INTRODUCTION
Many marine invertebrates have planktonic larvae which are dispersed by ocean currents (McEdward, Reference McEdward1995). These species are likely characterized by low levels of genetic differentiation (i.e. high homogeneity) among populations over broad spatial scales, but it has become apparent that significant genetic differentiation does exist among different populations in some species (see Palumbi, Reference Palumbi2003 for a review). This could be linked to a restricted larval dispersal ability (Tzeng et al., Reference Tzeng, Yeh and Hui2004), mostly by selection (Hedgecock, Reference Hedgecock1986) or behavioural differences (Burton & Feldman, Reference Burton, Feldman and Kennedy1982), and environmental factors, such as oceanic circulation patterns and hydrographic conditions, which create physical barriers and subsequently limit gene flow (Palumbi, Reference Palumbi1994; Perrin et al., Reference Perrin, Wing and Roy2004). Studies on the relative importance of these factors on population genetic structure are crucial to our understanding of the impact of environmental changes on the ecological functioning of different populations, as well as the management of marine resources (Palumbi, Reference Palumbi2003). In particular, the significance of intraspecific genetic diversity, including diversity among and within populations, has been emphasized with a view to conserving genetic resources (Kenchington et al., Reference Kenchington, Heino and Nielsen2003).
Stomatopods are widely distributed in both temperate and tropical regions (Ahyong, Reference Ahyong2001), and the planktonic stage of their larvae can last for 2–3 months (Provenzano & Manning, Reference Provenzano and Manning1978; Morgan & Goy, Reference Morgan and Goy1987). Whilst the dispersal potential of stomatopod larvae is expected to be high (Barber et al., Reference Barber, Moosa and Palumbi2002a), Reaka & Manning (Reference Reaka and Manning1981) believed that such larvae do not appear to disperse over long distances as reflected by their limited geographical ranges. Haptosquilla glyptocercus, for example, showed larval dispersal and recruitment over large spatio-temporal scales, resulting in high genetic diversity (Barber et al., Reference Barber, Moosa and Palumbi2002a). Conversely, Barber et al. (Reference Barber, Palumbi, Erdmann and Moosa2002b) used genetic techniques to infer dispersal history among populations of the benthic reef stomatopod, Haptosquilla pulchella, and detected significant genetic differentiation over relatively short distances (300 km) in the Indo-Pacific, despite an estimated long planktonic larval period (4–6 weeks). These aforementioned studies suggest that larval dispersal scales and genetic variation among populations are likely to be species specific in the Stomatopoda. However, with limited data, the genetic structure and larval dispersal potential of many stomatopods remains largely unknown.
Oratosquilla oratoria (Squillidae) is a common, commercially important, stomatopod in the Indo-West Pacific region (Ahyong, Reference Ahyong2001). A recent field study showed that it is one of the most abundant benthic crustaceans in Hong Kong waters (Lui et al., Reference Lui, Ng and Leung2007). In Hong Kong, the abundance of O. oratoria in western waters was consistently higher than in eastern waters, i.e. ~4-fold difference in average monthly abundance. The reduced abundance in eastern waters could be ascribed to episodic hypoxia events during August and October (Lui, Reference Lui2005). Laboratory-reared larvae of O. oratoria have a planktonic stage of between 36 and 59 days (Hamano & Matsuura, Reference Hamano and Matsuura1987). Larvae may be able to disperse over great distances during such a relatively long planktonic period and, thus, the degree of genetic differentiation among populations is expected to be low. There is, however, a lack of field-based population genetic studies to verify this postulation. Sequencing analysis of mitochondrial (mt) DNA is one of the most common methods to detect genetic differentiation (Bagley & Gall, Reference Bagley and Gall1998; Refseth et al., Reference Refseth, Nesbø, Stacy, Vøllestad, Fjeld and Jakobsen1998), and the mtDNA control region with a generally high rate of base substitutions makes it a suitable marker for studies of intraspecific population genetics (Meyer, Reference Meyer and Beaumont1994). The present study aimed to examine the genetic structure within and among six samples of O. oratoria in Hong Kong waters, using mtDNA control region sequences.
