


INTRODUCTION
Anthelmintic resistance is already a major problem for the control livestock parasites and an emerging concern for human parasites due to the increasing coverage and intensity of community-wide treatment regimes in the developing world (Kaplan, Reference Kaplan2004; Geary, Reference Geary2012). One of the most important research priorities for both human and veterinary parasitology is to identify the mutations in parasite genomes that give rise to anthelmintic resistance. This will help us understand the mechanisms involved, both at the cellular and population level, and develop much needed molecular diagnostic tests. Such tests have many important applications including routine diagnosis and surveillance, assessing the success and sustainability of control programmes and investigating the origin and spread of resistance. An understanding of the genetic basis of resistance may also lead to the development of drug synergists or novel resistance-breaking compounds.
In spite of a considerable amount of research over the last few decades, our understanding of the molecular mechanisms underlying anthelmintic resistance remains far from complete (Gilleard, Reference Gilleard2006). This is largely due to parasitic nematodes being difficult experimental subjects and a lack of tools for functional and genetic analysis. This has been compounded by the fact that genomic resources for parasitic nematodes have lagged behind most other pathogen groups due to a number of factors including limited financial investment, relatively large genome sizes and high levels of sequence polymorphism (Gilleard, Reference Gilleard2006). These factors have resulted in research being dominated by descriptive studies on Single Nucleotide Polymorphisms (SNPs) in a small number of candidate genes. Two approaches have generally been adopted. The first has been to investigate differences in the frequencies of candidate SNPs between resistant and susceptible parasite populations taken from the field (Prichard, Reference Prichard2001; Eng and Prichard, Reference Eng and Prichard2005; Gilleard and Beech, Reference Gilleard and Beech2007). The second has been to identify a loss of polymorphism in candidate gene sequences following selection for resistance during experimental infections (Kwa et al. Reference Kwa, Kooyman, Boersema and Roos1993; Blackhall et al. Reference Blackhall, Liu, Xu, Prichard and Beech1998; Prichard, Reference Prichard2001; Beech et al. Reference Beech, Skuce, Bartley, Martin, Prichard and Gilleard2011). These studies, often examine a small number of loci, sometimes only one, and generally only result in circumstantial evidence for an association between a sequence polymorphism and a drug resistance phenotype. Candidate gene studies are also inevitably based on prior assumptions about the possible mechanisms of resistance and so have major limitations in identifying novel or unsuspected mechanisms of resistance.
Recent progress in the genomic resources available for an increasing number of parasitic nematode species, together with rapid progress on genotyping technologies, makes the application of more global and systematic approaches increasingly possible. This review will discuss the use of Haemonchus contortus, the blood-feeding gastro-intestinal parasitic nematode of sheep, as a model parasite system in which to study anthelmintic resistance. The main focus will be on the potential for an integrated forward genetic and genomic approach to be developed in this parasite species with the goal of identifying resistance loci. The overall conceptual scheme of using H. contortus as a model system to identify causal mutations of anthelmintic resistance and investigate their significance is outlined in Fig. 1.

Fig. 1. Summary of the use of H. contortus as a model system in which to study the genetic basis of anthelmintic resistance.
ANTHELMINTIC RESISTANCE IS A COMPLEX QUANTITATIVE TRAIT
Drug resistance is an inherently quantitative trait; it is defined by numerical data from drug sensitivity or dose response assays with different parasite strains often having varying levels of resistance (Coles et al. Reference Coles, Jackson, Pomroy, Prichard, von Samson-Himmelstjerna, Silvestre, Taylor and Vercruysse2006; Demeler et al.; von Samson-Himmelstjerna et al. Reference von Samson-Himmelstjerna, Walsh, Donnan, Carriere, Jackson, Skuce, Rohn and Wolstenholme2009). Research has uncovered a variety of single mutations that are major determinants of drug resistance in a number of different parasite and pest species (Anderson et al. Reference Anderson, Nkhoma, Ecker and Fidock2011; Perry et al. Reference Perry, Batterham and Daborn2011). In the case of anthelmintic resistance, the best example is a SNP that results in a Phe200Tyr substitution in the isotype-1 β-tubulin polypeptide that occurs in a number of trichostrongyloid nematode species including H. contortus (Kwa et al. Reference Kwa, Veenstra and Roos1994, Reference Kwa, Veenstra, Van Dijk and Roos1995). It is apparent that in many cases, single mutations do not fully explain the variation detected in drug responses suggesting multiple mutations are often involved (Anderson et al. Reference Anderson, Nkhoma, Ecker and Fidock2011; Ghosh et al. Reference Ghosh, Andersen, Shapiro, Gerke and Kruglyak2012; Heckel, Reference Heckel2012). For example, mutations in multiple drug transporters contribute to the chloroquine resistance phenotype in Plasmodium falciparum (Mu et al. Reference Mu, Ferdig, Feng, Joy, Duan, Furuya, Subramanian, Aravind, Cooper, Wootton, Xiong and Su2003; Valderramos et al. Reference Valderramos, Valderramos, Musset, Purcell, Mercereau-Puijalon, Legrand and Fidock2010). Also, insecticide resistance is often due to a combination of drug target site mutations and over-expression of drug metabolizing enzymes and/or transporters (Ffrench-Constant et al. Reference Ffrench-Constant, Daborn and Le Goff2004; Perry et al. Reference Perry, Batterham and Daborn2011). Although, it was originally suggested that ivermectin resistance was determined by a single major dominant gene (Le Jambre et al. Reference Le Jambre, Gill, Lenane and Baker2000), it has been increasingly suspected that ivermectin resistance is multigenic in nature. The evidence for this has been indirect, being based on the complex dose-response phenotype of resistance and an inability to identify a clear cut association between various candidate loci and the resistance phenotype. However, recent work has provided more direct evidence of a multigenic basis for ivermectin resistance. Ghosh et al. (Reference Ghosh, Andersen, Shapiro, Gerke and Kruglyak2012) showed that ivermectin resistance occurs in natural populations of Caenorhabditis elegans isolated from the wild and that the resistance phenotype of these isolates is determined by multiple loci (Ghosh et al. Reference Ghosh, Andersen, Shapiro, Gerke and Kruglyak2012). That study reported a naturally occurring wild C. elegans isolate (CB4856) that was resistant to ivermectin compared to the wild type reference strain (N2). Quantitative trait locus (QTL) mapping was performed and determined that loci on chromosomes V and III additively accounted for approximately 26% and 6% of the phenotypic variance, respectively. The causal mutation for one of these loci was identified as a four amino acid deletion in the ligand-binding domain of the gene encoding the glutamate-gated chloride channel GLC-1 (a known ivermectin target). The glc-1 gene was sequenced in 53 diverse wild isolates and 16 of these carried alleles with the deletion, the presence of which was significantly associated with resistance. However, some isolates with the deletion were sensitive, and some isolates without it were resistant, indicating that other genetic factors influence drug sensitivity. Genetic complementation studies between a number of the isolates also suggested that additional loci were involved. Hence, this work clearly demonstrates the multigenic nature of ivermectin resistance in natural populations of a free-living nematode. Recent data from a backcrossing experiment also suggests that ivermectin resistance is multigenic in two different ivermectin resistant strains of H. contortus. In this experiment, discussed in more detail later in the review, after each backcross against the susceptible parental strain, only a low proportion of progeny survived a full in vivo therapeutic dose of ivermectin suggesting that multiple, independently segregating loci are responsible for the ivermectin resistance phenotype (Redman et al. Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). In the case of benzimidazole resistance in trichostrongyloid nematodes, the isotype-1 β-tubulin Phe200Tyr substitution is clearly very common and an important determinant of resistance (Prichard, Reference Prichard2001; Saunders et al. Reference Saunders, Wasmuth, Beech, Laing, Hunt, Naghra, Cotton, Berriman, Britton and Gilleard2013). Nevertheless, it does not appear to account for the full in vivo resistance phenotype in all cases suggesting that, even for benzimidazole resistance, multiple resistance loci may be involved in some isolates (von Samson-Himmelstjerna et al. Reference von Samson-Himmelstjerna, Walsh, Donnan, Carriere, Jackson, Skuce, Rohn and Wolstenholme2009; Barrere et al. Reference Barrere, Alvarez, Suarez, Ceballos, Moreno, Lanusse and Prichard2012).
