and NA strains, respectively (Table 2).
The SSCP results for the locus AC-4 are shown in Fig. 1. Only 2 alleles, A4 and B4, were detected. The homozygotes A4A4 and B4B4 and the corresponding heterozygote A4B4 were present in both strains. The allelic frequencies, which were higher in allele B4, were not statistically different between the two strains (Table 3). The most frequent SSCP pattern in both strains was the heterozygote A4B4 (Table 2).
Only one banding pattern was detected in the SSCP analysis of the locus AC-5 for both strains, the unique homozygote confirmed being identified as A5.
Four different alleles (A7, B7, C7 and D7) were detected in the SSCP analysis of locus GCP-7 (Fig. 2). Alleles C7 and D7 were exclusive to the SP and NA strains, respectively. Three homozygotes (A7A7, B7B7 and C7C7) and the corresponding heterozygotes (A7B7, A7C7 and B7C7) were identified in the SP strain (Fig. 2A). In the NA strain, the homozygotes A7A7 and D7D7 and the heterozygotes A7B7, A7D7 and B7D7 were found (Fig. 2B). The allelic frequencies ranged from 0·0480 (NA strain B7) to 0·6880 (NA strain A7) and were statistically different between the two strains (Table 3). The most frequent SSCP patterns were A7A7 and A7C7 for the SP strain, and A7A7 for the NA strain (Table 2).
Averaged across populations, the mean number of alleles was A=3·2, percentage of polymorphic loci P=80, and expected heterozygosity was He=0·39. No significant differences were found among populations for any of the polymorphic indices. Partitioning of the populations' genetic diversity showed that genetic diversity within populations, Hs=0·459, accounted for 85% of the total genetic diversity. Genetic diversity among populations, Ds=0·071, accounted for 15% of the total genetic diversity. This result was reflected in a Gst of 0·154 (Nei, 1978), which measures the proportion of the genetic diversity attributable to population differentiation. Based on Nei's (1978) genetic estimates a high mean identity of 0·885 was detected between the two H. contortus strains.
Nucleotide diversity and genetic relationships
Pairwise genetic distances (and S.E.) between alleles ranged from 0·0030 (0·0028) between alleles B3 and A3 to 0·1300 (0·0154) between alleles B7 and D7 (original data are available from authors). Other estimates of the nucleotide variability within and between strains are depicted in Table 4. Except for the locus AC-4, the number of polymorphic sites (S), the nucleotide diversity (π), the heterozygosity per nucleotide site (θ) and the mean distances within groups were different for each strain. The number of polymorphic sites fluctuated from 11 to 88, π from 0·0110 (0·0019) to 0·0555 (0·0026), θ from 0·0069 (0·0027) to 0·0291 (0·0082), and mean distances varied within groups from 0·0112 (0·0033) to 0·0595 (0·0072). Usually, the lowest and the highest values for all of these parameters corresponded to loci AC-4 and GCP-7, respectively. Similarly, the values of the mean distances between groups and the mean diversity for the entire population were highest in the GCP-7 locus, followed by the AC-3 and the AC-1 loci, and the lowest values corresponded to locus AC-4.
Table 4. Estimates of the nucleotide variability within and between strains (SP and NA strains) in 5 genes encoding Haemonchus contortus cysteine proteases
(Number of polymorphic (segregating) sites (S), nucleotide diversity (π) and heterozygosity per nucleotide site (θ). Standard errors are given in parentheses. SP: Spanish strain. NA: North American strain.)
Nucleotide sequences for the 20 alleles have been submitted to GenBank and their accession numbers are given in Table 1. Standard nucleotide–nucleotide BLAST analysis (NBC GenBank) was performed for all the alleles of each locus. In all cases, a high degree of identity was found between the genomic DNA sequences of the different alleles and the cDNA published sequences: AC-1 (M31112), AC-3 (M80388), AC-4 (M80386), AC-5 (M80385) and GCP-7 (AF046229). According to the alignments, 3 exons and 2 introns were present in each of the 5 loci analysed in this study. A general representation of the structure of the different alleles, indicating the exact point of coincidence with the cDNA reported sequences, is detailed in Table 1. Using BLASTX, the amino acid sequences of the different alleles were also inferred. Nucleotide variability was translated into amino acid changes in some alleles with differences ranging from 1·35 (E1vs. B1, C1 and D1) to 14·93% (B7 and C7vs. A7 and D7).