MATERIALS AND METHODS
Sample collection
Individuals of Oratosquilla oratoria were collected from the eastern (two stations in Tolo Harbour, E1 and E2, N = 34) and western (two stations off southern Lantau Island, W1 and W2, N = 35) waters of Hong Kong using a commercial shrimp trawler (beam size: 1.5 m; stretched mesh size: 2 cm), from June to July 2004 (Figure 1). Oratosquilla oratoria individuals were also collected from two locations in Hong Kong's surrounding waters (N = 30), i.e. near Neilingding Island (China-West, CW) and Dalu Wan (China-East, CE) (Figure 1). Samples were kept alive in the aquarium before DNA extraction.

Fig. 1. Sampling locations for Oratosquilla oratoria in Hong Kong and surrounding waters: eastern waters: E1, E2 and CE; western waters: W1, W2 and CW.
DNA extraction
Approximately 100 mg muscle tissues were dissected from the fifth abdominal somite of each individual. Tissues were washed with sterile distilled water, cut into small pieces, and transferred to 1.5 ml centrifuge tubes containing 500 µl lysis buffer (100 mM Tris–HCL, pH 8.0, 20 mM EDTA, 1.4 M NaCl, 2.0% hexadecyltrimethylammonium bromide (CTAB)). Each tissue sample was incubated and digested overnight at 55°C with 45 µl proteinase K (1 mg ml−1), and the total genomic DNA was then extracted using the phenol-chloroform extraction method (Hoelzel & Green, Reference Hoelzel, Green and Hoelzel1992). The extracted DNA pellets were resuspended in 50 µl TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, pH 8.0) and stored at 4°C before DNA amplification.
DNA amplification and sequencing
A DNA segment of ~1100-base pairs (bp; including the control region) was amplified by polymerase chain reaction (PCR) using a pair of primers, CR-F1 (5′–TAACCGCGACTGCTGGCAC–3′) and CR-F2 (5′–GGGTATGAGCCCATTAGCTT–3′) which were designed from the corresponding position of 13298 and 14772, respectively, of the blue crab Portunus trituberculatus mt sequence (from GenBank—Accession number: AB093006). PCR amplifications were carried out in a PTC-100 Thermocycler (MJ Research, USA). Thirty amplification cycles, each consisting of an initial DNA denaturation step of 2 minutes at 94°C, denaturation at 94°C for 1 minute, annealing at 56°C for 45 seconds, and extension at 72°C for 75 seconds, were performed. A 5 minute extension at 72°C was included in the final cycle. Reagents (50 µl in volume) contained 20 ng of template DNA, 1 unit of Taq DNA polymerase (InvitrogenTM Life Technologies Co.), 0.5 µM of each primer, 0.2 mM of each dNTPs (dATP, dCTP, dGTP and dTTP), 1.5 mM MgCl2 and 1×PCR buffer (200 mM Tris-HCl, pH 8.4, 500 mM KCl).
An aliquot of 2 µl of each amplification was electrophoresed on a 1.5% agarose gel to check for the correct fragment size, and to ensure only a single amplification product was obtained. PCR products were purified using the GFX PCR purification kit (GE Healthcare Bioscience, USA), following procedures outlined by the manufacturer. Cycle sequencings of the PCR product (~1100 bp) were performed using the sequencing BigDye® Terminator V 3.1 Sequencing Standard Kit (Applied Biosystems, USA), and conducted using the same pair of forward and reverse primers applied in the initial PCR reaction. The sequencing PCR products were then purified using AutoSeq 96TM Plate (GE Healthcare Bioscience, USA). Purified products were analysed by capillary electrophoresis using an Applied Biosystems Model 3730 automated DNA analyser (Perkin-Elmer, USA), in the Genome Research Centre of the University of Hong Kong.