The terms ‘dominant’ and ‘recessive’ are useful to describe the genetics of single gene traits but have limited value for quantitative traits that are determined by the additive effect of multiple loci. This is particularly the case for drug resistance phenotypes since the nature of the phenotype may depend on the concentration of the drug being used; an apparently dominant phenotype at one drug concentration may behave in a recessive manner at another. It is much more useful to consider drug resistance mutations as Quantitative Trait Loci (QTLs) that each contribute to the overall drug resistant phenotype in an additive manner. In some cases, a single QTL may account all of the observed resistance phenotype but in other cases, perhaps even the majority of cases, the phenotype may be determined by the additive effect of multiple loci.
The principles of mapping QTLs have been known for many decades and advances in genotyping technologies and statistical analyses have lead to a plethora of mapping studies for QTLs for human disease traits and production traits in domesticated animals and plants of agricultural importance (Mackay et al. Reference Mackay, Stone and Ayroles2009). The technical aspects of QTL mapping and recent developments in associated genotyping technologies are beyond the scope of this article but this topic is well reviewed elsewhere (Mackay et al. Reference Mackay, Stone and Ayroles2009). Application of QTL mapping approaches has not yet been widely exploited in anti-parasitic drug or pesticide resistance research. This is primarily due to a lack of genetic linkage maps and genome sequence resources for the important target species. There are now a number of successful examples (Mu et al. Reference Mu, Myers, Jiang, Liu, Ricklefs, Waisberg, Chotivanich, Wilairatana, Krudsood, White, Udomsangpetch, Cui, Ho, Ou, Li, Song, Li, Wang, Seila, Sokunthea, Socheat, Sturdevant, Porcella, Fairhurst, Wellems, Awadalla and Su2010; Witzig et al. Reference Witzig, Wondji, Strode, Djouaka and Ranson2013) and the recent explosion of genome sequence data together with the increasing availability of massively parallel sequencing technologies makes the application of QTL mapping approaches increasingly possible. Indeed, these approaches will be required in order to gain a detailed quantitative understanding of the genetic basis of anthelmintic resistance.
Consequently, there is a need to develop both forward genetic and genomic resources and methodologies for a number of key parasite species chosen as models for anthelmintic resistance research. The experimental advantages of H. contortus and the recent progress in the development of resources for such studies will be discussed in detail below.
THE TRACK RECORD OF H. CONTORTUS AS A MODEL SYSTEM IN ANTHELMINTIC DISCOVERY AND RESISTANCE RESEARCH
H. contortus is a parasitic nematode of sheep and goats and is of major socio-economic importance in many regions of the world (Urquhart et al. Reference Urquhart, Armour, Duncan, Dunn and Jennings1996). The L4 and adult stages reside on the surface of the host's abomasal (gastric) mucosa and feed on blood. The blood loss associated with parasite feeding and the haemorrhage from the associated mucosal lesions results in haemorrhagic anaemia. The severity of this is related to worm burden but it is often serious and not infrequently fatal (Urquhart et al. Reference Urquhart, Armour, Duncan, Dunn and Jennings1996). H. contortus has a very high propensity to develop resistance to anthelmintics and is the parasite species which has developed resistance most rapidly and in which resistance is most widespread (Kaplan, Reference Kaplan2004). Partly because of this, and partly because of its economic importance and experimental tractability, it is the helminth species for which we have the greatest understanding of mechanisms, genetics, diagnosis and management of anthelmintic resistance (Prichard, Reference Prichard2001; Gilleard, Reference Gilleard2006; Wolstenholme and Kaplan, Reference Wolstenholme and Kaplan2012). It is also one of the parasites for which we understand most about the modes of action of anthelmintics (Yates et al. Reference Yates, Portillo and Wolstenholme2003; Wolstenholme and Kaplan, Reference Wolstenholme and Kaplan2012; Saunders et al. Reference Saunders, Wasmuth, Beech, Laing, Hunt, Naghra, Cotton, Berriman, Britton and Gilleard2013). Consequently, it already provides a framework for much of the anthelmintic resistance and drug discovery research occurring in other parasite species. The extensive literature on anthelmintic resistance research on H. contortus has been covered in numerous reviews elsewhere and so will not be covered in detail here (Wolstenholme et al. Reference Wolstenholme, Fairweather, Prichard, von Samson-Himmelstjerna and Sangster2004; Gilleard, Reference Gilleard2006; Gilleard and Beech, Reference Gilleard and Beech2007; Wolstenholme and Kaplan, Reference Wolstenholme and Kaplan2012).
H. contortus has also been an extremely important organism in anthelmintic discovery research within the pharmaceutical industry for many years. An excellent recent example of the value of H. contortus for drug discovery and mode-of-action studies has resulted in the development of a new anthelmintic monepantel (Kaminsky et al. Reference Kaminsky, Ducray, Jung, Clover, Rufener, Bouvier, Weber, Wenger, Wieland-Berghausen, Goebel, Gauvry, Pautrat, Skripsky, Froelich, Komoin-Oka, Westlund, Sluder and Maser2008). The Novartis Animal Health group were exploring amino-acetonitrile derivatives (AAD) in their search for new anthelmintics. Six hundred AAD analogues were screened using an H. contortus in vitro larval development assay followed by in vivo validation in gerbils infected with H. contortus and Trichostrongylus colubriformis. Promising lead compounds were then tested by in vivo efficacy tests on H. contortus in sheep before other ruminant gastrointestinal nematodes were tested in the later stages of the development process. The group identified AAD-1566 for further development and this was ultimately launched for commercial use in sheep as the new anthelmintic monepantel.
H. contortus was not only central to the discovery of monepantel but also played a major role in the elucidation of its mode of action in parasites. Ce-acr-23, a nicotinic acetylcholine receptor subunit (nAChR) of the nematode-specific deg-3 family, was first identified as a monepantel target using a C. elegans forward genetic screen. H. contortus was then chosen as the experimental system in which to first investigate the target in parasites. A number of nAChR genes of the deg-3 subfamily were identified by mining the H. contortus genome database (http://www.sanger.ac.uk/resources/downloads/helminths/haemonchus-contortus.html) and the phylogenetically closest gene to the Ce-acr-23 gene was given the name Hco-mptl-1. In order to investigate the role of Hco-mptl-1 and of other closely related deg-3 subfamily members, two H. contortus strains that were highly resistant to monepantel were generated. A novel approach was taken to produce these monepantel-resistant strains; large numbers of free-living infective larvae (500 000) were exposed to drug prior to in vivo infection and eight rounds of such selection were performed with increasing doses of drug. The value of this approach was that it provided a much larger amount of genetic variation in the starting parental populations on which selection on could act, compared with the more widely used in vivo selection regimes. This approach is an excellent demonstration of how the high fecundity of H. contortus, together with the ability to store and manipulate the free-living infective stages, lends itself to the production of drug-resistant strains by experimental selection. Sequencing of the Hco-mptl-1 gene revealed a complex allelic series in the two monepantel resistant strains that was not present in the susceptible parental strains. These included intron–exon boundary deletions and premature stop codons resulting in shortened or truncated mRNA products. In addition, an insertion was found in the 5′UTR of the H. contortus des-2 homologue Hc-des2-2H. The significance of this is less clear because mutant C. elegans lacking functional DES-2 are not resistant to monepantel (Kaminsky et al. Reference Kaminsky, Ducray, Jung, Clover, Rufener, Bouvier, Weber, Wenger, Wieland-Berghausen, Goebel, Gauvry, Pautrat, Skripsky, Froelich, Komoin-Oka, Westlund, Sluder and Maser2008). Nevertheless, the results strongly suggest that Hc-mptl-1 is an important monepantel target in H. contortus. They also suggest that this may not be an essential gene raising the spectre of null mutations as a potential resistance mechanism in the field.