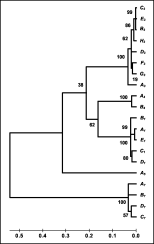
Genetic relationships among the different loci were examined further by a bootstrap test of phylogeny based on the Neighbour-Joining Tree construction method and are represented by a dendrogram (Fig. 3). The dendrogram separates the 5 loci into 2 primary clusters, one made up of the alleles from locus GCP-7 and the other formed by the alleles from loci AC 1-5. In the second cluster, the locus AC-5 is separated from the other 3 loci, two of them (loci AC-1 and AC-4) being closely associated and more distant from locus AC-3. These results are in accordance with previous data differentiating 2 distinct clades for cathepsin B-like cysteine proteases (Pratt et al. 1992; Rehman & Jasmer, 1998), Clade I containing the CBL AC-1 to AC-5 and Clade II containing GCP-7.
Fig. 3. Dendrogram of cysteine protease sequences of Haemonchus contortus from Spain and North America. The tree was obtained by 500 replicates using Jukes Cantor and Neighbour-Joining methods in MEGA 2.1 version. The numbers besides the branches are percentage support from the 500 bootstrap resamples. The scale represents the Jukes Cantor distances among the alleles of the different loci analysed.
DISCUSSION
The present study demonstrates genetic variation in cysteine protease genes between Spanish and North American strains of H. contortus by SSCP analysis and subsequent sequencing. Although genetic variation has already been reported in cathepsin B-like cysteine proteases (CBL) of H. contortus (see Cox et al. 1990; Pratt et al. 1990, 1992; Rehman & Jasmer, 1998; Skuce et al. 1999; Jasmer et al. 2001), the magnitude of this genetic diversity had not been measured previously within and among different strains of H. contortus for a same set of genes. Allelic variability for other H. contortus genes has also been investigated, which make this parasite an extremely diverse nematode at the genetic level. Nucleotide diversity has been demonstrated in the genes encoding 2 β-tubulins of the parasite (Kwa et al. 1993; Beech, Prichard & Scott, 1994), several P-glycoproteins (Pgps) (Blackhall et al. 1998a; Sangster et al. 1999), 2 glutamate-gated chloride channel subunits (GluCla and GluCl β subunits), an N-acetylcholine receptor and a phosphoenolpyruvate carboxykinase (Blackhall et al. 1998b). Genetic diversity has been proved as well in tandem-repeat-type galectins (Greenhalgh, Beckham & Newton, 1999), the transposable element Tc1 and transposon integration (Hoekstra et al. 1999, 2000) and in microsatellite analyses (Hoekstra et al. 1997; Otsen et al. 2000).
SSCP analysis and direct sequencing have been used in the characterization of DNA polymorphism in the 432-bp core region of the cruzipain gene, which encodes the active site of cathepsin L-like cysteine protease (De Leon et al. 1998). However, the usefulness of SSCP has not been previously demonstrated for detecting polymorphism in cathepsin B-like cysteine protease genes. SSCP banding patterns obtained for each of the 5 loci assessed in this study were all readily characterized once optimal conditions for the method were determined. Alleles which differed by 2 bases (A1vs. E1) could be distinguished. The variation in SSCP patterns among the 20 alleles reflected the sequence variability estimated by Jukes-Cantor pairwise distances. This evidence, together with the reproducibility of the SSCP results, indicates that PCR-linked SSCP provides a reliable method for displaying sequence variation in cysteine protease genes of H. contortus.