Data analysis
Chromatograms obtained from the automated analyser were read, and sequences were assembled using the sequence editing software SeqMan (DNASTAR Inc., Denmark). The resulting consensus sequences were aligned using Mega 3.1 (Kumur et al., Reference Kumur, Tamura and Nei2004). Haplotype diversity (h; Nei, Reference Nei1987) and nucleotide diversity (π; Tajima, Reference Tajima1983) were calculated using ARLEQUIN v. 3.1 (Excoffier et al., Reference Excoffier, Laval and Schneider2005). The same program was used to calculate genetic differentiation between the six samples using the pairwise F-statistics (Wright, Reference Wright1951) by performing 10,100 permutations.
Analysis of molecular variance (AMOVA) was performed using ARLEQUIN to examine hierarchical population structure by pooling the samples into two different groupings: (1) eastern (EW: CE, E1 and E2) and western (WW: CW, W1 and W2) waters; and (2) open-waters (OW: CE, CW) and sheltered bays (SB: W1, W2, E1 and E2). Sixteen thousand permutations were executed to guarantee <1% difference with the exact probability in 99% of cases (Guo & Thompson, Reference Guo and Thompson1992). Relationships among haplotypes were analysed in a parsimony network (TCS v. 1.12; Clement et al., Reference Clement, Posada and Crandall2000), using the statistical parsimony procedure (Crandall et al., Reference Crandall, Templeton, Sing, Scotland, Siebert and Williams1994). This method estimates the unrooted tree and provides a 95% plausible set for all sequence type linkages within the unrooted tree. A minimum spanning network tree was then produced. The relationship between geographical distance, which was estimated as the shortest pelagic distance between pairs of samples, and the pairwise F ST values was tested by Mantel's test for permutation procedure (ARLEQUIN v. 3.1).
RESULTS
Genetic diversity
Sequence analysis of a 923 bp region of the mtDNA control region showed there to be 27 unique haplotypes in the 99 Oratosquilla oratoria individuals, with a total of 14 polymorphic sites (Table 1). Haplotype (Hap) 1 of the sequences has been deposited in the GenBank database (Accession number: DQ120060). The haplotype diversity (h) ranged from 0 to 0.952 (Table 2). An overall high haplotype diversity was recorded (mean h = 0.886; Table 2). Across all six stomatopod samples, those from eastern waters showed high to moderate h values (0.588 to 0.952) with the number of haplotypes ranging from 3 to 6. However, extremely high and low h values were obtained for western waters. A high h (0.949) with sixteen haplotypes was recorded in the CW sample while only a single haplotype (Hap 27) with h = 0.000 was found in W1. Nucleotide diversity (π) ranged from 0 to 0.0035, although the within site value was quite low (mean π = 0.0026; Table 1). Relationships between haplotypes were shown on a haplotype minimum spanning network (MSN; Figure 2). The MSN displayed a star-like genealogy dominated by a few haplotypes connecting to other haplotypes by few mutations (Figure 2). The most common haplotype, Hap 21, was shared by a total of 24 individuals from E1, E2 and W2. No obvious pattern with haplotypes mixing from the eastern and western waters within the MSN was observed (Figure 2A). Total haplotype numbers from the eastern and western waters were 13 and 18, respectively. Among these, five of the haplotypes were shared.

Fig. 2. Minimum spanning network (MSN) of the mtDNA control region sequences based on the grouping of sites from (A), the six locations and (B), the open-waters (OW) and sheltered bays (SB). Each haplotype is defined by its corresponding number. The area of each circle is proportional to the frequency of that haplotype in the total sample. Shared haplotypes among sampling locations were represented as frequency diagrams. Substitution between haplotypes is represented by a line. Hypothetical missing haplotypes are represented as small filled circles.
Table 1. Variable sites of the 27 haplotypes and their frequencies in each sample of Oratosquilla oratoria. Dots represent the same base as haplotype 1.

Table 2. Genetic diversity of Oratosquilla oratoria from the six samples from Hong Kong and its surrounding waters.