This work is an excellent example of how H. contortus can be used in parallel with C. elegans to study the mechanisms of drug action and resistance and represents a notable success story in anthelmintic discovery and development. The monepantel drug has now been launched onto the market for parasite control in sheep in Australia, New Zealand and many European and South American countries (Epe and Kaminsky, Reference Epe and Kaminsky2013). Importantly, the research performed during the discovery process now provides us with a good, although not necessarily complete, understanding of the monepantel mode of action and important knowledge and tools with which to monitor the emergence of resistant parasites over time.
H. CONTORTUS POPULATION GENETICS AND ITS RELEVANCE TO THE DEVELOPMENT AND INVESTIGATION OF ANTHELMINTIC RESISTANCE
H. contortus is one of the parasitic nematodes for which we understand most about its genetics and population structure. The karyotype is 2n = 11 in males or 12 in females with the sex chromosomal karyotype of XO male and XX female (Redman et al. Reference Redman, Grillo, Saunders, Packard, Jackson, Berriman and Gilleard2008a). All five autosomes and the X-chromosome are of a similar size; an apparently identical karyotype to that of C. elegans. Unlike C. elegans, H. contortus has separate male and female adults with an obligate dioecious mode of sexual reproduction. Mating of adult worms in the host is polyandrous with a single females repeatedly mating with multiple males. Broods from single female worms can contain F1 progeny derived from at least four different paternal genotypes (Redman et al. Reference Redman, Grillo, Saunders, Packard, Jackson, Berriman and Gilleard2008a). This is also the case for Teladorsagia circumcincta suggesting it may be a common feature of the trichostrongyloid group (Gilleard and Grillo, unpublished data). This is an additional way in which these parasites generate genetic diversity and also has important practical implications for developing inbred strains (Redman et al. Reference Redman, Grillo, Saunders, Packard, Jackson, Berriman and Gilleard2008a).
The population genetics of anthelmintic resistance in trichostrongyloid nematodes has been reviewed in detail elsewhere (Gilleard and Beech, Reference Gilleard and Beech2007) but a number of key features are directly relevant to this review. H. contortus has a high level of genetic diversity predominantly due to its large population size (Prichard, Reference Prichard2001; Gilleard and Beech, Reference Gilleard and Beech2007). Genetic diversity in a population is a function of mutation rate (μ) and effective population size (Ne); in an idealized diploid population, the pairwise nucleotide diversity is equal to 4 μNe. Although we do not know the mutation rate in H. contortus, it has been accurately estimated for C. elegans and a similar rate recently determined for Caenorhabditis briggsae (Denver et al. Reference Denver, Morris, Lynch and Thomas2004, Reference Denver, Wilhelm, Howe, Gafner, Dolan and Baer2012). A rate of 2·1×10−8 mutations per site per generation has been calculated for the C. elegans nuclear genome (ten-fold lower than for the mitochondrial genome) which equates to 2·1 mutations per genome per generation. Since a single host can contain thousands of adult female worms, each producing thousands of eggs per day, massive numbers of new genotypes continually pass out onto the pasture. Although not all these genotypes will contribute to the next generation, the effective population size of H. contortus is potentially enormous resulting in extremely high levels of genetic diversity (see Fig. 2). One consequence of this is that the parasite has a remarkably high capacity to generate new mutations and any of these that are under positive selection – such as drug resistance mutations – may become fixed and increase in frequency. This has important implications for the origins of anthelmintic resistance in H. contortus, and other trichostrongyloid nematode species, and there is increasing evidence that new anthelmintic mutations arise frequently and recurrently in these parasite species (Silvestre and Humbert, Reference Silvestre and Humbert2002; Gilleard and Beech, Reference Gilleard and Beech2007; Skuce et al. Reference Skuce, Stenhouse, Jackson, Hypsa and Gilleard2010; E. Redman, F. Whitlow, P. Skuce, A. Tait and J. S. Gilleard, unpublished). These concepts are summarized in Fig. 2.

Fig. 2. The relevance of H. contortus population size to the origins of anthelmintic resistance mutations.
Early work on the population genetics of trichostrongyloid nematodes, including H. contortus, suggested that almost all genetic variation was partitioned within populations with essentially no geographical sub-structuring (Blouin et al. Reference Blouin, Yowell, Courtney and Dame1995; Braisher et al. Reference Braisher, Gemmell, Grenfell and Amos2004; Grillo et al. Reference Grillo, Jackson, Cabaret and Gilleard2007). This was ascribed to very large effective population sizes together with high gene flow due to animal movement but, as more work has been performed, it has become clear that this is an oversimplified view. A study of the global population genetic structure of H. contortus, using mitochondrial DNA and AFLP markers, has revealed a high level of genetic structure between continents with populations from each continent essentially forming monophyletic groups (Troell et al. Reference Troell, Engstrom, Morrison, Mattsson and Hoglund2006). This work was supported by the finding of extremely high levels of genetic divergence between laboratory strains derived from different continents, using microsatellite marker panels (Redman et al. Reference Redman, Packard, Grillo, Smith, Jackson and Gilleard2008b). These results are not entirely surprising since one would anticipate limited gene flow to occur between continents allowing genetic drift to result in genetic differentiation over time. Perhaps more surprising is the significant genetic differentiation between different H. contortus populations within the same country as has been reported in both Sweden and Australia (Troell et al. Reference Troell, Engstrom, Morrison, Mattsson and Hoglund2006; Hunt et al. Reference Hunt, Knox, Le Jambre, McNally and Anderson2008). We have also recently found measurable genetic differentiation between different H. contortus isolates in the UK in spite of short geographical distances and high levels of animal movement (E. Redman, F. Whitlow, P. Skuce, A. Tait and J. S. Gilleard, unpublished). We hypothesize that these findings are due to the complex epidemiology of H. contortus in different climatic regions and livestock production systems. For example, in the cold winters of Sweden and the UK and the dry summer months in parts of Australia, the vast majority of free-living H. contortus larvae in the environment will die. Consequently, parasite survival at these particular times of year is confined to adult worms and inhibited larvae inside the host. If anthelmintic drug treatments are applied at these times, then there is significant potential for population bottlenecks resulting in a reduction of overall genetic diversity and an increase in geographical genetic sub-structuring. The discovery that H. contortus populations can often have complex population genetic structures has a number of important implications for anthelmintic resistance research. Firstly, it cannot be assumed that the same anthelmintic resistance mutations will be present in resistant parasite populations in different locations (even within the same country). Secondly, a good understanding of the underlying population genetic structure is necessary to interpret candidate gene studies or to apply Genome Wide Association Studies (GWAS) to parasite populations in the field to identify areas of the genome under selection.
EVOLUTIONARY RELATIONSHIPS OF RELEVANCE TO THE USE OF H. CONTORTUS AS A MODEL SYSTEM IN ANTHELMINTIC RESISTANCE RESEARCH
The genus Haemonchus contains over a dozen different blood feeding parasite species that infect a large range of artiodactyl hosts (cloven-hooved mammals) (Anderson, Reference Anderson2000; Hoberg et al. Reference Hoberg, Kocan, Rickard, Samuel, Pybus and Kocan2001, Reference Hoberg, Lichtenfels and Gibbons2004). H. contortus is the most economically important species in the genus. Although it most commonly infects sheep and goats it can infect a wide range of other artiodactyl species including cattle, deer, antelope, bison, camelids and giraffes with varying degrees of efficiency (Hoberg et al. Reference Hoberg, Kocan, Rickard, Samuel, Pybus and Kocan2001, Reference Hoberg, Lichtenfels and Gibbons2004). The Haemonchus genus originated in Africa with basal diversification in antelopes followed by the development of other host-parasite species associations within the Antilopinae, Bovinae, Caprini and other artiodactyl groups (Hoberg et al. Reference Hoberg, Kocan, Rickard, Samuel, Pybus and Kocan2001, Reference Hoberg, Lichtenfels and Gibbons2004). Colonization of domesticated species and their subsequent spread around the globe by human transportation has led to the widespread translocation of H. contortus (and to a lesser extent H. placei and H. similis). Although originally a tropical parasite, H. contortus is now endemic throughout most of the Northern and Southern Hemispheres and is present as far north as Canada and Sweden even reaching the artic circle (Waller et al. Reference Waller, Rudby-Martin, Ljungstrom and Rydzik2004; Falzon et al. Reference Falzon, Menzies, Shakya, Jones-Bitton, Vanleeuwen, Avula, Stewart, Jansen, Taylor, Learmount and Peregrine2013).