According to the polymorphic indices (A, P and He) and the genetic variability statistics (Ht, Hs, Dst and Gst) based on the SSCP results, cysteine protease genes from H. contortus showed a considerable degree of polymorphism. Except for locus AC-5, all the loci were polymorphic, with a total of 20 alleles and a number of alleles per locus ranging from 2 to 8. Locus AC-3 was the most polymorphic, followed by locus GCP-7, then locus AC-1 and finally locus AC-4. The degree of allelic variation detected by SSCP agree with estimates of the nucleotide variability for all loci, except for locus GCP-7 in which the number of polymorphic sites (S), the nucleotide diversity (π) and the heterozygosity per nucleotide site (θ) had the highest values in both the Spanish (SP) and the North American strains. The discordance between the SSCP results and sequencing analyses for locus GCP-7 suggests that the number of regions of base pairing, rather than the primary structure of the molecule, is the main factor that determines the spatial conformation of the single-stranded DNA, since point mutations can be detectable by SSCP for fragments of >600 bp (Kukita et al. 1997).
The genetic diversity within populations (Hs) found in this study accounted for 85% of the total genetic diversity, while the genetic diversity between populations (Ds) accounted for 15% of the total genetic diversity. This is in accordance with Nei's (1978) Gst=0·154 which measures the proportion of genetic diversity attributable to population differentiation. In agreement with these data, studies in 4 species of trichostrongylid nematodes, including H. contortus, indicated that 96–99% of nucleotide diversity is found within populations (Blouin et al. 1992).
A number of interesting reports have demonstrated the human influence on the ecology, geographical distribution and genetic diversity among different organisms, including plants (Asimina tribola) (Huang, Layne & Kubisiak, 2000), animals (Ehhydra lutris) (Larson et al. 2002) and parasites including protozoa such as Plasmodium falciparum and diverse nematodes (Blouin et al. 1992, 1995; Read & Taylor, 2001; Wootton et al. 2002). International commerce in livestock animals between Europe and North America could have led to gene flow among livestock species and then among the parasites they harbour. Nevertheless, important differences were found to occur between the SP and NA strains of H. contortus at some of the loci. Allelic frequencies based on SSCP results were significantly different between the two populations for each of the loci AC-1, AC-3 and GCP-7 (G test), with some alleles being detected exclusively in the SP strain (F3, G3, H3 and C7) or the NA strain (E1 and D7). Accordingly, the estimation of the nucleotide variability between strains indicated a mean distance between groups ranging from 0·0121 (0·0036) (locus AC-4) to 0·0638 (0·0068) (locus GCP-7). Although inter-geographical variability may be a consequence of a moderate geographical isolation, the influence of host species (goats and sheep for the SP and the NA strains, respectively) on genetic divergence should also be taken into account, as discussed by Zhu et al. (2000) who studied the mitochondrial DNA polymorphism within and among species of Capillaria sensu lato from Australian marsupials and rodents. While these authors did not find significant variation in SSCP profiles within morphospecies within a particular host species, significant variation occurred between morphospecies originating from different host species.
Even though many investigations indicate that genetic variation in parasitic nematodes is an issue of considerable practical and theoretical significance, either from a morphological (Mendoza-Leon, Luis & Martinez, 2001), biological (Watkins & Fernando, 1984), therapeutic (Hejmadi et al. 2000) and immunological (Goyal & Wakelin, 1993) point of view, the extent to which genetically determined variation affects the host–parasite interaction is still poorly understood (Wakelin, Farias & Bradley, 2002). Much more work is needed to elucidate the functional implications of the genetic variability in cysteine protease genes from H. contortus measured in this and previous studies.
We cordially thank Dr Juan Manuel Afonso (University of Las Palmas de Gran Canaria, Spain) for critical reading and invaluable help with the manuscript. We also thank all the staff of the Dr Prichard's Lab and Dr Beech's Lab at the Parasitology Institute (McGill University, Canada). This study has been supported by the Canary Government, by the Spanish Ministry of Science and Technology, and by the FEDER Funds (Project AGL2002-03528).