When samples were classified into two groups, i.e. sheltered bays (SB) and open-waters (OW), Oratosquilla oratoria samples from the former had a relatively lower haplotype number and nucleotide diversity (range: h = 0–0.684, π = 0–0.0012) than those from the latter (range: h = 0.949–0.952; π = 0.0033–0.035; Table 2). Among the 27 haplotypes, samples from SB shared 9 haplotypes whilst those of OW shared 20 haplotypes (Figure 2B). Only two haplotypes (Hap 4 and Hap 15) overlapped between the two groups. Within the OW group, although the two samples (CE and CW) possessed high haplotype diversity, only two haplotypes (Hap 1 and Hap 5) were shared (Figure 2A).
Genetic differentiation within and among samples
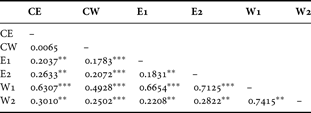
A significant genetic differentiation was detected between each pair of the six samples, except between the pair of CE and CW (pairwise F ST = 0.007, P > 0.05; Table 3). The highest pairwise F ST was found between W1 and W2, despite the fact that these regions were geographically close. Pairwise F ST values were, in general, higher between the two western SB samples (W1 and W2) and other samples (Table 3). The Mantel test showed that there was no significant correlation between linearized F ST and hydrographic distances between different samples (r =−0.078, P > 0.05; Figure 3).

Fig. 3. A plot of pairwise F ST between samples of Oratosquilla oratoria from different regions against their pairwise geographical distances. Letters next to each symbol represent the pairwise relationship of samples from the specified regions.
Table 3. Pairwise F ST values between samples of Oratosquilla oratoria from different regions. Significant differences between pairs, after 10,100 random permutations, are indicated by asterisk(s): **P < 0.01; ***P < 0.001.
Samples from the six localities were analysed using AMOVA by dividing them into groups according to hydrographic conditions: i.e. (1), eastern and western waters (EW and WW) and (2), open-waters and sheltered bays (OW and SB) as described above. Such partitioning did not reveal significant differentiation among the groups (Table 4). Instead, high and significant variation was present among samples within groups (38.8% and 39.8%, both P < 0.001) and within samples (63.6% and 64.2%, both P < 0.001).
Table 4. Analysis of molecular variance comparing all six samples of Oratosquilla oratoria based on the grouping of the six samples from: (1) eastern, EW (CE, E1 and E2) and western waters, WW (CW, W1 and W2); and (2) open-water, OW (CE and CW) and sheltered bay, SB (W1, W2, E1 and E2). Significant sources of variation, after 16,000 random permutations, are indicated by asterisk(s): ***P < 0.001.

DISCUSSION
In the present study, Oratosquilla oratoria generally displayed a meso-scale genetic homogeneity but had a micro-scale genetic divergence between samples collected from across Hong Kong waters. Apart from the fine-scale genetic patchiness, a larger difference in genetic diversity of O. oratoria was observed in samples obtained from open-waters than those obtained from sheltered bay sites, with a higher diversity recorded in the former.
Oratosquilla oratoria has a long planktonic larval phase (>1 month; Hamano & Matsuura, Reference Hamano and Matsuura1987), and the extensive transport and exchange of larvae across the South China Sea could be promoted by the Hainan Current (in summer) and the Kuroshio Current (in winter) (Morton & Morton, Reference Morton and Morton1983). Substantial larval dispersal between large-scale locations is expected and gene flow across Hong Kong and its surrounding waters within the South China Sea, therefore, does not appear to be restricted. As our findings demonstrated, a broad scale genetic homogeneity was observed (~100 km). Such low levels of genetic differentiation over a geographical scale have also been described for other stomatopods (Barber et al., Reference Barber, Moosa and Palumbi2002a; Lindstrom, Reference Lindstrom2003). Similar to O. oratoria, the coral Platygyra sinensis with planktonic larvae also exhibited a relatively homogeneous genetic structure across Hong Kong waters (Ng & Morton, Reference Ng and Morton2003).