The genus Haemonchus is in the order Strongylida within clade V of the nematode phylum based on the molecular evolutionary framework constructed from small subunit ribosomal DNA sequences (Blaxter et al. Reference Blaxter, De Ley, Garey, Liu, Scheldeman, Vierstraete, Vanfleteren, Mackey, Dorris, Frisse, Vida and Thomas1998). Members of the order Strongylida are all bursate nematodes which fall into four well-defined superfamilies; the Ancylostomatoidea (includes the hookworms of humans and domestic animals), the Strongyloidea (which includes many important equine parasites), the Metastrongyloidea (which includes some lungworm species of domestic animals) and the Trichostrongyloidea (which include many of the most economically important parasites of livestock) (Anderson, Reference Anderson2000). There are a number of important evolutionary relationships between H. contortus and other parasitic nematode groups that are worth highlighting with respect to its use as a model in which to study anthelmintic resistance.
Relationship to the trichostrongyloid nematodes of livestock
H. contortus is one of the Trichostrongyloidea – often referred to as the trichostrongyloid or trichostrongylid nematodes – which reside in the gastro-intestinal tracts of birds, lagomorphs, marsupials or ruminants (Durette-Desset, Reference Durette-Desset1985; Urquhart et al. Reference Urquhart, Armour, Duncan, Dunn and Jennings1996). There are many genera in this group that occur as mixed infections in domestic ruminants – including Haemonchus, Cooperia, Ostertagia, Trichostrongylus, Teladorsagia and Nematodirus. As a group, these are amongst the most economically important pathogens of grazing ruminants worldwide causing serious economic loss and animal welfare problems for the livestock industries in the industrialized world. For example, it has been estimated that this group of parasites causes over $2 billion dollars of economic loss to the North American cattle industry alone (Stromberg and Gasbarre, Reference Stromberg and Gasbarre2006). They are also a major contributor to worldwide poverty being ranked amongst the most important animal health problems for livestock producers in the developing world (Perry et al. Reference Perry, Randolph, McDermott, Sones and Thornton2002). Hence, H. contortus, as well as being an important livestock pathogen in its own right, serves as a model for a large number of closely related parasite species of huge economic importance for agriculture.
Relationship to the human hookworms
The human hookworms of the genera Necator and Ancylostoma are intractable experimental systems and it is challenging to get even small amounts of parasite material from the field. Although not as close as the trichostrongyloids, the hookworms are in the same phylogenetic clade as H. contortus and have a similar life cycle, host location and blood-feeding habit (Blaxter et al. Reference Blaxter, De Ley, Garey, Liu, Scheldeman, Vierstraete, Vanfleteren, Mackey, Dorris, Frisse, Vida and Thomas1998). Hence, mechanisms of anthelmintic resistance that have been elucidated in H. contortus may well be relevant to these important human pathogens. Thus studies on H. contortus are potentially a valuable source of candidate genes and hypotheses to be tested for roles in resistance in human hookworms in a targeted manner as illustrated by recent work on benzimidazole resistance in human helminths (Diawara et al. Reference Diawara, Schwenkenbecher, Kaplan and Prichard2013).
Relationship to C. elegans
H. contortus is in the closest phylogenetic clade of vertebrate parasites to the model nematode C. elegans. Comparative studies between H. contortus and C. elegans suggest that many aspects of their core biology are conserved, particularly when looking at basic developmental processes and neurobiology (Gilleard, Reference Gilleard2004). There are an increasing number of examples of functional expression of H. contortus genes in C. elegans (Couthier et al. Reference Couthier, Smith, McGarr, Craig and Gilleard2004; Glendinning et al. Reference Glendinning, Buckingham, Sattelle, Wonnacott and Wolstenholme2011). For example, drug sensitivity can be restored to either benzimidazole- or ivermectin-resistant C. elegans strains by heterologous expression of the relevant drug targets from H. contortus (Kwa et al. Reference Kwa, Veenstra, Van Dijk and Roos1995; Glendinning et al. Reference Glendinning, Buckingham, Sattelle, Wonnacott and Wolstenholme2011). This validates C. elegans as a heterologous expression system to test specific H. contortus mutations for causal relationship with resistance for the two most important classes of anthelmintics in current use. This ability to use C. elegans to test the function of H. contortus genes and mutations is an immensely important feature of H. contortus as a model system in which to study anthelmintic resistance.
PRACTICAL ASPECTS OF H. CONTORTUS AS AN EXPERIMENTAL SYSTEM
Parasitic nematodes are notoriously difficult experimental subjects due to the lack of in vitro culture systems and the limitations of laboratory animal hosts. This is particularly the case for most human parasites where experimentation is limited to in vitro work or via the use of ‘artificial’ laboratory animal hosts in which the parasite is usually poorly adapted or in which the life cycle is only partially completed (Lustigman et al. Reference Lustigman, Geldhof, Grant, Osei-Atweneboana, Sripa and Basanez2012). In contrast, it is relatively simple to maintain the lifecycle of H. contortus by experimental infection of its natural host, the sheep. Although this is true for a number of animal parasites, H. contortus affords a number of important experimental advantages over most other animal parasite species. It has a short pre-patent period (18–21 days) and burdens of several thousand worms per individual host can be established and maintained for many months from a single oral infection of L3 larvae. The parasite has an extremely high fecundity compared with other strongylid nematodes; a single adult female worm produces up to 4000 eggs per day (Coyne and Smith, Reference Coyne and Smith1992). Consequently, tens of millions of eggs can be produced from a single infected host over just few days. These can be developed to the infective L3 stage in faecal culture making it possible to obtain large numbers of any of the free-living stages – L1, L2 or L3s – by harvesting at the appropriate time. This level of production can be sustained from the same infected host over many weeks or even months. The adult worms are relatively large for a strongylid nematode (up to 2·5 cm in length) allowing grams of material of the L4 and adult parasitic stages to be obtained from a single sacrificed host. One particularly useful feature of H. contortus is the ability, in common with most of the other trichostrongyloids, to store large numbers of viable L3 larvae at 15–22 °C for many months with minimal mortality in the population. This makes it realistic for a well-organized laboratory to maintain multiple strains, since each strain only needs to be passaged through a single host once or twice per year. Furthermore, there are simple and efficient methods for L3 cryopreservation in which viability and infectivity is preserved (Van Wyk and Van Wyk, Reference Van Wyk and Van Wyk2002). The ability to permanently archive viable parasite strains and genetic cross progeny is a hugely important feature that allows genetic crossing and mapping strategies to be seriously contemplated.