On the other hand, an unexpected fine-scale, un-patterned genetic variability was recorded, and an extremely low haplotype diversity in Oratosquilla oratoria was observed in the two sheltered sites from western waters of Hong Kong. A small scale genetic variation was also reported for the stalked barnacle Ibla cumingi in Hong Kong waters, being attributed to the combined effect of a short lecithotrophic larval period and local hydrographic characters (Leung, Reference Leung2002). Those marine organisms that demonstrate substantial larval dispersal with large-scale genetic homogeneity, but have genetic heterogeneity among populations over a small spatial scale, have been identified as showing ‘chaotic genetic patchiness’ (Johnson & Black, Reference Johnson and Black1982; Watts et al., Reference Watts, Johnson and Black1990). Such genetic patchiness could be explained by several hypotheses. Firstly, it may relate to a ‘sweepstakes’-pattern of reproduction (Hellberg et al., Reference Hellberg, Burton, Neigel and Palumbi2002; Hedgecock et al., Reference Hedgecock, Launey, Pudovkin, Naciri, Lapègue and Bonhomme2007). Many high fecundity and free-spawning marine species, including O. oratoria, could experience extremely large variance in individual reproductive success when sweepstakes-chance reproductive activity matches oceanographic conditions (Li & Hedgecock, Reference Li and Hedgecock1998; Lundy et al., Reference Lundy, Rico and Hewitt2000). In such a case, the successful offspring of a population may be randomly contributed to by a limited number of reproductive adults, and the resulting low effective number of offspring may lead to the occurrence of small-scale patchy genetic structures. It has been reported that O. oratoria demonstrates high reproductive variability in Hong Kong waters (Lui, Reference Lui2005). For instance, the key local recruitment of O. oratoria first occurs during the summer months of May–September in western waters with a minor recruitment pulse in November. Conversely, eastern water individuals were mainly recruited during winter months from January–March with a minor summer recruitment in July (Lui, Reference Lui2005). During the monsoon rainy summer period (May–August); a lower percentage occurrence of ovigerous females was consistently observed in western waters than eastern ones and this might be attributed to low salinities (average salinity: ~10 to 20 ppt; Yin & Harrison, Reference Yin and Harrison2007) in the former due to the summer discharge of freshwater from the Pearl River. Under such harsh conditions, larvae of O. oratoria might suffer from hypo-osmotic stress and be restrained by the physical barrier of salinity stratification. If so, this may support the sweepstakes' concept.
Larval movement can be greatly influenced by variations in environmental variables such as current direction and hydrographic barriers, causing genetic differentiation among recruits (Hedgecock, Reference Hedgecock1986; Kordos & Burton, Reference Kordos and Burton1993; Morgan, Reference Morgan and McEdward1995). Although Oratosquilla oratoria larvae have high dispersal potential, freshwater runoff from the Pearl River Estuary in western Hong Kong may form a barrier to their dispersal and western SB sites (W1 and W2) may thus become a local retention zone. Estuarine circulation is known to act as a barrier to larval mixing (Roughgarden et al., Reference Roughgarden, Gaines and Possingham1988). The population genetic structures of the sea star, Coscinasterias muricata, for instance, were affected by freshwater influxes and distinctive estuarine circulations in New Zealand fiords (Perrin et al., Reference Perrin, Wing and Roy2004), and a similar pattern has also been observed in other estuarine organisms (amphipods, horseshoe crabs and polychaetes; Bilton et al., Reference Bilton, Paula and Bishop2002). Furthermore, larval dispersal profiles can be influenced by behavioural adaptations to oceanographic conditions (Young, Reference Young and McEdward1995), especially vertical migration (Dame & Allen, Reference Dame and Allen1996). Stomatopod larvae are strong swimmers and may not disperse as passive particles (Barber et al., Reference Barber, Palumbi, Erdmann and Moosa2002b). They also exhibit vertical migration (Reaka & Manning, Reference Reaka and Manning1981) which may further limit dispersal (Robichaux et al., Reference Robichaux, Cohen, Reaka and Allen1981). These aforementioned factors could concomitantly influence larval movement and shape the genetic patchiness pattern of O. oratoria in Hong Kong waters.