As discussed above, C. elegans has an important role as a heterologous expression system to study H. contortus gene function. Nevertheless, there are some limitations to this approach since gene function will not be conserved in all cases. Hence, there is a need to develop tools to directly examine gene function in H. contortus and significant progress has recently been made with RNAi-mediated gene knockdown. L1 larvae can be cultured to infective L3 on Escherichia coli (OP50) bacterial lawns grown on agar plates in the same way as C. elegans is cultured. Consequently, these stages are easily accessible to the application of RNAi by feeding on bacteria expressing double stranded RNA, soaking or electroporation. Early experiments had mixed results with the transcript level being successfully knocked down for only two of eleven genes targeted (Geldhof et al. Reference Geldhof, Murray, Couthier, Gilleard, McLauchlan, Knox and Britton2006). However, recent work in which genes were selected based on their being expressed in accessible tissues such as the intestine, excretory cell and amphids improved the success rate with the transcripts of four out of six target genes being successfully reduced by soaking in double-stranded RNA (Samarasinghe et al. Reference Samarasinghe, Knox and Britton2011). A recent study reported even greater success of RNAi in H. contortus using feeding rather than soaking or electroporation with transcript reduction being achieved for all nine genes tested using the feeding method (Zawadzki et al. Reference Zawadzki, Kotze, Fritz, Johnson, Hemsworth, Hines and Behm2012). Hence RNAi methodology in H. contortus is steadily being improved. Although, phenotypic analysis following gene knockdown remains challenging – since only the free-living larval stages can currently be targeted – drug sensitivity phenotypes are amongst the most amenable to analysis since drug dose response experiments can be easily performed on the free-living larval stages. Furthermore, there has been recent progress in targeting the parasitic stages of H. contortus in the host. This was achieved by applying RNAi to the infective L3 stage followed by examination of phenotype in infected hosts (Samarasinghe et al. Reference Samarasinghe, Knox and Britton2011). L3s were soaked for 24 h in dsRNA targeting the gut expressed aminopeptidase vaccine candidate, H11, prior to oral infection of lambs. Adult worm burdens and faecal egg counts were subsequently reduced by 40% and 57%, respectively compared to control groups and there was a corresponding 64% reduction in aminopeptidase activity (Samarasinghe et al. Reference Samarasinghe, Knox and Britton2011). There is significant potential to use this approach to determine the sensitivity of parasitic stages to drug treatment of experimentally infected hosts following RNAi knockdown of a candidate resistance genes in the infective larvae.
PROGRESS TOWARDS AN H. CONTORTUS REFERENCE GENOME SEQUENCE SUITABLE FOR FORWARD GENETICS AND GENOMICS RESEARCH
The use of H. contortus as a model for anthelmintic resistance research is entirely dependent on developing good genomic resources including a well assembled and annotated reference genome. In recent years there has been an explosion of activity in sequencing nematode genomes driven by the advent of ‘Next generation’ or ‘Highly parallel’ sequencing technologies. Most of the key resources are available through http://nematode.net/, http://www.nematodes.org/ and http://www.sanger.ac.uk/research/projects/parasitegenomics/. Although these projects have already provided tremendous resource for comparative genomics research, there are major limitations when using such draft genome sequences for functional genomics and genetic mapping approaches. Hence, it is imperative that there is iterative improvement in the genome assemblies and annotations for the most important parasite species of medical or veterinary importance and for those species being used as model experimental systems. In the case of H. contortus, there is now a good draft genome sequence available and work is continuing to improve this current draft (Laing et al. unpublished). The data are publicly available at (http://www.sanger.ac.uk/resources/downloads/helminths/haemonchus-contortus.html). H. contortus has presented some challenges for the production of a good quality draft reference genome and this is likely be common to the whole strongylid nematode group. It has a larger than anticipated genome size (currently estimated between 300 and 340 Mb), complex gene structures, high levels of sequence polymorphism and a high level of repetitive sequence (R. Laing et al. unpublished). Analysis of the current assembly and previous detailed annotation of two large regions of completely finished contiguous genome sequence (409 and 181 Mb respectively) has given a snapshot of genome structure in H. contortus. Gene density appears to be less in H. contortus than in C. elegans, with annotated H. contortus genes being an average of two-to-three times larger than their putative C. elegans orthologues due to a greater intron number and size (Laing et al. Reference Laing, Hunt, Protasio, Saunders, Mungall, Laing, Jackson, Quail, Beech, Berriman and Gilleard2011). Synteny appears high – the same genes are generally located on the same chromosomes in the two species – but gene order is generally poorly conserved. Nevertheless, areas of conserved microsynteny are apparent and C. elegans operons also appear to be partially conserved in H. contortus (Laing et al. Reference Laing, Hunt, Protasio, Saunders, Mungall, Laing, Jackson, Quail, Beech, Berriman and Gilleard2011).
One of the key challenges of producing a well assembled H. contortus genome sequence has been its high level of sequence polymorphism which complicates the genome assembly. MHco3(ISE) was originally chosen as the reference strain for the Wellcome Trust Sanger Institute H. contortus genome project because it was derived from an inbred strain: Inbred Strain Edinburgh, ISE (Roos et al. Reference Roos, Otsen, Hoekstra, Veenstra and Lenstra2004; Redman et al. Reference Redman, Grillo, Saunders, Packard, Jackson, Berriman and Gilleard2008a, Reference Redman, Packard, Grillo, Smith, Jackson and Gilleardb). The latter strain had been produced by using the L3 progeny derived from a single adult female worm to infect a recipient sheep and then repeating this process over multiple generations (Roos et al. Reference Roos, Otsen, Hoekstra, Veenstra and Lenstra2004). In spite of this inbreeding strategy, genetic analysis revealed that the strain has retained significant levels of sequence polymorphism (Redman et al. Reference Redman, Packard, Grillo, Smith, Jackson and Gilleard2008b). We hypothesized that this was at least partially due to polyandrous mating in H. contortus resulting in single mated adult female worms containing the sperm from multiple male worms (Redman et al. Reference Redman, Grillo, Saunders, Packard, Jackson, Berriman and Gilleard2008a). Consequently, we attempted to reduce further the polymorphism of the MHco3(ISE) strain by performing a single-pair mating. This was achieved by surgically transplanting 15 female worms, along with a single male worm, into the abomasum of a sheep and then subsequently recovering the female worms (N. Sargison, E. Redman and J. S. Gilleard, unpublished data). Broods from these females (which could only have mated with the single male) were then recovered and used to infect a recipient sheep to produce the MHCo3(ISE).N1 strain. Mapping a 6·2 Gb random sub-sample of 76 bp Hiseq Illumina reads from the MHco3(ISE) and the MHco3(ISE).N1 strains to a 319 Mb reference genome assembly revealed 204 248 and 99 747 SNPs in the two strains, respectively (H. Nahgra and J. Cotton, personal communication). This equates to 0·64 SNP/kb for the MHco3(ISE) strain compared to 0·31 SNP/kb for the MHco3.N1 inbred strain demonstrating a reduction in sequence polymorphism of 50% by the single pair mating step. The MHco3(ISE).N1 has subsequently been used to provide the DNA template for H. contortus genome sequencing currently being undertaken at the Wellcome Trust Sanger Institute.
The latest H. contortus genome assembly available at the time of writing has a total length of 345, 365, 851 bp. However, analysis suggests that approximately 20% of the scaffolds may be allelic with other contigs in the assembly. Hence the true haploid genome size may be nearer 300 Mb. The contig N50 length is 20 788 bp and the scaffold N50 length is 83 287 bp (J. Cottonand M. Berriman, personal communication). N50 length is defined as the length N for which half of all bases in the assembly are in contigs (or scaffolds) of lengths equal to or greater than N. CEGMA analysis (Parra et al. Reference Parra, Bradnam and Korf2007) reveals that of 248 eukaryotic genes, 93·15% are present at least partially and 90·73% are complete in this latest assembly. Although this latest assembly is a major step forward, it will be extremely important to ensure that efforts are continued to iteratively improve both the assembly and annotation in order to provide a resource suitable for functional genomics and genetic studies.
MONITORING THE INTEGRITY OF H. CONTORTUS STRAINS USED IN GENETIC STUDIES
The availability of genetically characterized strains is a prerequisite for undertaking genetic analysis and/or genetic mapping in any organism. This is a neglected area of study for parasitic nematodes and the lack of genetically characterized strains is a major limiting factor, not only for genetic studies, but also for many other types of research. Without the appropriate genetic markers and baseline data, it is impossible to ensure strain integrity or to meaningfully compare experimental results between laboratories and experiments. Isolates or strains that are assumed to be identical could have major genetic and phenotypic differences due the selective effects of experimental passage or due to human error in management. Conversely, strains assumed to be different could be closely related. It is commonplace for parasitic nematode strains to be transferred between laboratories and passaged by repeated experimental infection with no genetic monitoring, a practice that would be unthinkable for work with bacteria, protozoa or mammalian cell culture lines. The neglect of this issue in parasitic nematode research has, at least in part, been due to the lack of appropriate genetic markers and a relatively poor understanding of how genetic variation is partitioned within and between strains.
H. contortus is one of the few parasitic nematode species for which there has been detailed genetic characterization of a number of laboratory strains. There is a significant amount of data demonstrating a high level of genetic diversity both within and between many of the H. contortus strains used in various laboratories around the world (Hoekstra et al. Reference Hoekstra, Criado-Fornelio, Fakkeldij, Bergman and Roos1997; Hunt et al. Reference Hunt, Knox, Le Jambre, McNally and Anderson2008; Redman et al. Reference Redman, Packard, Grillo, Smith, Jackson and Gilleard2008b). Microsatellite markers have been developed and used by a number of groups to characterize strains and to investigate the genetic relationships between them (Hoekstra et al. Reference Hoekstra, Criado-Fornelio, Fakkeldij, Bergman and Roos1997; Hunt et al. Reference Hunt, Knox, Le Jambre, McNally and Anderson2008; Redman et al. Reference Redman, Packard, Grillo, Smith, Jackson and Gilleard2008b). Microsatellites are valuable markers for this purpose because they are highly polymorphic and multi-allelic allowing a relatively small number of markers to provide a high level of discrimination between strains (Redman et al. Reference Redman, Packard, Grillo, Smith, Jackson and Gilleard2008b). They can be amplified from single DNA samples prepared from populations of worms and multiplexed to allow strains to be quickly and cheaply ‘fingerprinted’ with just a few PCR reactions. These ‘genetic fingerprints’ are highly stable for a given strain, both over time and between individual hosts (Redman et al. Reference Redman, Packard, Grillo, Smith, Jackson and Gilleard2008b). We now use this approach to monitor H. contortus strains routinely by genetically fingerprinting every batch of worms produced by experimental infection to check identity and integrity before undertaking any experimental work or genetic analysis.
Strain integrity and quality assurance is also highly dependent on the use of an appropriate nomenclature system that unambiguously captures information about provenance and history. The ivermectin-resistant H. contortus CAVR strain, originally isolated from sheep at CSIRO Armidale in Australia, is just one of many strains that can be used to illustrate this issue (Le Jambre, Reference Le Jambre1993; Le Jambre et al. Reference Le Jambre, Gill, Lenane and Lacey1995). In the intervening years since its isolation, this strain has been distributed amongst many laboratories and widely used in experimental studies. Numerous publications refer to this strain as either ‘CAVR’ or ‘CAVRS’ (Tyrrell et al. Reference Tyrrell, Dobson, Stein and Walkden-Brown2002; Rowe et al. Reference Rowe, Gondro, Emery and Sangster2009). However, the way in which the strain has been passaged, selected and maintained is rarely recorded and, until recently, there has been a complete absence of genetic monitoring of integrity. Hence, it impossible to determine the relationship between the ‘CAVR’ strain used in different laboratories (or even in the same laboratory at different times) making it unwise to compare and extrapolate results between studies. Consequently, we have adopted the following approach for the strains that are maintained at the Moredun Research Institute, Edinburgh, UK and used for our genetic studies (Redman et al. Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). When a H. contortus strain is obtained from another laboratory, it is given the prefix ‘MHco’ followed by a unique number with the original strain designation in parentheses. This system allows that particular version of the strain to be distinguished from other versions in different laboratories; MHco10(CAVR) is the version of the CAVR strain that is maintained at the Moredun laboratory which has now been extensively characterized both phenotypically and genetically (Redman et al. Reference Redman, Grillo, Saunders, Packard, Jackson, Berriman and Gilleard2008a, Reference Redman, Packard, Grillo, Smith, Jackson and Gilleardb; Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). The strain is defined by a ‘genetic fingerprint’ based on a panel of neutral microsatellite markers which are routinely applied to test the integrity of the between passages and experiments (Redman et al. Reference Redman, Packard, Grillo, Smith, Jackson and Gilleard2008b). Whilst, the MHco10(CAVR) strain is historically related to the original ‘CAVR’ we do not assume identity with other versions used in other laboratories around the world.
The strains used in genetic crosses need to have sufficient genetic divergence to allow progeny genotypes to be unambiguously assigned to the alternate parental genotypes. Consequently, over the last few years, we have put a lot of effort into developing and applying robust panels of microsatellite markers to identify and characterize strains that are sufficiently genetically divergent to be used in genetic crosses (Redman et al. Reference Redman, Grillo, Saunders, Packard, Jackson, Berriman and Gilleard2008a, Reference Redman, Packard, Grillo, Smith, Jackson and Gilleardb). Current progress in using such strains for the genetic analysis of anthelmintic resistance is discussed in the next section.
FORWARD GENETIC APPROACHES CURRENTLY BEING USED TO INVESTIGATE MECHANISMS OF ANTHELMINTIC RESISTANCE IN H. CONTORTUS
Genetic crossing of parasitic nematodes is technically challenging because the adult stages are generally hidden inside the host and so are difficult to access and manipulate. Genetic crossing of trichostrongyloid nematodes has been practiced for over 30 years albeit to a limited extent (Le Jambre et al. Reference Le Jambre, Royal and Martin1979, Reference Le Jambre, Lenane and Wardrop1999, Reference Le Jambre, Gill, Lenane and Baker2000; Martin and McKenzie, Reference Martin and McKenzie1990; Sangster et al. Reference Sangster, Redwin and Bjorn1998). Most studies have used H. contortus, probably because its high fecundity makes it easier to recover cross progeny (Le Jambre et al. Reference Le Jambre, Royal and Martin1979, Reference Le Jambre, Gill, Lenane and Baker2000; Sangster et al. Reference Sangster, Redwin and Bjorn1998). The standard approach for H. contortus is to transplant immature male and female adult worms recovered from donor sheep directly into the abomasum of a recipient sheep. This is done either by surgery or through an abomasal cannula (Redman et al. Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). Eggs recovered in the faeces of the recipient sheep are the F1 progeny and these, following in vitro development to infective L3, are used to infect another sheep orally to develop to adult worms. These F1 adult worms can be recovered from the abomasum on autopsy following euthanasia and/or F2 progeny can be collected as eggs in the faeces. Single-pair matings, although challenging, are possible (N. Sargison, E. Redman and J. S. Gilleard, unpublished). However, most studies have generally involved mass matings of 50–100 males or females from a drug-resistant strain with 50–100 males or females from a susceptible strain. The main objective has typically been to assess the degree of dominance of the resistance phenotype and to investigate whether resistance is determined by a single locus or is multigenic (Le Jambre et al. Reference Le Jambre, Royal and Martin1979; Martin and McKenzie, Reference Martin and McKenzie1990; Sangster et al. Reference Sangster, Redwin and Bjorn1998). Undertaking crosses with H. contortus is technically challenging, labour intensive and time consuming (generally 1–2 backcross generations can be completed per year) and there is an ever-present risk of contamination. Hence, the importance of monitoring and validating genetic crosses using appropriately discriminatory genetic markers cannot be over emphasized. The main limitation of most studies to date has been that few, or often no, genetic markers were used to genetically validate crosses. Nevertheless, these were valuable ‘proof of concept’ experiments and have provided useful information (Gilleard, Reference Gilleard2006).
The most exciting aspect of developing genetic crossing strategies in parasitic nematodes is their potential to be used for mapping of the genetic loci underlying important traits such as anthelmintic resistance. Highly sophisticated and powerful mapping approaches have been developed in a number of model organisms and in domestic animal and plant breeding research (Fay and Bender, Reference Fay and Bender2008; Goddard and Hayes, Reference Goddard and Hayes2009; Jones et al. Reference Jones, Ougham, Thomas and Pasakinskiene2009). There are many technical challenges in applying some of these approaches in parasitic nematodes, including the lack of appropriate inbred and/or genetically divergent strains, difficulties of undertaking single pair matings and, in the case of anthelmintic resistance, difficulties in obtaining large numbers of reliably phenotyped susceptible and resistant progeny. The availability of an improved, albeit not yet finished, reference genome sequence for H. contortus, together with the use of highly parallel sequencing based genotyping strategies, makes it increasingly possible to adapt ‘conventional’ genetic mapping approaches to parasite systems. In essence, if parental strains that are sufficiently phenotypically and genetically divergent are crossed, it should be possible to apply genome wide analysis to identify SNPs that are over-represented in drug-resistant progeny compared with susceptible progeny (or unselected progeny if susceptible progeny cannot be obtained for practical reasons). Two different genetic crossing strategies aiming to do this in H. contortus have been recently published (Hunt et al. Reference Hunt, Kotze, Knox, Anderson, McNally and LF2010; Redman et al. Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). The first of these involved a classical F2 mapping cross (Hunt et al. Reference Hunt, Kotze, Knox, Anderson, McNally and LF2010) whereas the second involved a serial backcrossing approach to introgress anthelmintic resistance loci into a susceptible genetic background (Redman et al. Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). The principles of these two different approaches are shown in Fig. 3.