Both pre- and post-settlement mortalities may play roles in shaping the genetic structures of a marine species within a small spatial and/or temporal scale (Johnson & Black, Reference Johnson and Black1984; Johannesson et al., Reference Johannesson, Johannesson and Lundgren1995). Kodama et al. (Reference Kodama, Horiguchi, Kume, Nagayama, Shimizu, Shiraishi, Morita and Shimizu2006) reported that Oratosquilla oratoria juveniles in Tokyo Bay were sensitive to dissolved oxygen levels and thus massive mortalities occurred in shallow water areas when summer hypoxia events persisted. A similar large scale mortality may also happen in Hong Kong stomatopod settlers during summers when hypoxia events are more common, especially in the western waters (Yin et al., Reference Yin, Lin and Ke2004). Results of the present study, however, cannot conclude whether the observed genetic patchiness is a short-term pattern, i.e. when the unmixed portion of the population is not robust enough to sustain a lasting genetic structure (Selkoe et al., Reference Selkoe, Gaines, Caselle and Warner2006), or a long-term pattern, i.e. when genetic variations between cohorts accumulate over time (Maes et al., Reference Maes, Pujolar, Hellemans and Volckaert2006).
Haplotype sequences were closely related in all samples of Oratosquilla oratoria, and the minimum spanning network formed a star-shaped phylogeny around a few prevalent haplotypes. A star-shaped genealogy is often associated with demographic expansion of a single ancestral population (Slatkin & Hudson, Reference Slatkin and Hudson1991). High haplotype diversity but low nucleotide diversity was observed in samples of O. oratoria. This has also been recorded for other stomatopods (Barber et al., Reference Barber, Moosa and Palumbi2002a, Reference Barber, Palumbi, Erdmann and Moosab; Lindstrom, Reference Lindstrom2003) and echinoderms (Perrin et al., Reference Perrin, Wing and Roy2004), and is closely linked to recent, rapid, population expansion from a small, effective, ancestral population. When population size increases rapidly following environmental disturbances, haplotype diversity can usually recover via mutation, whereas nucleotide diversity remains low as there is not sufficient time to accumulate large sequence differences between haplotypes (Avise, Reference Avise2000).
The Hong Kong marine environment is highly disturbed in terms of pollution and fisheries activities such as trawling (Morton & Blackmore, Reference Morton and Blackmore2001; Leung, Reference Leung and Morton2003). Mantis shrimps including Oratosquilla oratoria are important commercial species and subject to high fishing pressures. It has been reported that severe overfishing could reduce the genetic diversity of marine species (Hauser et al., Reference Hauser, Adcock, Smith, Bernal Ramirez and Carvalho2002). A summer fishing moratorium (May–July) has been implemented in the South China Sea since 1999. Fish catch rate has consequently increased, and fish populations appear to be recovering in some of China's waters (AFCD, 2004). Hong Kong, however, has no fishing ban or other common fishery management tools such as a fishing quota system and regulations related to fishing net mesh size. While the problem of over-fishing persists, it is plausible that frequent bottom trawling activities could be one of the major driving forces leading to the observed fine-scale genetic patchiness of O. oratoria samples in Hong Kong waters.
In conclusion, this mtDNA sequence analysis has successfully revealed the first population genetic structure study of one of the most commercially important stomatopods, Oratosquilla oratoria, in Hong Kong waters. Genetic analysis has demonstrated an expected homogeneity of genetic structure in meso-scale distance, but revealed a high genetic variation between samples with a patchiness pattern at the fine-scale. Moreover, an extreme genetic diversity was found among different samples. The patchy patterns could be explained by the high variability in reproductive success of O. oratoria, locally dynamic hydrographic conditions and high post-settlement mortality in Hong Kong waters.
ACKNOWLEDGEMENTS
This research was partially supported by a seed grant from the University of Hong Kong (Project No. 1020-5213) to K.M.Y.L. The authors would like to thank Dr Jasmine Ng for her useful comments on the manuscript. The staff, undergraduates and postgraduates in the Division of Ecology & Biodiversity, School of Biological Sciences, University of Hong Kong are thanked for their help with field sampling.