Fig. 3. Schematic representation of two different crossing regimes applied to H. contortus. A single resistance conferring locus is represented by * on a single chromosome pair to illustrate the general concepts. In reality, there are likely to be multiple loci on multiple chromosomes acting as Quantitative Trait Loci in an additive manner. The parental chromosomes from the resistant parent are represented in red and from the susceptible parent in blue. (A) F2 mapping approach; (B) Serial backcrossing approach.
F2 mapping cross
The concept underlying this approach is illustrated in Fig. 3A. In a recent study, the Wallangra 2003 multi-drug resistant field-derived strain – resistant to four anthelmintic classes (benzimidazoles, macrocyclic lactones, levamisole and closantel) – was crossed with the McMaster 1931 susceptible strain (Hunt et al. Reference Hunt, Kotze, Knox, Anderson, McNally and LF2010). F2 progeny were then separately selected with the four anthelmintic drug classes to produce four drug-resistant F2 populations together with an unselected control population. Individual F2 worms were then analysed using six amplicon length polymorphism markers (ALPs) to validate the cross. The major limiting factor of this study was that, although there were high levels of genetic diversity within each of the parental strains, there was a lack of genetic differentiation between them. Only five of 50 alleles across the six genotyped loci displayed significant differences in frequency between the parental McMaster 1931 and Wallangra 2003 strains and none of these differences were significant after Bonferroni correction for multiple testing (Hunt et al. Reference Hunt, Kotze, Knox, Anderson, McNally and LF2010). This lack of genetic differentiation limited the ability to validate the crosses and to undertake further genetic analysis. Nevertheless, this study demonstrated the feasibility of an F2 mapping cross in H. contortus and the material generated may be a useful resource for further work. As genomic resources for H. contortus improve, a larger set of markers could be screened to identify those showing clearer differentiation between the parental McMaster 1931 and Wallangra 2003 strains. Such markers could then be used to re-interrogate the resistant F2 populations to identify alleles from the Wallangra 2003 strain that are over-represented in phenotypically resistant F2 progeny indicating linkage to a resistance conferring locus. There is also a strong case to repeat this type of cross using more genetically divergent strains. This F2 mapping approach ultimately has the potential to map resistance-conferring loci down to narrow genomic regions providing that a sufficiently large number of F2 progeny are genotyped with a sufficiently dense panel of discriminatory markers.
Serial backcrossing
We undertook a different approach in a serial backcrossing experiment that aimed to introgress ivermectin resistance genes from two independent ivermectin resistant strains, MHco4(WRS) and MHco10(CAVR), into the susceptible genome reference strain MHco3(ISE) (Redman et al. Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). The concept underlying this approach is illustrated in Fig. 3B and the experimental scheme in Fig. 4. In this case, the parental strains were chosen on the basis of their extreme genetic divergence in order to allow parental genotypes to be unequivocally discriminated in backcross populations and individual progeny (Redman et al. Reference Redman, Grillo, Saunders, Packard, Jackson, Berriman and Gilleard2008a, Reference Redman, Packard, Grillo, Smith, Jackson and Gilleardb, Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). The approach involved two independent serial backcrossing experiments in which two ivermectin-resistant strains – MHco4(WRS) and MHco10(CAVR) – were crossed with the susceptible MHco3(ISE). In each case, the resulting F1 progeny were then crossed with the susceptible parental MHco3(ISE) strain and the process repeated through four serial backcross generations in the face of selection with ivermectin (Fig. 4). The process aimed to increase the genetic background contributed by the MHco3(ISE) parental strain (and conversely reduce that of the resistant parental strain) at each backcross. Ivermectin treatment was applied at each generation to ensure that only phenotypically resistant worms survived and so resistance-conferring loci were retained throughout the backcrossing procedure. The overall aim was to produce a strain with a predominantly MHCo3(ISE) ‘susceptible’ genetic background into which ivermectin resistance-conferring loci from the resistant parental strain are introgressed (Fig. 5A). Analysis of backcross and parental strains using genome-wide markers which discriminate between the parental genotypes should then be able to identify regions of the genome that have been introgressed from the resistant parental strain indicating the location of important ivermectin resistance-conferring loci.
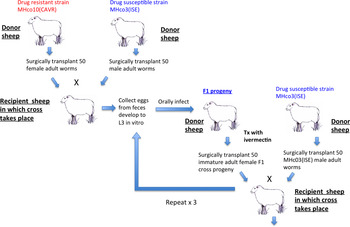
Fig. 4. Experimental design of the serial backcross. Crosses were performed by surgically transplanting immature adult female worms of an ivermectin resistant H. contortus strain, MHco10(CAVR), and immature adult male worms of the ivermectin susceptible reference strain, MHco3(ISE), into the abomasum of a recipient sheep. This methodology ensures that the only progeny produced are the product of a cross between the two different strains. After the first cross between the two parental strains, the F1 progeny of each cross is repeatedly backcrossed against the MHco3(ISE) susceptible reference strain using the same procedure.

Fig. 5. Genetic validation of the serial backcrossing procedure. (A) Diagrammatic representation the genetics of the serial backcross experiment. The parental strains are genetically divergent and so their genomes are represented by different colours; red for the MHco10(CAVR) ivermectin resistant strain and blue for the MHco3(ISE) susceptible reference strain. After four generations of serial backcrossing against the MHco3(ISE) strain, in the presence of ivermectin selection, regions of the MHco10(CAVR) genome containing ivermectin resistance loci have been introgressed into a predominantly MHco3(ISE) genetic background; (B and C): Genetic data validating the success of the serial backcross. These figures show the genetic validation of the of the serial backcross of the MHco10(CAVR) strain against the MHco3(ISE) susceptible strain. Thirty worms from each of the two parental strains, thirty worms from the fourth generation backcross progeny (no drug treatment) and thirty worms of the fourth generation backcross progeny that survived 0·1 mg/kg ivermectin treatment were genotyped with a panel of neutral microsatellite markers. Figure 5B shows pairwise FST estimates. Values below the diagonal are from the analysis of nine microsatellite loci and above the diagonal for eight microsatellite loci (when marker Hcms8a20 was excluded since it showed evidence of genetic linkage to the ivermectin resistance phenotype). Values showing genetic differentiation between strains at significance level P⩽0·01 are shown in red. The key results are circled that demonstrate the fourth generation backcross worms which survived ivermectin treatment are much more genetically similar to the susceptible MHco3(ISE) parental strain than to the resistant MHco10(CAVR) parental strain. Figure 5C shows the PCA analysis where each data point represents a single worm based on a multi-locus genotype of nine microsatellite markers. Note how the fourth generation backcross worms (with an ivermectin resistant phenotype) cluster with worms the MHco3(ISE) susceptible parental strain rather than with worms from the MHco10(CAVR) ivermectin resistant parental. These results show that fourth generation backcross worms with an ivermectin resistant phenotype have a genetic background predominantly from the MHco3(ISE) ivermectin susceptible parental strain.
A panel of 18 microsatellite loci (each with very different allelic profiles in the three parental strains) were used to monitor the procedure at each backcross generation using a genetic fingerprinting approach applied to bulk DNA preparations made from populations of worms (Redman et al. Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). The fourth generation backcross populations were then characterized in more detail by genotyping individual worms (>30 worms per population) with a sub-panel of 9 microsatellite markers (Redman et al. Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). This analysis clearly demonstrated that the worms from the fourth generation backcross that were phenotypically resistant to ivermectin (i.e. survived 0·1 or 0·2 mg/kg in vivo ivermectin treatment of the host) but had a genetic background very similar to the susceptible MHco3(ISE) parental strain and very different from the resistant parental strain (Figs. 5B and C). This analysis confirmed the success of the two backcrossing experiments and showed that ivermectin-resistant loci had been introgressed into the MHco3(ISE) susceptible genetic background from each of the resistant parental strains MHco4(WRS) and MHco10(CAVR).
Of the 18 microsatellite markers that were used to monitor the backcrossing procedure, there was only one, Hcms8a20, in which alleles specific to the resistant parental strain were retained in the fourth generation backcross progeny. Furthermore, these Hcms8a20 ‘retained alleles’ were present at a much higher frequency in phenotypically resistant backcross worms compared with unselected backcross worms. This was the case for both independent backcrosses; MHco4(WRS)/MHco3(ISE) and MHco10(CAVR)/MHco3(ISE), respectively (Redman et al. Reference Redman, Sargison, Whitelaw, Jackson, Morrison, Bartley and Gilleard2012). This represents strong evidence that the Hcms8a20 marker is genetically linked to a resistance-conferring polymorphism in both the MHco4(WRS) and MHco10(CAVR) parental strains.
We have recently undertaken deep sequencing of the parental and backcross strains; approximately 700–900 Mb of sequence per strain as 100 bp paired end reads, sequenced on Illumina Hiseq 2000 (J. Cotton, M. Berrimanand J. S. Gilleard, unpublished data). A preliminary genome-wide analysis of strain-specific SNPs confirms the microsatellite results; for most contigs in the genome assembly, the allelic profile generated from ivermectin resistant fourth generation backcross worms is similar to that of the MHco3(ISE) susceptible parental strain with a minority of contigs – about 10–20% – having an allelic profile distinct from thr MHco3(ISE) strain (A. Martinelli, MSc thesis, University of Edinburgh). This latter group, represents a set of genomic contigs with preliminary evidence of being located in genomic regions that have been introgressed into the MHco3(ISE) genetic background from the resistant parental strain. Consistent with the microsatellite analysis, the contig containing microsatellite Hcms8A20 is included in this group of contigs (A. Martinelli, MSc thesis, University of Edinburgh). We are currently undertaking further analysis of these data using the recently improved genome assembly to more accurately delineate the introgressed region(s). In addition, we have also passaged the fourth generation backcross populations through a further four generations of experimental infections with ivermectin selection (R. Laing, E. Devaney, D. Bartleyand J. S. Gilleard, unpublished data). Our hypothesis is that recombination should reduce the size of the introgressed region(s) at each generation and so interrogation of the resulting material should allow finer scale localization of the resistance-conferring loci.
FUTURE DIRECTIONS
The current revolution in sequencing and genotyping technologies, together with improving genomic resources, makes genetic approaches to identify drug resistance loci in H. contortus an increasingly realistic proposition. Approaches such as ‘bulk segregant analysis’ (Michelmore et al. Reference Michelmore, Paran and Kesseli1991; Ehrenreich et al. Reference Ehrenreich, Torabi, Jia, Kent, Martis, Shapiro, Gresham, Caudy and Kruglyak2010) or ‘linkage group selection’ (Culleton et al. Reference Culleton, Martinelli, Hunt and Carter2005; Borges et al. Reference Borges, Cravo, Creasey, Fawcett, Modrzynska, Rodrigues, Martinelli and Hunt2011) now allow QTL mapping to be undertaken using data generated from pooled DNA samples rather than individual DNA samples. This greatly increases the speed and scale with which mapping approaches can be applied, especially when combined with the new high-throughput genome sequencing methodologies. Furthermore, the cost of these approaches is rapidly decreasing making them increasingly affordable for many laboratories. However, as genomic technologies and resources improve, the limiting factor to the genetic mapping of anthelmintic resistance loci will be the practical challenges of undertaking genetic crosses in parasites. H. contortus is the nematode for which genetic crossing procedures are most established and practical in the strongylid nematode group. Nevertheless, these procedures are time consuming, labour intensive and require significant infrastructure and parasitological expertise. Consequently, the most feasible strategy will be to undertake a small number of carefully planned genetic crosses that can then be iteratively interrogated with the ever improving genomic resources. This has been a highly successful approach for studies on antimalarial resistance in P. falciparum, a parasite with even more practical challenges for undertaking genetic crosses than H. contortus (Anderson et al. Reference Anderson, Nkhoma, Ecker and Fidock2011). Just three genetic crosses have been performed with P. falciparum to date but the availability of excellent genomic resources has allowed each of these to be intensively analysed. A single genetic cross between the genetically divergent Dd2 (South East Asia) and HB3 (Central America) strains has led to the mapping and identification of mutations for a wide range of drug-resistant phenotypes (chloroquine, sulfadoxine, endogenous folate utilization and amodiaquine) (Anderson et al. Reference Anderson, Nkhoma, Ecker and Fidock2011). This provides an excellent paradigm for the application of the rapidly improving H. contortus genomic resources to forward genetic analysis and mapping.
Another approach that holds great promise for the identification of anthelmintic resistance loci is the application of GWAS to parasite populations in the field. This approach relies on the following principle; as drug selection increases the frequency of resistance mutations in parasite populations, the flanking neutral polymorphisms in the genome ‘hitchhike’ to high frequency as well. This can lead to reduced variation and increased linkage disequilibrium around resistance loci. The GWAS approach essentially searches the genome for these ‘footprints’ of selection by genotyping polymorphic marker loci at intervals throughout the genome and using them to identify locations of the drug resistance loci. These approaches are being applied to study the genetic basis of drug resistance in an increasing number of organisms including Plasmodium parasites and Anopheles mosquitoes (Jones et al. Reference Jones, Liyanapathirana, Agossa, Weetman, Ranson, Donnelly and Wilding2012; Volkman et al. Reference Volkman, Neafsey, Schaffner, Park and Wirth2012). Amongst the parasitic nematodes, H. contortus is well placed to apply GWAS approaches due to the availability of well-characterized resistant field populations and our increasing understanding its population genetic structure (Troell et al. Reference Troell, Engstrom, Morrison, Mattsson and Hoglund2006; Gilleard and Beech, Reference Gilleard and Beech2007). Nevertheless, we need to understand much more about the population genetics of this parasite and its response to selection before we can understand how to apply GWAS approaches in an appropriate manner. This issue has been previously reviewed in some detail and so will not be covered further here (Gilleard and Beech, Reference Gilleard and Beech2007).
In summary, H. contortus has already contributed a huge amount to our understanding of anthelmintic resistance both at the molecular and population levels. In this review, the case has been made for its further development as a model system for both forward genetic mapping and the application of GWAS approaches to elucidate the molecular genetic basis of anthelmintic resistance and to study the evolution and spread of resistance in the field. These approaches are highly dependent on the availability of a well-finished reference genome sequence. Consequently, there is a need for the quality of the genome resources for H. contortus to be improved beyond that which is currently typical for draft genomes of non-model organisms. If this is achieved, we can expect the next few years to yield major progress in our understanding of the basic processes by which parasitic nematodes become resistant to anthelmintic drugs.
ACKNOWLEDGEMENTS
The previous and on going research that underlies this review is very much a team effort. In particular, Prof Neil Sargison (University of Edinburgh), Dr Elizabeth Redman (University of Calgary), Dr Dave Bartley and Dr Frank Jackson (Moredun Research Institute) have been instrumental in developing and applying the methodologies around H. contortus genetics. Similarly, the sequencing of the H. contortus genome has been led by Dr Matt Berriman and Dr James Cotton of the Parasite Genomics Group at the Sanger Genome Institute. Prof Eileen Devaney and Dr Roz Laing (University of Glasgow) also continue to provide invaluable input and support to this work. I am also grateful to Dr Dave Bartley and Neil Sargison for some of the images used in Figs 1 and 3 and also to Dr Roz Liang for the artwork in Fig. 4.





